

Abstract

Nirmatrelvir (PF-07321332) is a nitrile-bearing small-molecule inhibitor that, in combination with ritonavir, is used to treat infections by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). Nirmatrelvir interrupts the viral life cycle by inhibiting the SARS-CoV-2 main protease (Mpro), which is essential for processing viral polyproteins into functional nonstructural proteins. We report studies which reveal that derivatives of nirmatrelvir and other Mpro inhibitors with a nonactivated terminal alkyne group positioned similarly to the electrophilic nitrile of nirmatrelvir can efficiently inhibit isolated Mpro and SARS-CoV-2 replication in cells. Mass spectrometric and crystallographic evidence shows that the alkyne derivatives inhibit Mpro by apparent irreversible covalent reactions with the active site cysteine (Cys145), while the analogous nitriles react reversibly. The results highlight the potential for irreversible covalent inhibition of Mpro and other nucleophilic cysteine proteases by alkynes, which, in contrast to nitriles, can be functionalized at their terminal position to optimize inhibition and selectivity, as well as pharmacodynamic and pharmacokinetic properties.
Introduction
In late 2021, the small-molecule active pharmaceutical ingredient (API) of paxlovid, i.e., nirmatrelvir (PF-07321332, 1; Figure 1a),1 was approved for emergency use in humans to treat COVID-19 (in combination with ritonavir). Nirmatrelvir is a selective inhibitor of the SARS-CoV-22 main protease (Mpro or 3C-like protease, 3CLpro).1 Mpro is a nucleophilic cysteine protease that, together with the papain-like cysteine protease (PLpro), catalyzes hydrolysis of viral polyproteins to give functional nonstructural proteins (nsps), a process essential for the viral life cycle, rendering Mpro and PLpro attractive drug targets.3−8 The therapeutic use of nirmatrelvir is precedented by many small-molecule viral protease inhibitors that are used to treat infections by human immunodeficiency virus (HIV) and hepatitis C virus (HCV).9
Figure 1.

Nirmatrelvir inhibits SARS-CoV-2 Mpro by reversible covalent reaction with the nucleophilic Cys145. (a) Reversible covalent reaction of nirmatrelvir (PF-07321332, 1) with the nucleophilic thiolate of SARS-CoV-2 Mpro Cys145 (deprotonated by His41).1 (b) View from a crystal structure of SARS-CoV-2 Mpro (gray cartoon) in complex with nirmatrelvir (1; green carbon backbone) (PDB ID: 7TE0(22)). The thioimidate is located in an oxyanion hole (involving the main chain amide NHs of Cys145 and Gly143).
SARS-CoV-2 Mpro may be a more viable medicinal chemistry target than PLpro because its fold and substrate selectivities are reported to be different from human proteases;10,11 Mpro is highly conserved among coronaviruses, including SARS-CoV-2 variants of clinical concern.12 Indeed, nirmatrelvir inhibits SARS-CoV-2 variants, including omicron13−17 and other coronaviruses,1,18−20 but does not substantially inhibit most tested human cysteine proteases in vitro.1,21
Considerable efforts for the development of SARS-CoV-2 Mpro inhibitors have focused on substrate-based inhibitors, which react covalently with the nucleophilic thiolate of the catalytically active Cys145 of Mpro, a strategy that is well precedented for inhibiting viral (and other) proteases.9 One noncovalently binding Mpro inhibitor, i.e., Shionogi’s ensitrelvir (S-217622, 2; Figure 2a), has been recently approved for clinical use.23
Figure 2.

SARS-CoV-2 Mpro and DPP4 inhibitors. (a) Ensitrelvir (S-217622, 2),23 (b) GC376 (3),25,26,31 (c) MI-09 (4),27 (d) vildagliptin (5),44 and saxagliptin (6), and (e) selected reported nirmatrelvir derivatives (7a–d).19
Nirmatrelvir (1) is particular amongst reported covalent SARS-CoV-2 Mpro inhibitors because it employs a nitrile group as an electrophilic warhead. The nitrile of 1 covalently reacts with Cys145, as shown by crystallography (Figure 1b),18 likely to give a complex in which a thioimidate electron pair occupies the Mpro oxyanion hole (which is formed by the main chain amide NHs of Cys145 and Gly143; Figure 1b), mimicking binding by tetrahedral intermediates in catalysis.24 By contrast, other covalently reacting Mpro inhibitors employ different electrophilic warheads such as, e.g., α-ketoamides,11,25,26 aldehydes27−30 and precursors,25,26,31 α-hydroxymethylketones and α-acyloxymethylketones,32−35 and Michael acceptors,10 among others (Figure 2b,c).6,7,20,36−43
The use of a nitrile group as a warhead was apparently key to the successful development of nirmatrelvir (1), in part because the nonspecific reactivity of nitriles with off-target nucleophiles may be lower than that of many other available electrophilic warheads and in part because the covalent reaction of 1 with the thiolate of Cys145 is reversible.1 The use of nitriles as electrophilic warheads in protease inhibitors has previously been demonstrated to be of clinical use, as shown by vildagliptin and saxagliptin, which are the APIs of clinically used type 2 diabetes therapeutics that inhibit human dipeptidylpeptidase-4 (DPP4) by a reversible covalent reaction of their nitrile group with the active site serine residue of DPP4 (Figure 2d).44 Nitriles are also present in investigational therapeutics45 and have inter alia been employed as electrophilic warheads in substrate-derived inhibitors of SARS-CoV Mpro in 2013.46
