

Key Points
-
•
Deleterious germ line predisposition variants are shared in patients with MDS and their related donors and occur at all ages.
-
•
Based on the significant frequency of germ line variants in MDS, genetic testing is recommended for all patients.
Visual Abstract
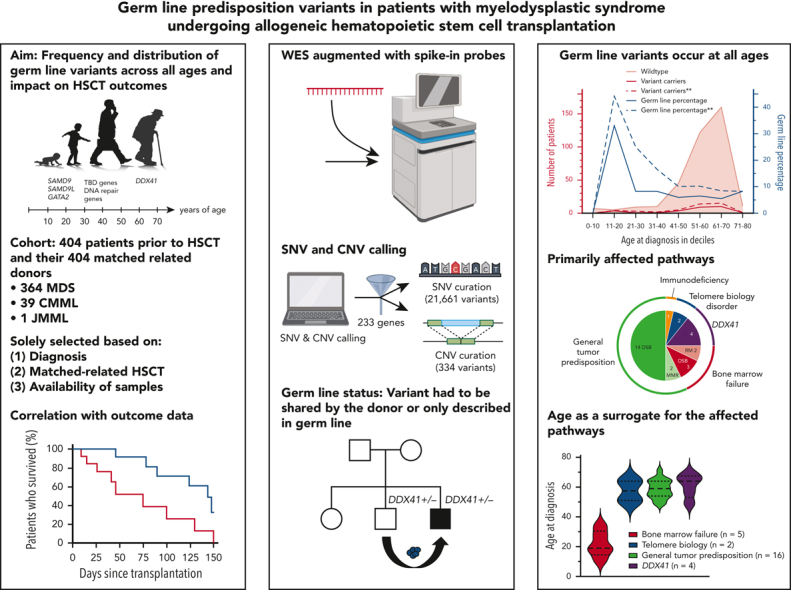
Abstract
The frequency of pathogenic/likely pathogenic (P/LP) germ line variants in patients with myelodysplastic syndrome (MDS) diagnosed at age 40 years or less is 15% to 20%. However, there are no comprehensive studies assessing the frequency of such variants across the age spectrum. We performed augmented whole-exome sequencing of peripheral blood samples from 404 patients with MDS and their related donors before allogeneic hematopoietic stem cell transplantation. Single-nucleotide and copy number variants in 233 genes were analyzed and interpreted. Germ line status was established by the presence of a variant in the patient and related donor or for those seen previously only as germ line alleles. We identified P/LP germ line variants in 28 of 404 patients with MDS (7%), present within all age deciles. Patients with P/LP variants were more likely to develop higher-grade MDS than those without (43% vs 25%; P = .04). There was no statistically significant difference in outcome parameters between patients with and without a germ line variant, but the analysis was underpowered. P/LP variants in bone marrow failure syndrome genes were found in 5 patients aged less than 40 years, whereas variants in DDX41 (n = 4), telomere biology disorder genes (n = 2), and general tumor predisposition genes (n = 17) were found in patients aged more than 40 years. If presumed germ line variants were included, the yield of P/LP variants would increase to 11%, and by adding suspicious variants of unknown significance, it would rise further to 12%. The high frequency of P/LP germ line variants in our study supports comprehensive germ line genetic testing for all patients with MDS regardless of their age at diagnosis.
Widespread adoption of next-generation sequencing in myeloid malignancies is revealing the importance of inherited germ line predisposition disorders (GPDs). Feurstein and colleagues demonstrate that approximately 7% of patients with myelodysplastic dis-orders carry a pathogenic or likely pathogenic germ line variant associated with GPDs. This surprisingly high frequency has implications as to when germ line testing should be considered in this disease.
Introduction
Pathogenic/likely pathogenic (P/LP) germ line variants are recognized increasingly as driving hematopoietic malignancies,1 and testing for germ line susceptibility to myeloid malignancies is now included within the 2016 revision of the World Health Organization (WHO) classification of myeloid malignancies2 as well as in clinical guidelines such as those provided by the National Comprehensive Cancer Network and the European LeukemiaNet.3,4 Numerous clinical criteria are used to prioritize individuals for germ line predisposition testing, including a positive personal and family history, and diagnosis of aplastic anemia (AA), bone marrow failure (BMF), or myelodysplastic syndrome (MDS) at age 40 years or less.5, 6, 7, 8
Studies examining the germ line predisposition alleles present in individuals diagnosed with AA/MDS at a particular age suggest that the age at presentation is a surrogate for the molecular pathway driving the disease. Testing of children and adolescents with MDS diagnosed at less than age 18 years indicated that 17% of cases were associated with deleterious variants in SAMD9 and SAMD9L, and 7% were being caused by variants in GATA2.10, 11, 9 Among 179 children and young adults with a likely inherited BMF based on physical abnormalities, family history, and/or consanguinity, or presentation at less than 2 years of age, 48% had a detectable germ line predisposition allele, most often in genes associated with BMF, telomere biology disorder (TBD), or DNA repair.8 Of patients with hereditary MDS and acute leukemia, 18% to 37% carry causative germ line variants.12, 13, 14 Children and young adults up to age 45 years undergoing hematopoietic stem cell transplantation (HSCT) and patients with AA/MDS diagnosed between ages 18 and 40 years carry a disease-causing germ line variant in 13% to 19% of all MDS cases and 5% to 13% of all AA cases, mostly in genes involved in DNA repair and TBD,5 a finding that was independently verified by the largest case series of families with hereditary MDS/acute myeloid leukemia (AML).15 Contrary to the widely held notion that individuals with germ line predisposition variants always present at a young age, germ line variants in DDX41 are associated with a median age at onset of 65 to 69 years,16, 17, 18 which is similar to the age at onset seen in sporadic MDS cases. These prior studies highlight the fact that germ line predisposition to AA/MDS is much more common than previously thought. However, prior work focused specifically on younger age ranges or on subsets of patients with a positive family or personal medical history.
Thus, the frequency of germ line predisposition to MDS across all ages remains unknown. Moreover, HSCT is currently the only potential cure for MDS and is routinely performed in younger patients, with a preference for related donors. Identification of an underlying germ line predisposition syndrome is paramount before considering related donors. There are no comprehensive studies addressing the presence of deleterious germ line variants in donors, or the risk and consequences of reintroduction of the causative variant through HSCT. Outcomes of such transplants are reported to date only through case reports and suggest failure or delay in engraftment,19, 20, 21 poor immune function post-HSCT,19, 20, 21 early relapse,21 donor-derived leukemias in recipients22, 23, 24, 25, 26, 27 as well as poor HSC mobilization by the donor,19,26,28 and development of leukemia in the donor after stem cell mobilization using granulocyte colony-stimulating factor.23,27 To address the gap in knowledge regarding the frequency of deleterious germ line variants in patients of all ages with MDS, we used data provided by a large cohort from the Center for International Blood and Marrow Transplant Research (CIBMTR) comprising 404 patients with MDS who spanned the age spectrum and who received allogeneic HSCs from their related donors.
Methods
CIBMTR cohort
Our CIBMTR cohort was assembled from 364 patients with MDS, 39 with chronic myelomonocytic leukemia, and 1 with juvenile myelomonocytic leukemia who underwent a first related HSCT between 2008 and 2016 and for whom peripheral blood samples were available from them and their donors, for a total of 808 participants. The patients/donors were selected solely on the basis of (1) diagnosis, (2) matched-related HSCT, and (3) the availability of samples from the MDS/HSCT recipient and his/her related donor. All participants had given informed consent to contribute to the CIBMTR database and sample repository. Peripheral blood samples were collected from patients with MDS before conditioning and from their donors at the time of HSC collection.
Augmented whole-exome sequencing
Details regarding DNA sequencing are available in the supplemental Methods on the Blood website, and the overall workflow is illustrated in Figure 1. DNA was extracted from peripheral blood samples stored at −80°C, using a QIAcube robotic workstation (Qiagen, Hilden, Germany). Whole-exome sequencing (WES) was augmented with custom-designed spike-in probes covering noncoding regions known to contain inherited risk alleles, including the following: the 5′ untranslated region (UTR) of ANKRD26 (NM_014915), the 5′ UTR of DKC1 (NM_001363), intron 31 of FANCI (NM_001113378), intron 4 of GATA2 (NM_032638), and the entire RNA gene TERC (NR_001566). Sequencing was performed at the Yale Center for Genome Analysis at 40× coverage. Variant calling and annotation were performed with an in-house custom pipeline (https://github.com/LucyGodley/Pipeline/tree/main/Variant_Calling/WES/hg).
Figure 1.