Researchers at Pfizer have reported structure–activity relationship (SAR) studies on nirmatrelvir, which focused on investigating the effect of the P4 substrate-equivalent residue and of ketobenzothiazole groups substituting the nitrile on inter alia inhibitor potency.1 Indeed, only a few additional SAR studies on nitrile warhead-bearing covalent SARS-CoV-2 Mpro inhibitors have been reported to date, with these mainly focusing on altering the nirmatrelvir P4 substrate-equivalent residue,24 on introducing nitrile electrophilic warheads to other inhibitor scaffolds than nirmatrelvir,109 and on using azanitriles as electrophilic warheads for the predicted covalent reaction with the thiolate of Cys145.47 Recently, studies on nirmatrelvir derivatives in which the nitrile group has been substituted with alternative electrophilic warheads, including aldehydes, benzyloxy/hydroxymethylketones, Michael acceptors, and α-ketoamide groups, which have been previously used in Mpro inhibitors other than nirmatrelvir, have been reported with the derivatives being shown to retain high potency in vitro and in cells (Figure 2e).19
The substitution of the nirmatrelvir nitrile group for the isoelectronic alkyne group has not yet been explored. Alkynes are particularly attractive (latent) electrophiles for covalent reactions with nucleophilic cysteine proteases, in part because they are isoelectronic with nitriles and share their linear geometry; however, unlike nitriles, they can be functionalized at their terminal position with substituents that could, in principle, bind the protease S′ sites and thus alter inhibitor binding kinetics and be used to improve selectivity or pharmacokinetic properties. The irreversible covalent reaction of both nonactivated terminal and internal alkynes with the active site cysteine thiols of human and viral cysteine proteases has been reported,48−52 but such studies have not been reported with SARS-CoV-2 Mpro. Amino acid-derived activated bromo alkynes have been identified as a result of a fragment screen that covalently react with Cys145 through a yet unidentified mechanism involving loss of bromide.53
It is proposed that the efficiency of the addition of thiols to alkynes is largely proximity-driven, i.e., is promoted by preorganization of the reactants.48 The reaction of deubiquitinases (DUBs), including SARS-CoV-2 PLpro, with ubiquitin and/or other ubiquitin-like protein modifiers (e.g., ISG15) bearing C-terminal nonactivated terminal alkynes has been extensively studied.49−51,54 Such activity probes react efficiently with the DUB active site cysteine thiolate, in part, likely because of the small size of the alkyne group, which fits well into the relatively tight DUB active sites and because of the efficient (allosteric) binding of ubiquitin or ubiquitin-like modifiers by DUBs, which position the alkyne electrophile close to the nucleophilic cysteine thiolate.50 The use of C-terminally alkynylated ubiquitin and ISG15 activity probes has found widespread application, in part because of the relatively low electrophilicity of alkynes, minimizing the reaction with off-target nucleophiles lacking a templating effect. It has been recently shown that the template effect required to enable efficient reactions of alkynes with nucleophilic thiols is not limited to relatively large protein components, but extends to small-molecule enzyme inhibitors, i.e., an alkyne derivative of the nitrile-bearing investigational drug odanacatib was shown to irreversibly react with the nucleophilic thiolate of a human cathepsin.48
Here, we report studies which reveal that derivatives of nirmatrelvir (1) bearing activated and nonactivated alkynes, as well as alkyne derivatives of other substrate-derived peptidomimetic Mpro inhibitors, are efficient covalent Mpro inhibitors, both against isolated Mpro enzyme and Mpro in cells. The mechanism of inhibition was investigated using crystallography and mass spectrometry (MS) and was shown to involve covalent reactions of the thiolate of the Mpro active site Cys145 with the alkynes.
Results
Nirmatrelvir Derivatives with a Nonactivated Terminal Alkyne Inhibit Mpro
To investigate the capacity of using nonactivated terminal alkynes as electrophilic warheads to covalently inhibit SARS-CoV-2 Mpro, the nirmatrelvir-derived alkyne 13 was synthesized by COMU55-mediated amide coupling of the reported acid 12(1) with alkyne 11 (17% yield following HPLC purification; Scheme 1). Note that the comparatively low yield is a result of material loss during HPLC purification, which was required to obtain sufficiently pure materials when reactions were performed on a laboratory scale; the purification process can likely be optimized to avoid the use of HPLC when reactions are performed on a larger scale, as done by Pfizer researchers during their synthesis of 1.1
Scheme 1. Synthesis of Alkyne-Bearing Nirmatrelvir Derivative 13.
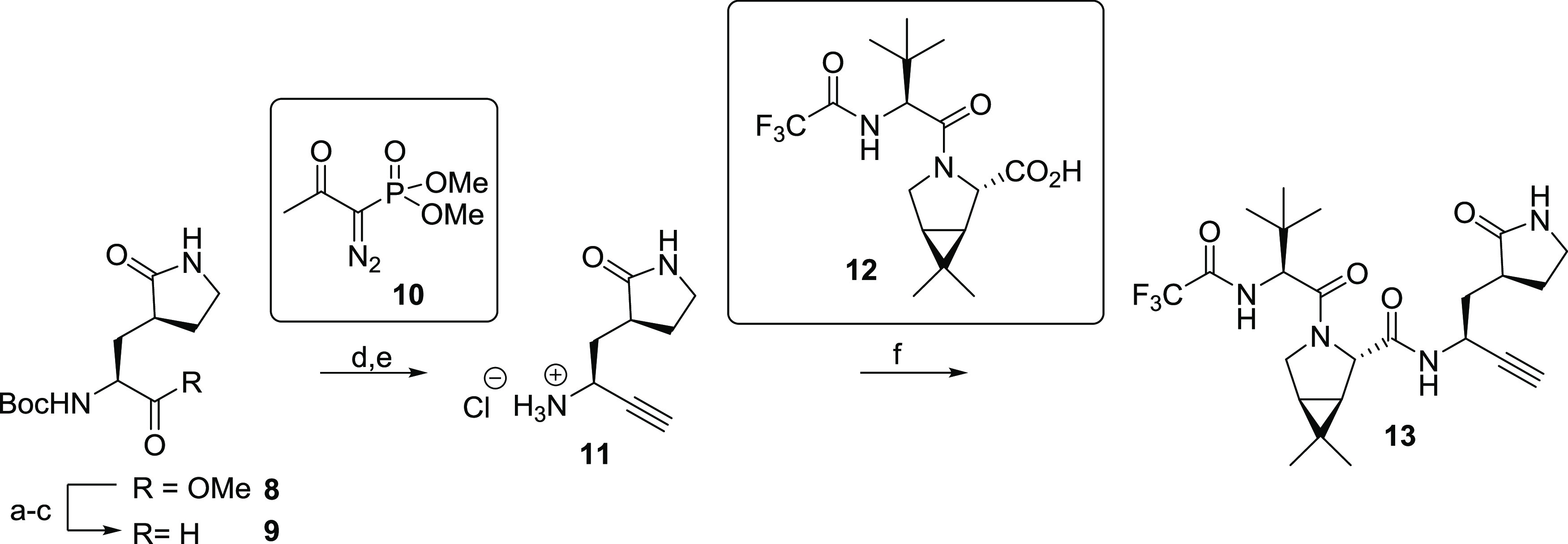
Reagents and conditions: (a) LiOH, THF/H2O, 0 °C to room temperature (rt), 83%; (b) Me(OMe)NH·HCl, 1,1′-carbonyldiimidazole (CDI), iPr2NEt, CH2Cl2, rt, 58%; (c) LiAlH4, THF/Et2O, −78 °C, 93%; (d) 10,59,60 K2CO3, MeOH, rt, 43%; (e) HCl (4 M in dioxane), CH2Cl2, rt, app. quant.; (f) 12,1 COMU,55N-methylmorpholine (NMM), DMF/CH2Cl2, 0 °C to rt, 17%.
Alkyne 11 was synthesized in five steps from the commercially sourced methylester 8, i.e., saponification, Weinreb amide formation,56 LiAlH4-mediated reduction to the reported aldehyde 9,57,58 alkynylation of 9 using the Ohira–Bestmann reagent 10,59,60 and Boc deprotection (Scheme 1). An alternative four-step synthesis of 11 involving the LiBH4-mediated reduction of 8 to the corresponding alcohol, followed by oxidation to 9(57,58) using Dess–Martin periodinane,61 alkynylation of 9 using 10, and Boc deprotection was less reproducible and resulted in higher levels of C-α epimerization at the aldehyde stage, as evidenced by 1H NMR analysis.
The half-maximum inhibitory concentrations (IC50 values) of nirmatrelvir (1) and nirmatrelvir alkyne 13 were determined with purified recombinant SARS-CoV-2 Mpro using a reported solid-phase extraction coupled to mass spectrometry (SPE-MS)-based Mpro inhibition assay that directly monitors Mpro-catalyzed hydrolysis of a synthetic 37-mer oligopeptide substrate based on the sequence of the N-terminal Mpro self-cleavage site (ALNDFSNSGSDVLYQPPQTSITSAVLQ/SGFRKMAFPS-NH2; “/” indicates the cleavage site).43,62 Note that the N-terminally acetylated C-terminal product peptide (Ac-SGFRKMAFPS-NH2) was employed as an internal standard to avoid the identification of false positive hits due to inhibitor-induced suppression of ionization of the product peptide (Supporting Figures S1 and S2).
The results reveal that alkyne 13 inhibits Mpro approximately 5-fold less efficiently than nirmatrelvir (1), highlighting the potential of nonactivated terminal alkynes to efficiently inhibit Mpro (IC50 ≤ 0.025 μM for 1 and ∼0.14 μM for 13; Table 1, entries i and ii). Note, however, that the reduction in potency of 13 compared to 1 may be more pronounced because 1 inhibits Mpro at concentrations close to the lowest detection limit of the SPE-MS assay.
Table 1. Inhibition of SARS-CoV-2 Mpro by Nirmatrelvir Derivatives Bearing a Terminal Alkyne.