Work flow diagram for the identification of P/LP germ line variants. DNA was extracted from thawed peripheral blood from 404 patients with MDS and was sequenced using an augmented whole-exome sequencing platform. SNVs and CNVs were called in 233 genes, using custom bioinformatic pipelines. A total of 21,661 SNVs and 334 CNVs were manually curated according to the ACMG/AMP and ClinGen SVI-WG guidelines. P/LP germ line variants were validated by Sanger sequencing, subcloning, and/or RT-qPCR. ACMG, American College of Medical Genetics; AMP, Association for Molecular Pathology; ClinGen SVI-WG, Clinical Genome Sequence Variant Interpretation Working Group; CNV, copy number variant; LP, likely pathogenic; MDS, myelodysplastic syndrome; P, pathogenic; RT-qPCR, quantitative real-time PCR; SNV, single-nucleotide variant; WES, whole-exome sequencing.
Variants in 233 genes associated with inherited hematopoietic malignancies, BMF syndromes, TBD, DNA repair deficiency, immunodeficiency, RASopathies, cancer predisposition syndromes, and congenital cytopenias were analyzed (supplemental Table 1). DNA from 9 of the patients was unavailable for augmented WES; however, sequencing data from a next-generation sequencing panel including 77 of these 233 genes (supplemental Table 1) were available for our use.29 Somatic variants in all 404 recipients and 25 donors sharing a germ line variant were assessed in an additional 167 genes known to be somatically mutated in MDS based on large MDS data sets from cBioPortal30 and the Catalogue of Somatic Mutations in Cancer31 (supplemental Table 2).
Variant interpretation
Details regarding variant calling, interpretation, validation, and the multiple-criteria decision analysis are available in the supplemental Methods. Briefly, germ line single-nucleotide variants (SNVs) and copy number variants (CNVs) were analyzed manually according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP), and Clinical Genome Sequence Variant Interpretation Working Group guidelines.32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48 P/LP germ line variants were validated by custom-designed polymerase chain reaction (PCR) amplification and Sanger sequencing. For diseases with recessive inheritance, subcloning of PCR amplicons/sequencing was done to confirm the variants were in trans. Validation of germ line CNVs was performed by quantitative real-time PCR.
Germ line status was determined by the presence of the identical P/LP variant within both the patients with MDS and their related donors, and/or variants with a variant allele frequency (VAF) of 40% to 60% seen previously only as germ line alleles,49 including DDX41 (NM_016222.4) p.Asp140Glyfs∗2, and p.Arg53Alafs∗16 (Figure 2). Presumed germ line status was determined for P/LP variants with a VAF within germ line range (0.4-0.6 or 1.0) that were not shared in the donor sample, with no other potential P/LP variants within germ line range in the same recipient, and a combination of the following criteria based on a multiple-criteria decision analysis: (1) presence and frequency of somatic variants in the gene of question; (2) previous report of the identical variant in the somatic setting; (3) additional somatic variants and their VAF; (4) reported predisposing condition; (5) MDS subtype with higher bone marrow blast count (>5%) or advanced disease; and (6) high pretest probability as has been described for genes, such as BRCA1/2, DDX41, GATA2, RUNX1, and others (supplemental Table 3).49, 50, 51, 52 The interpretation of somatic variants was conducted using the ACMG/AMP and American Society of Clinical Oncology guidelines.53
Figure 2.

Variant subgroups based on donor status. This schematic identifies subgroups of patients with MDS/HSCT recipients based on the presence or absence of a predisposing variant, the manner of inheritance, whether the variant is shared by the donor and would be disease-causing, as well as subgroups chosen for statistical analyses of outcome parameters. †Healthy carriers refer to recipients with a P/LP variant who received donor stem cells lacking an AD P/LP variant or cells with a heterozygous P/LP allele with an AR mode of inheritance. AD, autosomal dominant; AR, autosomal recessive; het, heterozygous; HSCT, hematopoietic stem cell transplantation; LP, likely pathogenic; MDS, myelodysplastic syndrome; mut, mutated; P, pathogenic; wt, wild-type.
Statistical analysis
Details regarding statistical analysis are given in the supplemental Methods. Briefly, baseline characteristics, including existence of a predisposing condition, age at diagnosis and HSCT, race, disease classification according to WHO criteria and revised International Prognostic Scoring System (IPSS-R),54 cytogenetic abnormalities, bone marrow cellularity, disease status at the time of HSCT, conditioning regimen, donor type, and mobilization parameters, were evaluated using row percentage, Fisher exact, and Kruskal-Wallis tests. HSCT outcomes analyzed included time to neutrophil engraftment and platelet recovery, primary graft failure, nonrelapse mortality, relapse, disease-free and overall survival, and grade of acute and chronic graft-versus-host disease (GVHD). Univariate outcomes were calculated by the Kaplan-Meier method, and cumulative incidence estimates were used to accommodate for competing risks. Univariable analyses for patient, disease, and transplant-related characteristics by recipient P/LP germ line variant status and donor status (affected, healthy, or heterozygotes) (Figure 2) were performed using the Pearson χ2 test or Fisher exact test for categorical variables. For continuous variables, the Kruskal-Wallis test was performed. Comparison of somatic patterns across different groups was carried out by odds ratio analysis.
Results
Patient series and baseline characteristics
The CIBMTR patients with MDS included 404 individuals diagnosed at a median age of 59 years (range, 11-75 years), and a predominantly male sex distribution (n = 248; 61%) (Table 1; supplemental Tables 4 and 5). Most patients were White (n = 357; 88%). MDS subtypes were classified according to the revised 2008 WHO classifications.55
Table 1.
Characteristics of patients with MDS undergoing related HSCT with and without P/LP germ line variants
| Characteristic | No P/LP variant | P/LP variant | P value |
|---|---|---|---|
| Patients, No. | 376 | 28 | |
| Median age at diagnosis, y (range) | 59 (1-75) | 57 (12-72) | .44∗ |
| Age at diagnosis, No. (%†) | |||
| 0-10 | 7 (100) | 0 (0) | |
| 11-20 | 6 (67) | 3 (33) | |
| 21-30 | 11 (92) | 1 (8) | |
| 31-40 | 11 (92) | 1 (8) | |
| 41-50 | 47 (94) | 3 (6) | |
| 51-60 | 127 (93) | 9 (7) | |
| 61-70 | 156 (94) | 10 (6) | |
| 71-80 | 11 (92) | 1 (8) | |
| Sex, No. (%) | .69‡ | ||
| Male | 232 (62) | 16 (57) | |
| Female | 144 (38) | 12 (43) | |
| Recipient race, No. (%) | .94§ | ||
| White | 330 (88) | 27 (96) | |
| Black or African American | 21 (6) | 1 (4) | |
| Asian | 14 (4) | 0 (0) | |
| Native Hawaiian or other Pacific Islander | 1 (0) | 0 (0) | |
| American Indian or Alaska Native | 3 (1) | 0 (0) | |
| Unknown | 7 (2) | 0 (0) | |
| Disease classification at diagnosis, No. (%) | .04§ | ||
| RA | 20 (5) | 2 (7) | |
| RARS | 16 (4) | 0 (0) | |
| RAEB-1 | 79 (21) | 2 (7) | |
| RAEB-2 | 93 (25) | 12 (43) | |
| RCMD | 68 (18) | 3 (11) | |
| RCMD-RS | 4 (1) | 0 (0) | |
| 5q– syndrome | 1 (0) | 2 (7) | |
| CMML | 37 (10) | 2 (7) | |
| JMML | 1 (0) | 0 (0) | |
| MDS, unclassifiable | 57 (15) | 5 (18) | |
| Bone marrow cellularity | .86§,∥ | ||
| Decreased (hypocellular) | 30 (8) | 3 (11) | |
| Normal | 51 (13) | 7 (25) | |
| Increased (hypercellular) | 127 (34) | 14 (50) | |
| Unknown | 168 (45) | 4 (14) | |
| Disease status before HSCT¶, No. (%) | .83§,∥ | ||
| Early | 88 (23) | 9 (32) | |
| Advanced | 187 (50) | 17 (61) | |
| Unknown | 101 (27) | 2 (7) | |
| Cytogenetics, No. (%) | .57§,∥ | ||
| Favorable | 134 (36) | 15 (54) | |
| Intermediate | 17 (5) | 1 (4) | |
| Poor | 28 (7) | 5 (18) | |
| Unknown | 197 (52) | 7 (25) | |
| IPSS-R, No. (%) | .61§,∥ | ||
| Very low | 8 (2) | 2 (7) | |
| Low | 46 (12) | 4 (14) | |
| Intermediate | 59 (16) | 9 (32) | |
| High | 46 (12) | 4 (14) | |
| Very high | 13 (3) | 2 (7) | |
| Unknown | 204 (54) | 7 (25) | |
| Predisposing condition, No. (%) | .044§,∥ | ||
| No predisposing condition | 252 (67) | 23 (82) | |
| Fanconi anemia | 0 (0) | 1 (4) | |
| Aplastic anemia | 6 (2) | 0 (0) | |
| Other | 8 (2) | 2 (7) | |
| Unknown | 110 (29) | 2 (8) | |
| Conditioning regimen intensity, No. (%) | .93§ | ||
| MAC | 203 (54) | 15 (54) | |
| RIC/NMA | 171 (45) | 13 (46) | |
| Unknown | 2 (1) | 0 (0) | |
| Graft source, No. (%) | .75‡ | ||
| Bone marrow | 48 (13) | 3 (11) | |
| Peripheral blood | 328 (87) | 25 (89) | |
| Donor type, No. (%) | .99§ | ||
| HLA-identical sibling | 315 (84) | 24 (86) | |
| Twin | 2 (1) | 0 (0) | |
| Haploidentical | 46 (12) | 3 (10) | |
| Other related: Not haploidentical | 13 (3) | 1 (4) |
CMML, chronic myelomonocytic leukemia; HLA, human leukocyte antigen; HSCT, hematopoietic stem cell transplantation; IPSS-R, revised International Prognostic Scoring System; JMML, juvenile myelomonocytic leukemia; MAC, myeloablative conditioning; MDS, myelodysplastic syndrome; NMA, nonmyeloablative conditioning; RA, refractory anemia; RAEB-1, RAEB-2, refractory anemia with excess blasts types 1 and 2; RARS, refractory anemia with ring sideroblasts; RCMD, refractory cytopenia with multilineage dysplasia; RCMD-RS, refractory cytopenia with multilineage dysplasia and ring sideroblasts; RIC, reduced intensity conditioning.
Kruskal-Wallis test.
Row percentage.
Fisher exact test.
Fisher exact test via Monte Carlo simulation.
P values based on data excluding missing categories.
Early: RA, RARS, RCMD, RCMD-RS, 5q– syndrome. Advanced: RAEB-1, RAEB-2, CMML.
Yield and variant spectrum of confirmed P/LP germ line variants
We identified P/LP germ line variants in 28 of 404 patients with MDS (7%), present in all age deciles from ages 11 to 71 years (Table 2; supplemental Tables 6 and 7). The yield was highest in the 11- to 20-year age group at 33%, followed by 8% in the age deciles 21 to 30, 31 to 40, and 71 to 80 years; 7% in the age decile 51 to 60 years; and 6% in the age deciles 41 to 50 and 61 to 70 years (Figure 3). Twenty-three of the patients were identified with a germ line predisposition syndrome with autosomal dominant (AD) inheritance, and of those, variants were shared with the related donor in 20 cases (Figure 2; Table 2). Three patients with MDS with truncating DDX41 variants (p.Asp140Glyfs∗2 [Nos. 12R and 13R] and p.Arg53Alafs∗16 [No. 15R]) did not share the variant with their donors (Table 2). However, these variants were considered germ line based on the VAF within germ line range (0.4-0.6) and the historical absence of truncating DDX41 variants in tumor tissue (Figure 2).49 In the 5 patients with autosomal recessive (AR) conditions, 3 were homozygous for a P/LP variant in ERCC6L2 (No. 16R), FANCA (No. 17R), and FANCG (No. 18R), respectively, and 2 individuals (Nos. 24R and 25R) were confirmed to have 2 different SBDS variants in the compound heterozygous state (Table 2). The related donors of these 5 patients with AR disease were heterozygotes for 1 of the P/LP variants (Figure 2). One patient with MDS (No. 3R) had a shared LP BLM variant as well as a second LP BLM variant with a VAF in germ line range that was not shared (Table 2), but the phase of these alleles could not be determined due to technical limitations. If both variants were present in the compound heterozygous state, the patient would be diagnosed with AR Bloom syndrome.
Table 2.
Overview of all P/LP confirmed germ line variants detected in this cohort of patients with MDS
| ID | Shared | Gene | Inheri-tance | Variant type | cDNA | Protein | Transcript | VAF | ACMG/AMP classifi-cation | Age at diagnosis (y) | MDS WHO 2008 classification | Somatic driver variants | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1R∗ | Yes | BARD1 | AD | Frameshift | c.1935_1954dup | p.Glu652Valfs∗69 | NM_000465.4 | 0.61 | LP | 64 | RAEB-2 | — | Alive |
| 2R∗ | Yes | BLM | AD | Frameshift | c.2506_2507del | p.Arg836Glyfs∗18 | NM_000057.4 | 0.52 | LP | 71 | CMML |
MPL p.Ser204Pro TET2 p.Arg544Ter TET2 p.Gln1053Ter |
Relapse, death from infection |
| 3R∗ | Yes | BLM | AD | Nonsense | c.1642C>T | p.Gln548∗ | NM_000057.4 | 0.43 | LP | 57 | MDS, unclassifiable† | — | Alive |
| 4R∗ | Yes | BRCA2 | AD | Frameshift | c.5946delT | p.Ser1982Argfs∗22 | NM_000059.4 | 0.51 | P | 52 | RAEB-1† |
RAD50 p.Asn934Lysfs TP53 p.Lys132Glu |
Relapse, death from primary disease |
| 5R∗ | Yes | BRCA2 | AD | Frameshift | c.5946delT | p.Ser1982Argfs∗22 | NM_000059.4 | 0.58 | P | 59 | RCMD† | — | Relapse, death from primary disease |
| 6R∗ | Yes | BRCA2 | AD | Nonsense | c.5217_5223del | p.Tyr1739∗ | NM_000059.4 | 0.61 | P | 56 | RAEB-2 |
TP53 c.783-1G>A PPM1D p.Val465Aspfs SMC1A p.Lys752Argfs |
Relapse, death from primary disease |
| 7R∗ | Yes | BRIP1 | AD | Frameshift | c.1236delA | p.Val413Phefs∗10 | NM_032043.3 | 0.53 | LP | 63 | RAEB-2 | — | Relapse, death from infection |
| 8R∗ | Yes | BRIP1 | AD | Frameshift | c.3196delT | p.Ser1066Hisfs∗12 | NM_032043.3 | 0.45 | LP | 64 | MDS, unclassifiable† |
CBL p.Cys404Tyr DNMT3A p.Arg882His NRAS p.Gly12Asp SETBP1 p.Asp868Asn ASXL1 p.Tyr591Ter |
Relapse, death from primary disease |
| 9R∗ | Yes | BRIP1 | AD | Frameshift | c.2392C>T | p.Arg798∗ | NM_032043.3 | 0.69 | LP | 60 | RAEB-2 | NF1 c.3198-1G>T | Relapse, death from primary disease |
| 10R∗ | Yes | CHEK2 | AD | Frameshift | c.1229del | p.Thr410Metfs∗15 | NM_001005735.2 | 0.39 | P | 49 | MDS, unclassifiable | — | Relapse, alive |
| 11R∗ | Yes | CHEK2 | AD | Frameshift | c.1392delT | p.Ser465Valfs∗15 | NM_001005735.2 | 0.53 | LP | 54 | MDS associated with isolated del(5q) |
NFE2 p.Arg323Alafs CSNK1A1 p.Asp140Ala |
Alive |
| 12R | No | DDX41 | AD | Nonsense | c.415_418dup | p.Asp140Glyfs∗2 | NM_016222.4 | 0.43 | P | 66 | MDS, unclassifiable | ASXL1 p.Gly646Trpfs | Alive |
| 13R | No | DDX41 | AD | Nonsense | c.415_418dup | p.Asp140Glyfs∗2 | NM_016222.4 | 0.49 | P | 57 | RAEB-2 | — | Alive |
| 14R∗ | Yes | DDX41 | AD | Nonsense | c.415_418dup | p.Asp140Glyfs∗2 | NM_016222.4 | 0.42 | P | 49 | RAEB-2 | — | Alive |
| 15R | No | DDX41 | AD | Frameshift | c.155dupA | p.Arg53Alafs∗16 | NM_016222.4 | 0.49 | P | 69 | RAEB-2 | — | Relapse, death from primary disease |
| 16R | Yes | ERCC6L2 | AR | Nonsense | c.19C>T | p.Gln7∗ | NM_001010895.4 | 1 | LP | 34 | RAEB-1 |
SF3B1 p.Arg238Cys TP53 p.Arg175His |
Relapse, death from primary disease |
| 17R | Yes | FANCA | AR | Indel | c.3791_3793del | p.Ser1264del | NM_000135.4 | 1 | P | 11 | RAEB-2 | — | Nonrelapse mortality, cause of death not specified |
| 18R | Yes | FANCG | AR | Frameshift | c.907_908dup | p.Glu304Trpfs∗4 | NM_004629.2 | 0.93 | LP | 27 | RA | ASXL1 p.Arg693Ter | Alive |
| 19R∗ | Yes | MRE11 | AD | Nonsense | c.1516G>T | p.Glu506∗ | NM_005591.4 | 0.54 | LP | 54 | RCMD† | — | Death from infection |
| 20R∗ | Yes | MSH6 | AD | Frameshift | c.1634_1635del | p.Lys545Argfs∗17 | NM_000179.3 | 0.46 | P | 49 | RA† | — | Nonrelapse mortality, death from secondary malignancy |
| 21R∗ | Yes | NBN | AD | Splicing | c.2071-1G>A | na | NM_002485.5 | 0.62 | P | 55 | CMML |
TET2 p.His667Ilefs GATA2 p.Met388_Lys389del RUNX1 p.Leu161_Arg162insSer CBL p.Leu380Pro SRSF2 p.Pro95Leu TET2 p.Lys875Ter |
Primary graft failure/rejection, alive |
| 22R∗ | Yes | PALB2 | AD | Copy number variant |
na‡ | na | NC_000016.10 | 0.52 | LP | 65 | RAEB-2 | — | Relapse, death from primary disease |
| 23R∗ | Yes | PMS2 | AD | Nonsense | c.11C>G | p.Ser4∗ | NM_001322015.2 | 0.41 | LP | 60 | RCMD | — | Nonrelapse mortality, death from GVHD |
| 24R | Yes/no§ | SBDS | AR | Nonsense/splicing | c.184A>T/c.258+2T>C | p.Lys62∗/na | NM_016038.4 | 0.38/0.43 | P/P | 18 | RAEB-2 | — | Relapse, death from primary disease |
| 25R | Yes/no§ | SBDS | AR | Splicing/splicing | c.258+1G>C/c.258+2T>C | na/na | NM_016038.4 | 0.7/0.26 | P/P | 19 | MDS associated with isolated del(5q) | TP53 c.559+1G>A | Alive |
| 26R∗ | Yes | TERT | AD | Nonsense | c.951G>A | p.Trp317∗ | NM_198253.3 | 0.43 | LP | 51 | RAEB-2 | — | Alive |
| 27R∗ | Yes | TERT | AD | Nonsynonymous | c.193C>G | p.Pro65Ala | NM_198253.3 | 0.63 | LP | 64 | RAEB-2 |
TP53 p.Arg248Gln TP53 p.Thr125Thr |
Relapse, death from primary disease |