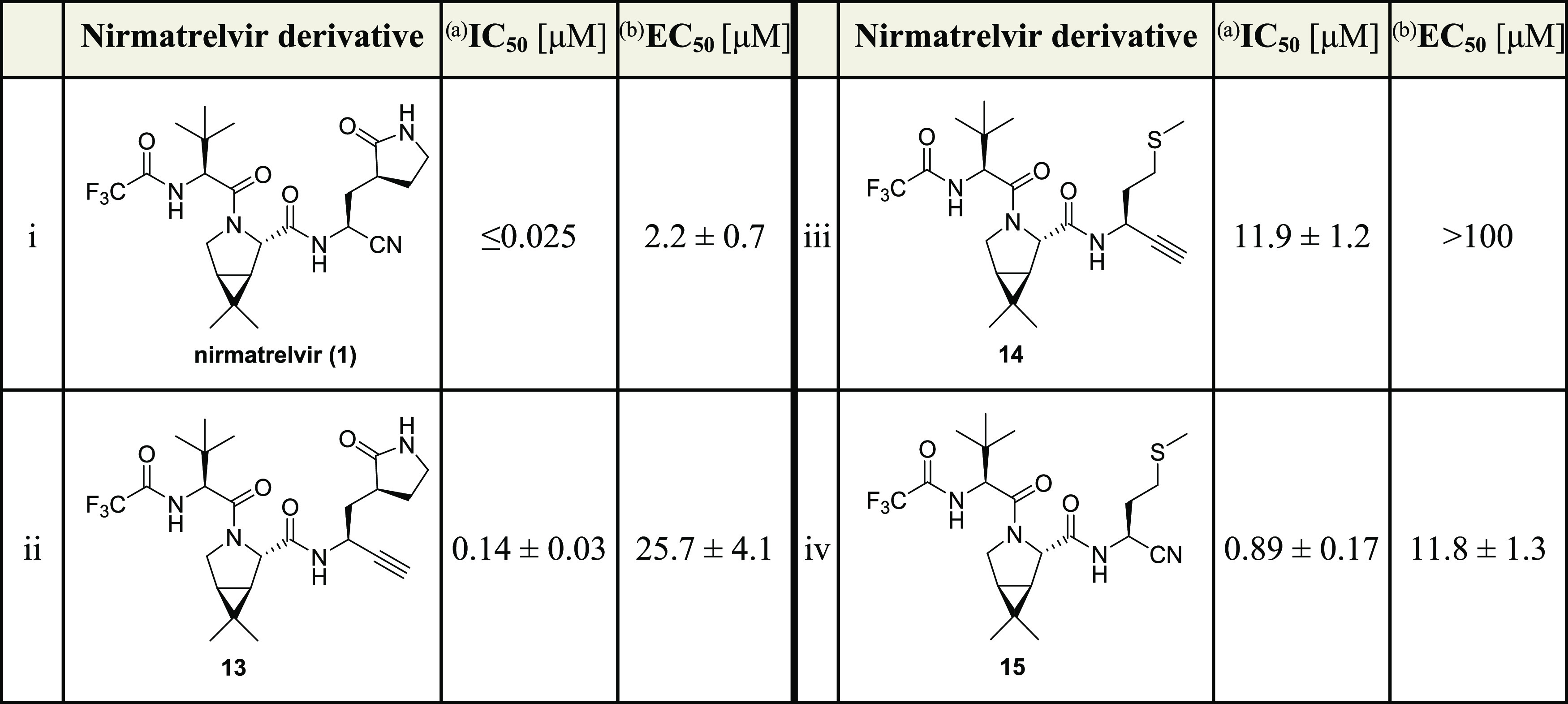
Inhibition assays were performed using SPE-MS as described employing SARS-CoV-2 Mpro (0.05 μM) and ALNDFSNSGSDVLYQPPQTSITSAVLQ/SGFRKMAFPS-NH2 as a substrate (2.0 μM) in the presence of an internal standard (Supporting Figures S1 and S2).43,62 The results are means of three independent repeats, each composed of technical duplicates (n = 3; mean ± standard deviation, SD). Representative dose–response curves are shown in Figure 3a.
Representative dose–response curves for cell-based assays are shown in Supporting Figure S3a. The results are means of three independent repeats (n = 3; mean ± SD).
The ability of nirmatrelvir alkyne derivative 13 to inhibit SARS-CoV-2 progression in infected VeroE6 cells was assessed in the absence of CP-100356, which is reported to inhibit the efflux pump-mediated removal of 1 from cells.1 Importantly, the half-maximum effective concentrations (EC50 values) revealed that alkyne 13 inhibits SARS-CoV-2 progression in cells, however, ∼11-fold less efficiently than nirmatrelvir (EC50 ∼ 2.2 μM for 1 and ∼25.7 μM for 13; Table 1, entries i and ii), a difference consistent with the studies with isolated Mpro. Alkyne 13 was apparently not cytotoxic by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay (Supporting Table S1). Note that further biological investigations are required because the interpretation of cell-based SARS-CoV-2 progression inhibition data can be challenging and metabolism of alkynes in animals may influence toxicity (Figure 3).63
Figure 3.