| 28R∗ | Yes | TNFRSF13B | AD | Frameshift | c.204dupA | p.Leu69Thrfs∗12 | NM_012452.3 | 0.46 | P | 66 | MDS, unclassifiable | — | Relapse, alive |
ACMG, American College of Medical Genetics and Genomics; AD, autosomal dominant; AMP, Association for Molecular Pathology; AR, autosomal recessive; GVHD, graft-versus-host disease; indel, insertion/deletion; na, not applicable; VAF, variant allele frequency; WHO, World Health Organization. Other abbreviations are explained in Table 1.
Donor shares AD germ line variant.
Therapy related.
NC_000016.10:g.(?_23607316)_(23641590_?)del.
Donor only shares 1 of the variants in the heterozygous state.
Figure 3.

Age distribution of pathogenicknownand presumed germ line variants in patients with MDS. The number of patients with MDS (left y-axis, red) with a confirmed germ line variant (red, solid line), a presumed germ line variant (red, dashed line), or wild-type (ie, without a germ line variant; peach, solid line with shading) is plotted per age decile (x-axis). The percentage (right y-axis, blue) of confirmed germ line variants (blue, solid line) and presumed germ line variants∗ (blue, dashed line) is plotted per age decile. ∗Presumed germ line variants based on a multiple-criteria decision analysis.
Among the 28 P/LP variants, 27 were SNVs. One CNV, a PALB2 deletion including exons 1 to 12, was found. The vast majority of variants were truncating variants (n = 26), followed by 1 small in-frame deletion and 1 missense variant (Table 2). P/LP variants affected genes involved in BMF syndromes [ERCC6L2 (n = 1), FANCA (n = 1), FANCG (n = 1), SBDS (n = 2)], TBD [TERT (n = 2)], general tumor predisposition syndromes [BARD1 (n = 1), BLM (n = 2), BRCA2 (n = 3), BRIP1 (n = 3), CHEK2 (n = 2), MRE11 (n = 1), MSH6 (n = 1), NBN (n = 1), PALB2 (n = 1), PMS2 (n = 1)], and DDX41 (n = 4). One P/LP variant was found in an immunodeficiency-related gene, TNFRSF13B (Figure 4A-B).56 The subgroup of patients with BMF syndromes was younger at the time of diagnosis (median age, 20 years) compared with patients with TBD and general tumor predisposition syndromes, who were diagnosed at a median age of 58 and 59 years, respectively. Patients with DDX41 variants presented with the oldest median age at disease onset, 65 years (Figure 4C), consistent with prior literature.16, 17, 18 This pattern is also upheld when comparing the age of confirmed and presumed germ line variant carriers in our cohort and other larger cross-sectional studies on germ line variants in patients with MDS using either panel-based or WES covering most genes of interest. Deleterious variants in DDX41 were associated with an age at onset greater than 40 years, whereas those in GATA2 were associated with an age at onset less than 40 years (Figure 4D).5, 57, 6, 7, 8,12,15,57
Figure 4.

Spectrum and distribution of deleterious germ line variants among subgroups and association with age at diagnosis. (A) Bar chart depicting the number of confirmed P/LP germ line variants per gene identified in 28 patients with MDS. Compound heterozygous variants in the AR gene SBDS were counted as 1 variant per patient. Color schematic: purple, DDX41; green, general tumor predisposition genes; blue, telomere biology disorders; red, bone marrow failure genes; gray, others. (B) Nested pie chart showing the distribution of gene subgroups as well as biological functions. (C) Violin plots of the age at diagnosis of the 4 most common groups of predisposition syndromes identified in this study. (D) Association between the affected gene in the germ line and the age at MDS diagnosis. The data are derived from our study (confirmed/presumed germ line, n = 44) and other larger cross-sectional studies on germ line variants in patients with MDS using either panel-based or WES covering most genes of interest.5, 57, 6, 7, 8,12,15,57 Each circle represents a gene, and the circle size corresponds to the frequency of variants within this gene in the total cohort. Genes in the upper left quadrant were associated with an age at diagnosis of less than 40 years, whereas genes in the upper right quadrant were associated with an age at diagnosis greater than 40 years, based on calculated odds ratio. Color schematic: purple, DDX41; green, general tumor predisposition genes; blue, telomere biology disorders; red, bone marrow failure genes; gray, others. DSB, double-strand break repair; MMR, mismatch repair; RM, ribosome maturation.
In total, 20 of the 28 donors (71%) shared the P/LP AD germ line variant with the recipients. Five of the 28 donors were heterozygous carriers for a P/LP germ line variant with AR inheritance (Nos. 16D, 17D, 18D, 24D, and 25D) (Table 2; supplemental Table 7A). One donor was found to have a LP germ line DDX41 variant (NM_016222.4: c.3G>A, p.?) that was found only in the donor and not the patient with MDS, although it was confirmed that these 2 individuals were related based on the presence of shared variants (No. 29D) (supplemental Tables 7A and 8). This donor was aged 54 years, which is lower than the average age at onset for DDX41-related malignancies (65-69 years), for which the penetrance is known to be incomplete.16, 17, 18 Of the 404 related donors, 21 (5%) were heterozygous for a LP/P variant in a gene with AD inheritance with a VAF within the germ line range (supplemental Figure 1; supplemental Table 9).
Patients with MDS with P/LP germ line variants were more likely to have a reported predisposing condition, compared with those patients without such disorders (11% vs 4%, respectively; P < .04) (supplemental Table 5). Moreover, patients with MDS with P/LP germ line variants were more likely to have higher-grade MDS than those without such variants (43% vs 25%, respectively; P = .04) (Table 1). There was no difference in the age at onset, sex, ethnicity, cytogenetic abnormalities, IPSS-R classification, bone marrow cellularity, graft source, or donor type between patients with or without a P/LP germ line variant (Table 1; supplemental Table 5). The power to detect differences in cytogenetic abnormalities or IPSS-R classification was limited because of missing data.
Germ line variants of unknown significance (VUSs) were detected frequently in this study. Fifty-six shared germ line VUSs were found, and among these, 6 (10%) were deemed highly suspicious by calculating posterior probabilities in a Bayesian framework38 based on ACMG/AMP criteria (posterior probabilities between 67.5% and 90%) (supplemental Table 10). These highly suspicious VUSs were identified in MLH1 (n = 2), SRC (n = 1), and TERT (n = 3) (supplemental Tables 7B and 10). The median age of the patients with MDS with these variants was 53 years (range, 45-69 years). If these highly suspicious VUSs are considered P/LP, it would raise the total yield of germ line variants to 12%.
Yield and variant spectrum of presumed P/LP germ line variants
Two hundred fifty-one other P/LP variants were identified among the 404 patients with MDS, but could not be confirmed as germ line variants because they were not shared between the patient and the related donor (supplemental Table 7A). By performing a multiple-criteria decision analysis, we determined that 16 variants were most likely germ line, including 5 GATA2 variants, 3 RUNX1 variants, 2 DDX41 variants, 2 TP53 variants, and single variants in ACD, CSF3R, NBN, and TUBB1, respectively (supplemental Figure 2; supplemental Tables 3 and 11). The penetrance of recognized phenotypes, such as the presence of cytopenias, in patients with P/LP germ line GATA2 or RUNX1 variants, would be expected to result in rejection of related donors who share the germ line variant in most cases, explaining why these variants were not identified as shared in our study.
Somatic variants
Because our methodology used sequencing of peripheral blood samples from patients with MDS, we were able to use the data to determine the somatic variants in the malignant cells and to test whether particular somatic variants were associated with specific predisposition disorders. However, sequencing was performed at an average depth of 40× for germ line variant calling, giving us the ability to identify relatively large somatic clones with a VAF greater than 10%. Smaller clone sizes were rarely detected. Variants were only considered somatic variants when: (1) they occurred below a population frequency of 0.5% in the general Genome Aggregation Database population or continental subpopulations; (2) they were not shared between donor and recipient; and (3) they were not presumed germ line based on the multiple-criteria decision analysis.
Among all patients with MDS and 167 genes analyzed, we identified 652 driver variants. Forty-seven of those 167 genes were also analyzed for germ line variants (supplemental Table 2), with 58 presumed somatic variants detected within a germ line VAF range (8.9%). The top 10 genes most commonly somatically mutated were as follows: ASXL1 (19.1%), TP53 (11.9%), DNMT3A (11.6%), TET2 (10.4%), SRSF2 (9.7%), RUNX1 (9.2%), U2AF1 (7.2%), SF3B1 (5.7%), SETBP1 (4.7%), and STAG2 (3.7%) (Figure 5A). Variants were found mostly in genes affecting DNA methylation (27.7%), chromatin modification (25.5%), splicing (24.5%), transcription (24%), checkpoint/cell cycle regulation (13.1%), and Ras signaling (11.1%) (Figure 5B; supplemental Table 12A). The somatic variants in individuals with confirmed/presumed germ line variants were grouped by broad gene ontology and biological pathways and are shown in Figure 5C, together with the age at diagnosis, MDS subgroup, cytogenetics, and outcomes. We found no differences in the pattern of somatic variants in the group with confirmed/presumed germ line variants (n = 44) against the group with no germ line predisposition (n = 360), either gene-wise or by affected gene groups, suggesting that the pathways leading to MDS were similar (supplemental Table 13). Two of 25 donors sharing a germ line variant had clonal hematopoiesis of indeterminate potential, with 1 variant in ARID1A (No. 20D at a VAF of 23%) and another variant in U2AF1 (No. 26D at a VAF of 19%) (supplemental Table 12B).
Figure 5.

Spectrum and distribution of germ line variants, co-occurring somatic variants, and clinical data. (A) Bar chart displaying the top 20 most commonly affected genes for somatic variants (dark blue, wild-type; light blue, confirmed or presumed germ line variant). Percentages are given at the end of each bar. (B) Bar chart showing the top 10 most commonly affected gene groups (by broad gene ontology and biological pathways) (dark red, wild-type; light red, confirmed or presumed germ line variant). Percentages are given at the end of each bar. (C) Mutational grid with all 28 patients with MDS with confirmed germ line P/LP variants (left) and the 16 patients with MDS with presumed germ line variants (right). Patients are grouped by age at diagnosis in both panels. (Top) Clinical data such as age, MDS subtype, cytogenetic analysis, and outcome parameters, including death, relapse and nonrelapse mortality, and primary graft failure/rejection as well as co-occurring somatic variants (grouped by broad gene ontology and biological pathways) are included. (Bottom) The legend indicates the colors used for age deciles, MDS subgroups, cytogenetic analysis, variant status, and missing data. CMML, chronic myelomonocytic leukemia; MDS, myelodysplastic syndrome; RA, refractory anemia; RAEB, refractory anemia with excess blasts; RARS, refractory anemia with ring sideroblasts; RCMD, refractory cytopenia with multilineage dysplasia; RCMD-RS, refractory cytopenia with multilineage dysplasia and ring sideroblasts.
Patient outcomes
There were no differences in clinical measurements of HSCT outcomes, including neutrophil engraftment, platelet recovery, primary graft failure, overall- and disease-free survival, nonrelapse mortality and relapse, new malignancies, or cause of death between patients with MDS with a shared P/LP variant (n = 20), patients with MDS whose donor was a healthy carrier (donors lacking an AD P/LP variant or with a single P/LP allele with an AR mode of inheritance), and patients with MDS without a P/LP germ line variant (Figures 2 and 5; Table 2; supplemental Tables 8, 13, 14, and 15). Among the 20 patients whose donor shared an AD P/LP variant, 11 patients experienced a relapse (55%). Of those, 7 died of primary disease (35%) and 2 of infections post-HSCT (10%). Non-relapse-related mortality (therapy-related secondary malignancy and GVHD, respectively) was the cause of death in 2 patients (10%), and another 2 patients died of infections (10%). One patient experienced early graft failure/rejection, but is still alive (Table 2; supplemental Tables 8, 14, and 15; Figure 5C). Based on the available data, donor-derived malignancies could not be distinguished from a relapse of primary disease. Compared with patients with no germ line variants or patients who have received a transplant from a donor with an AR germ line variant in the heterozygous state, no statistically significant differences in clinical measurements of HSCT outcomes were detected. Acute GVHD, grades II through IV, was more common in patients with MDS who received a transplant from healthy carriers compared to MDS patients with no P/LP variant and MDS patients with a shared P/LP variant (P = .009; supplemental Table 14), although levels of grade III-IV acute GVHD and chronic GVHD were similar.
Discussion
This is the first study to examine the frequency of P/LP germ line variants leading to MDS across the entire age spectrum, regardless of other criteria, such as a positive personal or family history. Using conservative manual variant analysis, we identified confirmed P/LP germ line variants in 7% of all patients with MDS. When divided into age deciles, we observed P/LP germ line variants in 6% or more of patients with MDS in every decile, establishing for the first time that germ line predisposition to MDS occurs frequently in patients of all ages. The age group from 11 to 20 years had the highest yield of P/LP germ line variants, at 33%.
Prior studies examined the frequency of germ line predisposition in patients with MDS diagnosed at an age less than 45 years.5,7 Taken together with these studies, we see that the age at which MDS is diagnosed is merely a surrogate for the biological pathway affected (Figure 4). MDS develops in the youngest children, aged less than 5 years, most commonly because of germ line variants in SAMD9/SAMD9L and in teenagers and young adults because of variants in GATA2, especially those with loss of chromosome 7.10, 11, 9 Based on our study and others,5,7 adults aged less than 40 years often carry P/LP variants in genes involved in DNA repair, TBD, and BMF, such as CTC1, DKC1, ERCC6L2, Fanconi genes, PARN, RTEL1, SBDS, TERC, TERT, and TINF2. Patients with MDS diagnosed between ages 40 and 70 years are often identified with P/LP germ line variants in genes associated with TBD and general tumor predisposition genes, such as BARD1, BLM, BRCA2, BRIP1, CHEK2, MRE11, MSH6, NBN, PALB2, PMS2, and TERT. Most elderly patients have P/LP DDX41 germ line variants. The fact that presumably healthy related donors who shared the P/LP germ line variant were used for HSCT highlights the incomplete penetrance of these disorders and the potential older age at disease onset that is inherent to some of the well-known germ line syndromes.16, 17, 18,58,59
Although this is the largest study to date of patients with MDS and their related donors, we nonetheless lacked statistical power to detect differences between the outcomes of patients with and without a P/LP germ line variant. This analysis may also have been confounded by differences in disease status and/or treatments received before HSCT between the 2 groups. Advanced MDS was more common in patients with germ line predisposition and the median follow-up was 62 months, thereby potentially missing later-onset relapse, which is inherently more common in advanced MDS post-HSCT.29,60 In addition, we would anticipate different effects on the outcomes depending on the genes/pathways affected, but the genetic landscape of genes with P/LP variants in our study was too diverse and the numbers too small to distinguish effects from single-gene variants. The study population was also predominantly composed of White patients (88%), and therefore, additional studies of patients from more diverse backgrounds are needed.