Dose–responses observed for nitrile- or alkyne-bearing SARS-CoV-2 Mpro inhibitors. Representative dose–response curves of Mpro inhibitors shown in (a) Table 1, (b) Table 2, and (c) Table 3. High Z′-factors66 (>0.5 for each inhibition plate) indicate excellent solid-phase extraction coupled to mass spectrometry (SPE-MS) assay quality (Supporting Figure S5).
Nirmatrelvir alkyne derivatives with a methionine at the substrate P1 equivalent position were of particular interest for SAR studies, because efficient SARS-CoV-2 Mpro inhibitors with a methionine at this position have been reported.25,64 Thus, the alkyne 14 (Table 1, entry iii) was synthesized from the reported methionine alkyne building block corresponding to 11(65) to probe the effect of the P1 substrate-equivalent substituent on inhibitor potency (Supporting Figure S4). To enable comparison with nirmatrelvir (1), the corresponding methionine nitrile analogue of nirmatrelvir (15; Table 1, entry iv) was synthesized from commercially sourced methionine amide by adapting Pfizer’s reported synthesis of 1 (Supporting Figure S4).1
Inhibition assays reveal that substituting the P1 substrate-equivalent glutamine derivative of nirmatrelvir (1) for a methionine derivative, as in 15, results in a ∼35-fold decrease in inhibition potency vs isolated SARS-CoV-2 Mpro (IC50 ≤ 0.025 μM for 1 and ∼0.9 μM for 15; Table 1, entries i and iv) but only in a ∼5-fold decrease in inhibiting SARS-CoV-2 progression in infected VeroE6 cells compared to 1 (EC50 ∼ 2.2 μM for 1 and ∼11.8 μM for 15; Table 1, entries i and iv). These observations highlight the importance of the cycloglutamine P1 Gln analogue at the P1 substrate-equivalent residue present in 1 and other inhibitors for efficient inhibition of isolated Mpro. Interestingly, nitrile 15 inhibited SARS-CoV-2 progression in infected cells ∼2-fold more efficient than alkyne 13 (EC50 ∼ 11.8 μM for 15 and ∼25.7 μM for 13; Table 1, entries iv and ii), despite its ∼6-fold reduced potency in inhibiting isolated Mpro (Table 1). Note that the alkyne methionine derivative 14 was ∼85-fold less efficient in inhibiting isolated Mpro than 13 (IC50 ∼ 11.9 μM, Table 1, entry iii), indicating that nonactivated terminal alkynes may be of interest as electrophilic warheads in Mpro inhibitors other than those related to 1.
Alkyne Derivatives of Investigational COVID-19 Therapeutics Inhibit Mpro
To investigate the potential of alkynes as electrophilic warheads of substrate-derived SARS-CoV-2 Mpro inhibitors other than those closely related to nirmatrelvir (1), we synthesized alkyne derivatives of the aldehyde MI-09 (4, Figure 2c), which has been reported to efficiently inhibit Mpro both in vitro and in animal model studies.27 Note that the structure of MI-09 (4) differs from that of nirmatrelvir (1), both with respect to its electrophilic warhead and its P3/P4 substrate-equivalent substituents, while both molecules have the same P1 and P2 substrate-equivalent residues. The MI-09 alkyne derivatives 18 and 19 were synthesized by COMU55-mediated amide coupling of alkyne 11 with the reported acid 16(27) or 17 in 14 and 13% yield, respectively, following HPLC purification (Scheme 2). For comparison, the corresponding nitrile-bearing MI-09 derivatives 20 and 21, which have not been previously reported, were synthesized according to procedures for the synthesis of 1 (Table 2 and Supporting Figure S6).1
Scheme 2. Synthesis of Alkyne Derivatives of the Investigational COVID-19 Therapeutics GC376 (3) and MI-09 (4).

Reagents and conditions: (a) 16(27) or 17, COMU,55 NMM, DMF/CH2Cl2, 0 °C to rt, 14 and 13%, respectively; (b) COMU,55 NMM, DMF/CH2Cl2, 0 °C to rt, 33%.
Table 2. Inhibition of SARS-CoV-2 Mpro by Alkyne Derivatives of Investigational COVID-19 Small-Molecule Therapeuticsa,b.

Inhibition assays were performed using SPE-MS as described employing SARS-CoV-2 Mpro (0.05 μM) and ALNDFSNSGSDVLYQPPQTSITSAVLQ/SGFRKMAFPS-NH2 as a substrate (2.0 μM) in the presence of an internal standard (Supporting Figures S1 and S2).43,62 The results are means of three independent repeats, each composed of technical duplicates (n = 3; mean ± SD). Representative dose–response curves are shown in Figure 3b.
Representative dose–response curves for cell-based assays are shown in Supporting Figure S3b. The results are means of three independent repeats (n = 3; mean ± SD). Cbz: −C(O)OCH2C6H5.
The alkyne derivative 23 of GC376 (3, Figure 2b), which is a broad-spectrum pan-coronavirus Mpro inhibitor,25,26,31 was synthesized by COMU55-mediated amide coupling of commercially sourced Z-Leu-OH (22) with alkyne 11 in 33% yield following HPLC purification as a 5:1 mixture of diastereomers (Scheme 2).