Our identification of germ line predisposition alleles in 7% of all patients with MDS is an underestimate for several reasons. Given the design of our study and lack of availability of true germ line tissue (eg, cultured skin fibroblasts), we determined the germ line nature of a variant based on its presence in the peripheral blood of both the patient with MDS and the related donor. We recognize that hematopoietic tissues, including blood, undergo somatic reversion easily, especially for genes associated with BMF syndromes, immunodeficiency, and SAMD9/SAMD9L,49,61, 62, 63, 64 and hence, it is possible that germ line variants in these genes went undetected. In addition, based on the independent segregation of chromosomes into germ cells and the AD inheritance that characterize most of these variants, we would expect that about one-half of potential related donors would not have inherited the P/LP germ line variant. Moreover, it is likely that some fraction of relatives with P/LP germ line variants were excluded as HSC donors based on physical findings, abnormal peripheral blood cell counts, and/or bone marrow biopsies. The presence of multiple patients with MDS with GATA2 and RUNX1 presumed P/LP germ line variants and the absence of the variant in the related donors may reflect exclusion of those donors based on phenotypes apparent at the time of donor selection. Unfortunately, the CIBMTR does not collect information about related donors who were excluded as HSC donors. Patients with MDS with a positive family history may also have been more likely to receive HSCs from an unrelated donor. Finally, we used conservative variant calling based strictly on ACMG/AMP criteria, but suspect that some of the VUSs identified are actually deleterious in nature and may be upgraded to P/LP in the future, when more evidence becomes available. Thus, with time, we anticipate that additional studies using germ line material from patients with MDS will allow a more precise estimate of the frequency of germ line predisposition alleles, well above 7%. By performing a multiple-criteria decision analysis based on somatic variants affecting the gene in question, somatic variants in other genes and their VAF, a high pretest probability, reported predisposing condition, and expected number of circulating blasts, we were able to show that the overall yield would increase to 11% including presumed germ line variants. With the addition of suspicious VUSs, the yield would further rise to 12%. Given that one-half of potential related donors will not share the variant in AD disease, a yield of 20% to 25% is a more realistic estimate.
Another bias of the study was the fact that only patients who underwent HSCT were included and their median age at diagnosis (59 years) was less than the median age at MDS diagnosis in the United States (71 years),65 because high rates of nonrelapse mortality and morbidity often prevent older patients from being eligible for HSCT. This may lead to slightly lower rates of germ line predisposition in patients diagnosed in their 80s or 90s, although we have shown a germ line percentage of 8% in patients between 71 and 80 years, and DDX41 variants are highly prevalent in this age group.16, 17, 18
The overall pattern of somatic variants identified in all 404 patients with MDS was similar to other large MDS data sets consisting of high-risk patients with MDS.29,66 Compared with an unselected cohort, variants affecting DNA methylation and splicing, in particular variants in TET2 and SF3B1, were clearly underrepresented in our study, which is based on the association of these variants with a more favorable outcome.67 Our overall yield is slightly lower than the average of 2 driver variants per patient with MDS,68,69 which is likely due to our germ line sequencing depth of 40× and sample collections at any point before HSCT, which may include patients who were in remission. There was no difference in the pattern of somatic variants between the groups of patients with confirmed/presumed germ line variants and no germ line predisposition. However, the sample size for the germ line group was small, and its heterogeneity may confound potential effects by variants in single genes.
This study establishes that at least 7% of all patients with MDS across the age spectrum have a P/LP germ line variant. The National Comprehensive Cancer Network recommends germ line screening in the setting of hereditary breast, ovarian, and pancreatic cancer when the pretest probability is >5%,70 and the ACMG list of 59 medically actionable genes for reporting of secondary findings from clinical exome and genome sequencing71 has a pretest probability of 3.1% to 5.2%.72 Our confirmed germ line frequency of 7%, and likely germ line frequency of 11% to 12% and higher, is similar to that seen in hereditary breast and colon cancers, where germ line predisposition variants are found in approximately 5% to 10% of unselected patients and up to 20% in high-risk groups.73, 74, 75, 76
Therefore, with this study, we surpass this minimal threshold and recommend that germ line genetic testing become standard clinical practice for all patients with MDS undergoing HSCT regardless of their age at diagnosis or other features, such as a positive personal or family history. Identification of germ line predisposition to hematopoietic malignancies is critical in the clinical setting for several reasons: (1) There may be relevant implications of the choice of treatment, conditioning, and intensity.18,77, 78, 79 (2) Related donors may share the P/LP germ line variant, as we show in this study, and their use should be avoided.19,21, 22, 23, 24, 25, 26, 27,80 (3) The presence of a germ line syndrome can co-occur with increased risk for other malignancies or other nonhematopoietic manifestations requiring surveillance and can be associated with excessive treatment-related toxicities from standard treatments used for AA/MDS.78,79,81 We hope that there is rapid uptake in the hematology community of our recommendation for universal germ line testing for all patients with MDS undergoing HSCT.
Conflict-of-interest disclosure: L.A.G. receives royalties from UptoDate, Inc for a co-authored article on germ line predisposition to hematopoietic malignancies. R.N. reports the following conflicts of interest: consulting fees from Bluebio, Sanofi, Omeros; payment or honoraria for lectures, presentations, speakers, bureaus, manuscript writings, or educational events from Viracor, Magenta, Kadmon; member of NCCN Panel for Hematopoietic Cell Transplantation; and research support from Miyarisan. T.N. has received financial support for a clinical trial from Novartis and financial support for drug-only supply for a clinical trial from Karyopharm. A.S. reports consulting fees from Magenta Therapeutics, Incyte Pharmaceuticals, CareDx; and patents planned, issued, or pending from In8Bio therapeutics. B.L.S. reports compensation from his role as advisor (Celgene, Alexion), funding received from Novartis; and payments from the BMS advisory committee; American Society of Hematology government affairs committee; and Taihoe Oncology (payment of honoraria for lectures, presentations, speaker’s bureaus, manuscript writing or educational events). B.O. reports research funding from ASTEX and AROG pharmaceuticals. R.S. discloses his participation on the advisory board for CareDx. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the patients and their donors for participating in research through the CIBMTR. The authors acknowledge all participating centers and researchers from the CIBMTR and thank the members of the Chronic Leukemia Writing Committee, especially for their critical review of this work and their helpful suggestions: Mahmoud Aljurf, Ulrike Bacher, Amer Beitinjaneh, Jean-Yves Cahn, Jan Cerny, Edward Copelan, Miguel A. Diaz, Shahinaz Gadalla, Robert Peter Gale, Siddhartha Ganguly, Michael Grunwald, Nasheed M. Hossain, Mohamed A. Kharfan-Dabaja, Tania Jain, Mark Juckett, Hillard M Lazarus, Mark Litzow, Sunita Nathan, Richard F. Olsson, Attaphol Pawarode, David A. Rizzieri, Lynn Savoie, Kirk R. Schultz, Sachiko Seo, Melhem Solh, Celalettin Ustun, David Valcárcel, and Baldeep Wirk.
Funding for this work was provided by the Department of Defense (W81XWH-19-1-0241) (L.A.G.). S.F. receives funding from the Olympia Morata Program of the Medical Faculty Heidelberg. A.M.T. was supported by generous donors from the QEII Health Sciences Centre Foundation. M.W.D. was supported by the Damon Runyon Cancer Research Foundation, the Edward P. Evans Foundation, and the National Institutes of Health Paul Calabresi K12 Program in Oncology.
The CIBMTR is supported primarily by Public Health Service U24CA076518 from the National Cancer Institute (NCI), the National Heart, Lung, and Blood Institute (NHLBI), and the National Institute of Allergy and Infectious Diseases (NIAID); HHSH250201700006C from the Health Resources and Services Administration (HRSA); and N00014 to 20-1-2705 and N00014 to 20-1-2832 from the Office of Naval Research. Support is also provided by Be The Match Foundation, the Medical College of Wisconsin, the National Marrow Donor Program, and the following commercial entities: AbbVie; Accenture; Actinium Pharmaceuticals, Inc; Adaptive Biotechnologies Corporation; Adienne SA; Allovir, Inc; Amgen, Inc; Astellas Pharma US; bluebird bio, Inc; Bristol Myers Squibb Co; CareDx; CSL Behring; CytoSen Therapeutics, Inc; Daiichi Sankyo Co, Ltd; Eurofins Viracor, DBA Eurofins Transplant Diagnostics; Fate Therapeutics; Gamida-Cell, Ltd; Gilead; GlaxoSmithKline; HistoGenetics; Incyte Corporation; Iovance; Janssen Research & Development, LLC; Janssen/Johnson & Johnson; Jasper Therapeutics; Jazz Pharmaceuticals, Inc; Kadmon; Karius; Karyopharm Therapeutics; Kiadis Pharma; Kite Pharma Inc; Kite, a Gilead Company; Kyowa Kirin International plc; Kyowa Kirin; Legend Biotech; Magenta Therapeutics; Medac GmbH; Medexus; Merck & Co; Millennium, the Takeda Oncology Co; Miltenyi Biotec, Inc; MorphoSys; Novartis Pharmaceuticals Corporation; Omeros Corporation; OncoImmune,Inc; Oncopeptides, Inc; OptumHealth; Orca Biosystems, Inc; Ossium Health, Inc; Pfizer, Inc; Pharmacyclics, LLC; Priothera; Sanofi Genzyme; Seagen, Inc; Stemcyte; Takeda Pharmaceuticals; Talaris Therapeutics; Terumo Blood and Cell Technologies; TG Therapeutics; Tscan; Vertex; Vor Biopharma; Xenikos BV.