The MI-09-derived nitrile 20 inhibits isolated SARS-CoV-2 Mpro less efficiently than nirmatrelvir (1), but with a similar efficiency as the nirmatrelvir alkyne derivative 13 (IC50 ∼ 0.16 μM, Table 2, entry i). By contrast, the MI-09 alkyne derivative 18 was ∼17-fold less potent than 20 and ∼20-fold less potent than 13 (IC50 ∼ 2.8 μM, Table 2, entry ii), in accord with a similar trend observed for 1 and 13, which is about 5-fold less efficient than 1 (Table 1). Similar to nitrile 21, which inhibits Mpro with comparable potency as nitrile 20 (IC50 ∼ 0.16 μM, Table 2, entry iii), its alkyne derivative 19 was as potent as the corresponding alkyne 18, within experimental error (IC50 ∼ 3.0 μM, Table 2, entry iv). Although alkynes 18 and 19, as well as nitriles 20 and 21, were potent inhibitors of isolated SARS-CoV-2 Mpro, they did not efficiently inhibit SARS-CoV-2 progression in infected VeroE6 cells (Table 2). The ability of nitriles 20 and 21 to inhibit SARS-CoV-2 progression in infected VeroE6 cells was ∼20- to ∼30-fold reduced than that of nirmatrelvir (1), while alkynes 18 and 19 did not show any inhibitory activity over the tested concentration range, highlighting the importance of the P3 and P4 substrate-equivalent positions for efficient inhibition, in accord with previous observations by Pfizer.1
The GC376-derived alkyne 23 inhibited Mpro about 2-fold less efficiently than the MI-09-derived alkynes 18 and 19 (IC50 ∼ 4.7 μM, Table 2, entry i) and more than a 100-fold less efficiently than GC376 (Table 2, entry vi). GC376-derived alkyne 23 inhibited Mpro also about ∼30-fold less efficient than the nirmatrelvir-derived alkyne 13, an observation which may in part reflect its less rigid structure compared to nirmatrelvir (1), rendering it less optimized for efficient binding to the Mpro active site, and in part its reduced purity (5:1 diastereomeric mixture). Note that while GC376 (3) inhibited SARS-CoV-2 progression in infected VeroE6 cells with similar efficiency to that reported (EC50 ∼ 2.5 μM, Table 2, entry vi),25,26 alkyne 23 did not show any inhibitory activity over the tested concentration range (Table 2, entry v). Nonetheless, the results clearly illustrate the general utility of nonactivated terminal alkynes as electrophilic warheads for inhibitors of isolated Mpro, including those which are attached to less tight-binding inhibitor scaffolds than that of 1.
Nirmatrelvir Derivatives with C-Terminal-Derivatized Alkynes Inhibit Mpro
The reactivity of alkynes with nucleophiles may be altered by introducing substituents other than a proton at the terminal position. In the case of Mpro inhibition, substituents of interest include sterically bulky groups that may bind in the S′ pockets and those which alter the electronic properties of the alkyne and thus its reactivity and selectivity profile with respect to the reaction with nucleophiles, e.g., electron-withdrawing or -donating groups.
Initially, we investigated derivatives 27a and 27b bearing aryl-substituted alkynes as SARS-CoV-2 Mpro inhibitors, in part because previous work has shown that the S2′ pocket can accommodate aromatic groups. Derivatives 27a and 27b were synthesized by Sonogashira coupling67,68 of 24, an intermediate in the synthesis of building block 11 (Scheme 1), with aryl iodides, followed by Boc deprotection, by COMU55-mediated amide coupling with the reported acid 12,1 and then HPLC purification (Scheme 3).
Scheme 3. Synthesis of Nirmatrelvir Derivatives Bearing an Internal Alkyne.