Authorship
Contribution: S.F., A.M.T., L.A.G., and W.S. designed and coordinated the study; S.F., A.M.T., K.S., and M.P. contributed to DNA extraction, sample preparation, and augmented WES; S.F. and A.M.T. analyzed and interpreted SNVs; S.F. performed variant validations by Sanger sequencing and subcloning and analyzed, interpreted, and validated CNVs; N.E.-M. and S.K. conducted statistical analyses and contributed to data collection; S.F., A.M.T., M.P., K.M., M.W.D., B.R., and A.L.K. were in charge of bioinformatic pipelines for variant filtering and calling and downstream bioinformatic analyses; R.C.L. provided sequencing data; B.L.S., B.O., T.N., V.A., A.S., R.N., Z.H., R.S., S.S., and W.S. contributed to patient recruitment and data collection and provided conceptional input; S.F., A.M.T., and L.A.G. wrote the manuscript; and all authors approved the final version.
Footnotes
The CIBMTR supports accessibility of research in accord with the National Institutes of Health (NIH) Data Sharing Policy and the National Cancer Institute (NCI) Cancer Moonshot Public Access and Data Sharing Policy. The CIBMTR only releases de-identified data sets that comply with all relevant global regulations regarding privacy and confidentiality.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Trottier AM, Godley LA. Inherited predisposition to haematopoietic malignancies: overcoming barriers and exploring opportunities. Br J Haematol. 2021;194(4):663–676. doi: 10.1111/bjh.17247. [DOI] [PubMed] [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg PL, Stone RM, Al-Kali A, et al. Myelodysplastic Syndromes, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw. 2017;15(1):60–87. doi: 10.6004/jnccn.2017.0007. [DOI] [PubMed] [Google Scholar]
- 4.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keel SB, Scott A, Sanchez-Bonilla M, et al. Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica. 2016;101(11):1343–1350. doi: 10.3324/haematol.2016.149476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singhal D, Hahn CN, Feurstein S, et al. Targeted gene panels identify a high frequency of pathogenic germline variants in patients diagnosed with a hematological malignancy and at least one other independent cancer. Leukemia. 2021;35(11):3245–3256. doi: 10.1038/s41375-021-01246-w. [DOI] [PubMed] [Google Scholar]
- 7.Feurstein S, Churpek JE, Walsh T, et al. Germline variants drive myelodysplastic syndrome in young adults. Leukemia. 2021;35(8):2439–2444. doi: 10.1038/s41375-021-01137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bluteau O, Sebert M, Leblanc T, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018;131(7):717–732. doi: 10.1182/blood-2017-09-806489. [DOI] [PubMed] [Google Scholar]
- 9.Sahoo SS, Kozyra EJ, Wlodarski MW. Germline predisposition in myeloid neoplasms: Unique genetic and clinical features of GATA2 deficiency and SAMD9/SAMD9L syndromes. Best Pract Res Clin Haematol. 2020;33(3):101197. doi: 10.1016/j.beha.2020.101197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz JR, Ma J, Lamprecht T, et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun. 2017;8(1):1557. doi: 10.1038/s41467-017-01590-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wlodarski MW, Hirabayashi S, Pastor V, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–1397. doi: 10.1182/blood-2015-09-669937. [DOI] [PubMed] [Google Scholar]
- 12.Churpek JE, Pyrtel K, Kanchi K-L, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood. 2015;126(22):2484–2490. doi: 10.1182/blood-2015-04-641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guidugli L, Johnson AK, Alkorta-Aranburu G, et al. Clinical utility of gene panel-based testing for hereditary myelodysplastic syndrome/acute leukemia predisposition syndromes. Leukemia. 2017;31(5):1226–1229. doi: 10.1038/leu.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiNardo CD, Bannon SA, Routbort M, et al. Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC) Clin Lymphoma Myeloma Leuk. 2016;16(7):417–428.e2. doi: 10.1016/j.clml.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rio-Machin A, Vulliamy T, Hug N, et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat Commun. 2020;11(1):1044. doi: 10.1038/s41467-020-14829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheah JJC, Hahn CN, Hiwase DK, Scott HS, Brown AL. Myeloid neoplasms with germline DDX41 mutation. Int J Hematol. 2017;106(2):163–174. doi: 10.1007/s12185-017-2260-y. [DOI] [PubMed] [Google Scholar]
- 17.Lewinsohn M, Brown AL, Weinel LM, et al. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood. 2016;127(8):1017–1023. doi: 10.1182/blood-2015-10-676098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sébert M, Passet M, Raimbault A, et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood. 2019;134(17):1441–1444. doi: 10.1182/blood.2019000909. [DOI] [PubMed] [Google Scholar]
- 19.Fogarty PF, Yamaguchi H, Wiestner A, et al. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet. 2003;362(9396):1628–1630. doi: 10.1016/S0140-6736(03)14797-6. [DOI] [PubMed] [Google Scholar]
- 20.Galera P, Hsu AP, Wang W, et al. Donor-derived MDS/AML in families with germline GATA2 mutation. Blood. 2018;132(18):1994–1998. doi: 10.1182/blood-2018-07-861070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owen CJ, Toze CL, Koochin A, et al. Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood. 2008;112(12):4639–4645. doi: 10.1182/blood-2008-05-156745. [DOI] [PubMed] [Google Scholar]
- 22.Berger G, van den Berg E, Sikkema-Raddatz B, et al. Re-emergence of acute myeloid leukemia in donor cells following allogeneic transplantation in a family with a germline DDX41 mutation. Leukemia. 2017;31(2):520–522. doi: 10.1038/leu.2016.310. [DOI] [PubMed] [Google Scholar]
- 23.Buijs A, Poddighe P, van Wijk R, et al. A novel CBFA2 single-nucleotide mutation in familial platelet disorder with propensity to develop myeloid malignancies. Blood. 2001;98(9):2856–2858. doi: 10.1182/blood.v98.9.2856. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi S, Kobayashi A, Osawa Y, et al. Donor cell leukemia arising from preleukemic clones with a novel germline DDX41 mutation after allogenic hematopoietic stem cell transplantation. Leukemia. 2017;31(4):1020–1022. doi: 10.1038/leu.2017.44. [DOI] [PubMed] [Google Scholar]
- 25.Gibson CJ, Kim HT, Zhao L, et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. J Clin Oncol. 2022;40(2):189–201. doi: 10.1200/JCO.21.02286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rojek K, Nickels E, Neistadt B, et al. Identifying inherited and acquired genetic factors involved in poor stem cell mobilization and donor-derived malignancy. Biol Blood Marrow Transplant. 2016;22(11):2100–2103. doi: 10.1016/j.bbmt.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao H, Shi J, Luo Y, et al. First report of multiple CEBPA mutations contributing to donor origin of leukemia relapse after allogeneic hematopoietic stem cell transplantation. Blood. 2011;117(19):5257–5260. doi: 10.1182/blood-2010-12-326322. [DOI] [PubMed] [Google Scholar]
- 28.Churpek JE, Nickels E, Marquez R, et al. Identifying familial myelodysplastic/acute leukemia predisposition syndromes through hematopoietic stem cell transplantation donors with thrombocytopenia. Blood. 2012;120(26):5247–5249. doi: 10.1182/blood-2012-09-457945. [DOI] [PubMed] [Google Scholar]
- 29.Lindsley RC, Saber W, Mar BG, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376(6):536–547. doi: 10.1056/NEJMoa1611604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data: figure 1. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tate JG, Bamford S, Jubb HC, et al. COSMIC: the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019;47(D1):D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feurstein S, Hahn CN, Mehta N, Godley LA. A practical guide to interpreting germline variants that drive hematopoietic malignancies, bone marrow failure, and chronic cytopenias. Genet Med. 2022;24(4):931–954. doi: 10.1016/j.gim.2021.12.008. [DOI] [PubMed] [Google Scholar]
- 33.Wu D, Luo X, Feurstein S, et al. How I curate: applying American Society of Hematology-Clinical Genome Resource Myeloid Malignancy Variant Curation Expert Panel rules for RUNX1 variant curation for germline predisposition to myeloid malignancies. Haematologica. 2020;105(4):870–887. doi: 10.3324/haematol.2018.214221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biesecker LG, Harrison SM. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med. 2018;20(12):1687–1688. doi: 10.1038/gim.2018.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walsh MF, Ritter DI, Kesserwan C, et al. Integrating somatic variant data and biomarkers for germline variant classification in cancer predisposition genes. Hum Mutat. 2018;39(11):1542–1552. doi: 10.1002/humu.23640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feurstein S, Zhang L, DiNardo CD. Accurate germline RUNX1 variant interpretation and its clinical significance. Blood Adv. 2020;4(24):6199–6203. doi: 10.1182/bloodadvances.2020003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tavtigian SV, Greenblatt MS, Harrison SM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20(9):1054–1060. doi: 10.1038/gim.2017.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fortuno C, Lee K, Olivier M, et al. Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants. Hum Mutat. 2021;42(3):223–236. doi: 10.1002/humu.24152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross JE, Zhang BM, Lee K, et al. Specifications of the variant curation guidelines for ITGA2B/ITGB3: ClinGen Platelet Disorder Variant Curation Panel. Blood Adv. 2021;5(2):414–431. doi: 10.1182/bloodadvances.2020003712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) Genet Med. 2020;22(2):245–257. doi: 10.1038/s41436-019-0686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo X, Feurstein S, Mohan S, et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv. 2019;3(20):2962–2979. doi: 10.1182/bloodadvances.2019000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh R, Harrison SM, Rehm HL, Plon SE, Biesecker LG. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum Mutat. 2018;39(11):1525–1530. doi: 10.1002/humu.23642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strande NT, Brnich SE, Roman TS, Berg JS. Navigating the nuances of clinical sequence variant interpretation in Mendelian disease. Genet Med. 2018;20(9):918–926. doi: 10.1038/s41436-018-0100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39(11):1517–1524. doi: 10.1002/humu.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brnich SE, Rivera-Muñoz EA, Berg JS. Quantifying the potential of functional evidence to reclassify variants of uncertain significance in the categorical and Bayesian interpretation frameworks. Hum Mutat. 2018;39(11):1531–1541. doi: 10.1002/humu.23609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gelb BD, Cavé H, Dillon MW, et al. ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet Med. 2018;20(11):1334–1345. doi: 10.1038/gim.2018.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brnich SE, Abou Tayoun AN, Couch FJ, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2020;12(1):3. doi: 10.1186/s13073-019-0690-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feurstein S, Drazer M, Godley LA. Germline predisposition to hematopoietic malignancies. Hum Mol Genet. 2021;30(R2):R225–R235. doi: 10.1093/hmg/ddab141. [DOI] [PubMed] [Google Scholar]
- 50.Bannon SA, Routbort MJ, Montalban-Bravo G, et al. Next-generation sequencing of DDX41 in myeloid neoplasms leads to increased detection of germline alterations. Front Oncol. 2021;10 doi: 10.3389/fonc.2020.582213. 5822131582216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kraft IL, Godley LA. Identifying potential germline variants from sequencing hematopoietic malignancies. Hematology. 2020;2020(1):219–227. doi: 10.1182/hematology.2020006910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simon L, Spinella J-F, Yao C-Y, et al. High frequency of germline RUNX1 mutations in patients with RUNX1-mutated AML. Blood. 2020;135(21):1882–1886. doi: 10.1182/blood.2019003357. [DOI] [PubMed] [Google Scholar]
- 53.Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer. J Mol Diagn. 2017;19(1):4–23. doi: 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–2465. doi: 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 56.Tangye SG, Al-Herz W, Bousfiha A, et al. Human Inborn Errors of Immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24–64. doi: 10.1007/s10875-019-00737-x. [published correction appears in J Clin Immunol. 2020;40(1):65]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang MY, Keel SB, Walsh T, et al. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica. 2015;100(1):42–48. doi: 10.3324/haematol.2014.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Churpek JE, Godley LA. How I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood. 2016;128(14):1800–1813. doi: 10.1182/blood-2016-05-670240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43(5):598–608. doi: 10.1053/j.seminoncol.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 60.Shaffer BC, Ahn KW, Hu Z-H, et al. Scoring system prognostic of outcome in patients undergoing allogeneic hematopoietic cell transplantation for myelodysplastic syndrome. J Clin Oncol. 2016;34(16):1864–1871. doi: 10.1200/JCO.2015.65.0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen D-H, Below JE, Shimamura A, et al. Ataxia-pancytopenia syndrome is caused by missense mutations in SAMD9L. Am J Hum Genet. 2016;98(6):1146–1158. doi: 10.1016/j.ajhg.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hockings C, Gohil S, Dowse R, et al. In trans early mosaic mutational escape and novel phenotypic features of germline SAMD9 mutation. Br J Haematol. 2020;188(4):e53–e57. doi: 10.1111/bjh.16322. [DOI] [PubMed] [Google Scholar]
- 63.Shima H, Koehler K, Nomura Y, et al. Two patients with MIRAGE syndrome lacking haematological features: role of somatic second-site reversion SAMD9 mutations. J Med Genet. 2018;55(2):81–85. doi: 10.1136/jmedgenet-2017-105020. [DOI] [PubMed] [Google Scholar]
- 64.Tesi B, Davidsson J, Voss M, et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood. 2017;129(16):2266–2279. doi: 10.1182/blood-2016-10-743302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma X. Epidemiology of myelodysplastic syndromes. Am J Med. 2012;125(7):S2–S5. doi: 10.1016/j.amjmed.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ogawa S. Genetics of MDS. Blood. 2019;133(10):1049–1059. doi: 10.1182/blood-2018-10-844621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bernard E, Tuechler H, Greenberg PL, et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022;1(7):1–14. doi: 10.1056/EVIDoa2200008. [DOI] [PubMed] [Google Scholar]
- 68.Makishima H, Yoshizato T, Yoshida K, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49(2):204–212. doi: 10.1038/ng.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–3627. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Daly MB, Pilarski R, Yurgelun MB, et al. NCCN guidelines insights: genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 1.2020. J Natl Compr Cancer Netw. 2020;18(4):380–391. doi: 10.6004/jnccn.2020.0017. [DOI] [PubMed] [Google Scholar]
- 71.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–255. doi: 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- 72.Haverfield EV, Esplin ED, Aguilar SJ, et al. Physician-directed genetic screening to evaluate personal risk for medically actionable disorders: a large multi-center cohort study. BMC Med. 2021;19(1):1–10. doi: 10.1186/s12916-021-01999-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Susswein LR, Marshall ML, Nusbaum R, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2016;18(8):823–832. doi: 10.1038/gim.2015.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun J, Meng H, Yao L, et al. Germline mutations in cancer susceptibility genes in a large series of unselected breast cancer patients. Clin Cancer Res. 2017;23(20):6113–6119. doi: 10.1158/1078-0432.CCR-16-3227. [DOI] [PubMed] [Google Scholar]
- 75.Stoffel EM, Koeppe E, Everett J, et al. Germline genetic features of young individuals with colorectal cancer. Gastroenterology. 2018;154(4):897–905.e1. doi: 10.1053/j.gastro.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Momozawa Y, Iwasaki Y, Parsons MT, et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat Commun. 2018;9(1):4083. doi: 10.1038/s41467-018-06581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tawana K, Wang J, Renneville A, et al. Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood. 2015;126(10):1214–1223. doi: 10.1182/blood-2015-05-647172. [DOI] [PubMed] [Google Scholar]
- 78.Dietz AC, Orchard PJ, Baker KS, et al. Disease-specific hematopoietic cell transplantation: nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant. 2011;46(1):98–104. doi: 10.1038/bmt.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ebens CL, MacMillan ML, Wagner JE. Hematopoietic cell transplantation in Fanconi anemia: current evidence, challenges and recommendations. Expet Rev Hematol. 2017;10(1):81–97. doi: 10.1080/17474086.2016.1268048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Churpek JE, Nickels E, Marquez R, et al. Identifying familial myelodysplastic/acute leukemia predisposition syndromes through hematopoietic stem cell transplantation donors with thrombocytopenia. Blood. 2012;120(26):5247–5249. doi: 10.1182/blood-2012-09-457945. [DOI] [PubMed] [Google Scholar]
- 81.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113(26):6549–6557. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.