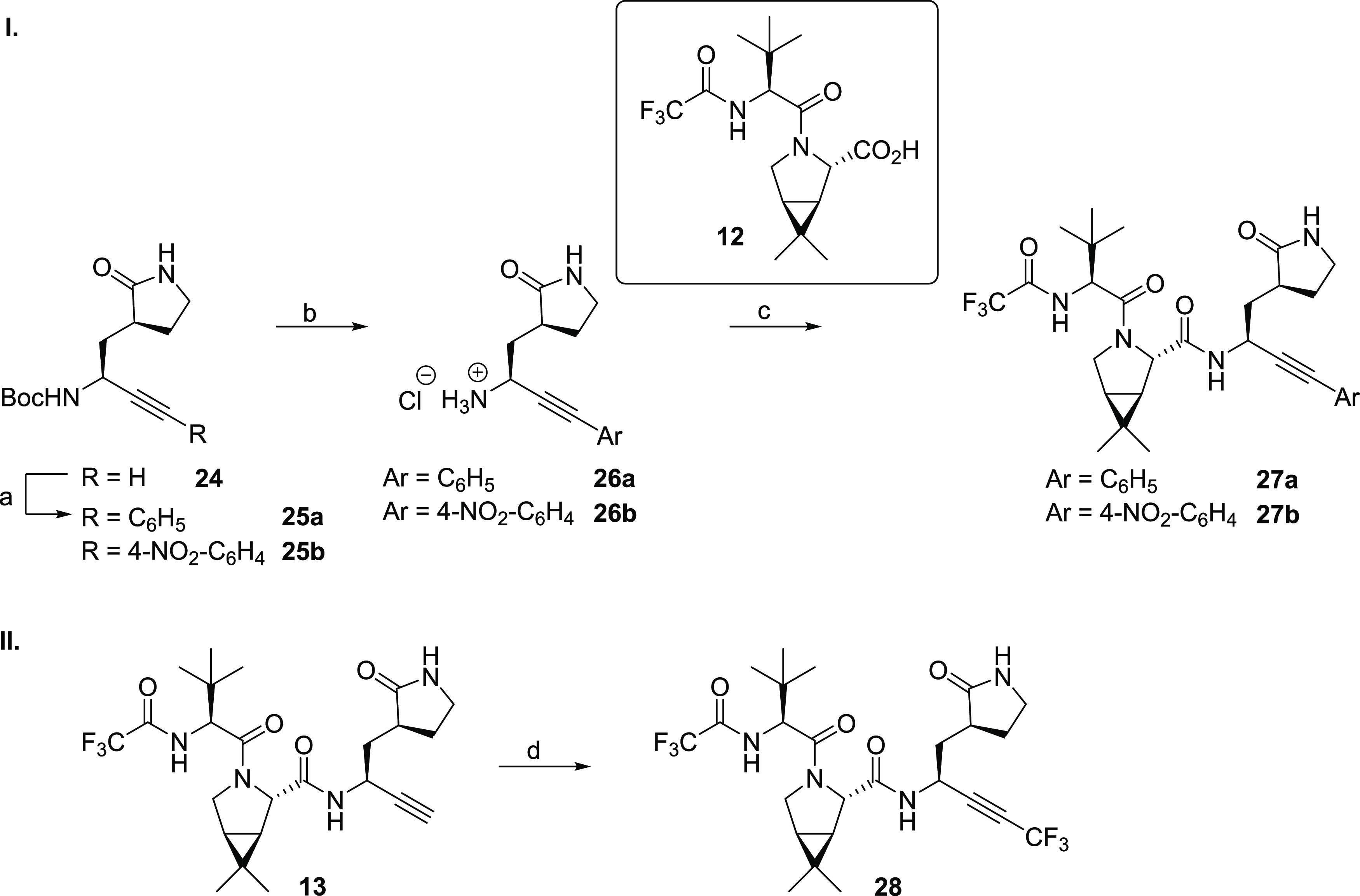
Reagents and conditions: (a) aryl iodide, CuI (5 mol %), PdCl2(PPh3)2 (2.5 mol %), NEt3/THF, 80 °C, 46% (25a) or 44% (25b); (b) HCl (4 M in dioxane), CH2Cl2, rt, app. quant.; (c) 12,1 COMU,55 NMM, DMF/CH2Cl2, 0 °C to rt, 17% (27a) or 11% (27b); (d) TMSCF3, CuI, K2CO3, N,N,N′,N′-tetramethylethylenediamine (TMEDA), DMF, rt, air, 26%.
In addition to the nirmatrelvir derivatives 27a and 27b bearing aryl-substituted internal alkynes, the terminal alkyne of 13 was capped with an electron-withdrawing CF3 group to enhance its electrophilicity; the presence of an N-trifluoroacetyl group in nirmatrelvir was also important in its in vivo optimization.1 The synthesis of 28 was achieved by employing a reported Cu(I)-mediated CH activation reaction (Scheme 3),69 which was performed at a late stage in the synthesis, as it was challenging to carry a CF3-capped alkyne derivative of 11 through the entire synthesis, as done for the aryl-capped alkynes 27a and 27b. Thus, the reaction of 13 with TMSCF3 afforded 28 in 26% yield following HPLC purification.
The unoptimized phenyl-capped nirmatrelvir alkyne derivatives 27a and 27b inhibit isolated SARS-CoV-2 Mpro ∼80- to ∼50-fold less efficiently than the terminal nirmatrelvir alkyne derivative 13 (IC50 ∼ 11.0 and ∼7.1 μM, respectively; Table 3, entries ii and iii). By contrast, the CF3-capped alkyne 28 inhibits Mpro with a similar potency as the underivatized terminal alkyne 13 (IC50 ∼ 0.22 μM, Table 3, entry iv), within experimental error, an observation which is remarkable considering that the size of the CF3 group ranges in between those of isopropyl and tert-butyl groups.70,71 Thus, the loss of inhibition potency upon capping the alkyne of 13 with the tested phenyl groups is likely to be, in a substantial part, a result of electronic effects.
Table 3. Inhibition of SARS-CoV-2 Mpro by Nirmatrelvir Derivatives Bearing a C-Terminal Derivatized Alkyne.

Inhibition assays were performed using SPE-MS as described employing SARS-CoV-2 Mpro (0.05 μM) and ALNDFSNSGSDVLYQPPQTSITSAVLQ/SGFRKMAFPS-NH2 as a substrate (2.0 μM) in the presence of an internal standard (Supporting Figures S1 and S2).43,62 The results are means of three independent repeats, each composed of technical duplicates (n = 3; mean ± SD). Representative dose–response curves are shown in Figure 3c.
Representative dose–response curves for cell-based assays are shown in Supporting Figure S3c. The results are means of three independent repeats (n = 3; mean ± SD).
The phenyl-capped alkyne derivatives 27a and 27b inhibit SARS-CoV-2 progression in infected VeroE6 cells ∼2- to ∼3-fold less efficiently than the terminal alkyne 13 (EC50 ∼ 65.2 and ∼88.2 μM, respectively; Table 3, entries ii and iii). By contrast, the CF3-capped alkyne 28 inhibits SARS-CoV-2 progression in infected VeroE6 cells ∼5-fold more efficiently than the terminal alkyne 13 (EC50 ∼ 5.1 μM, Table 3, entry iv), even though both 13 and 28 inhibit isolated SARS-CoV-2 Mpro with similar potency; this observation may reflect the enhanced electrophilicity of 28 or other, currently unknown, factors such as, e.g., improved pharmacodynamic and/or pharmacokinetic properties. Importantly, the results reveal that capping the terminal alkyne of 13 (and likely other terminal alkynes) appears to be a suitable strategy to improve the activity of alkyne Mpro inhibitors in cells. Note that while alkyne 28 inhibits SARS-CoV-2 progression in infected VeroE6 cells ∼2-fold less efficiently than nirmatrelvir (1) and GC376 (3), its potency is ∼2-fold higher than that of nirmatrelvir methionine nitrile 15, suggesting that the structure of 13 or 28 can be further optimized to increase cellular activity further, potentially to achieve similar or better inhibitory activity in cells than observed for 1 and 3.
Alkyne-Bearing Mpro Inhibitors Inhibit Ser144Ala SARS-CoV-2 Mpro Catalysis
The Ser144Ala SARS-CoV-2 Mpro variant has been observed in humans and is reported to reduce the inhibition efficacy of nirmatrelvir by >10-fold.72,73 Ser144 is located on the loop that forms the Mpro oxyanion hole and is adjacent to the nucleophilic Cys145, the backbone NH of which, along with those of Gly143 and Ser144, helps stabilize the tetrahedral oxyanionic intermediates during catalysis. Ser144 variations can reduce the potency of nirmatrelvir, and it has thus been proposed that Ser144 Mpro variations may enable SARS-CoV-2 to develop resistance toward nirmatrelvir treatment.72,73 We therefore investigated the efficacy of the nirmatrelvir alkyne derivatives against the Ser144Ala Mpro variant using SPE-MS inhibition assays (Supporting Table S2).
The results show that the Ser144Ala variant is catalytically less active than wildtype (WT) Mpro and that nirmatrelvir (1) inhibits Ser144Ala Mpro less efficiently than WT Mpro (∼4-fold), in accord with previous reports,72,73 likely in both cases because of impaired oxyanion stabilization. Importantly, the results show that the nirmatrelvir alkyne derivatives 13, 14, 27a, 27b, and 28 also inhibited Ser144Ala Mpro, however, ∼2- to ∼4-fold less efficiently than WT Mpro (Supporting Table S2). Interestingly, the MI-0927-derived nitrile 20 inhibits Ser144Ala Mpro ∼20-fold less efficiently than WT Mpro, whereas the corresponding alkyne 18 inhibits Ser144Ala Mpro only ∼3-fold less efficiently than WT Mpro. Although further work is required, these results reveal the effect of catalytically relevant Mpro active site variations, in particular substitution of an oxyanion hole-related residue (Ser144), to differentially impact on the relative inhibition efficiency of alkyne- and nitrile-based inhibitors and, by implication, of other electrophilic warheads.
Alkyne-Bearing Mpro Inhibitors Do Not Affect SARS-CoV-2 PLpro Catalysis
Since the covalent reaction of the active site cysteine Cys111 of SARS-CoV-2 PLpro with alkyne electrophiles has been described,51,54 the selectivity of the synthetic alkyne SARS-CoV-2 Mpro inhibitors 13, 14, 18, 19, 23, 27a, 27b, and 28 was determined using reported SPE-MS PLpro inhibition assays, which directly monitor PLpro-catalyzed hydrolysis of a substrate-derivate oligopeptide.74 In addition, the nitrile Mpro inhibitors 15, 20, and 21 were investigated for PLpro inhibition. None of the tested Mpro inhibitors inhibited SARS-CoV-2 PLpro, in accord with the reported lack of PLpro inhibition by nirmatrelvir (Supporting Table S3).74
Nirmatrelvir Alkyne Derivative 13 Inhibits Mpro by Covalent Reaction with Cys145
The mechanism by which the alkyne 13 inhibits SARS-CoV-2 Mpro was next investigated by crystallography. Twelve microcrystals, which were obtained by cocrystallization of Mpro with 13, were analyzed using the VMXm microfocus beamline at the Diamond Light Source synchrotron;75,76 the diffraction data were merged, and a structure of Mpro in complex with 13 was solved by molecular replacement using a reported Mpro structure (PDB ID: 6YB7) as a search model (P21212 space group, 1.9 Å resolution; Supporting Figure S7 and Table S4). The overall Mpro protein folds in the Mpro:13 complex and the reported Mpro:1 complex (PDB ID: 7TE0(22)) structures are very similar (RMSD = 0.313 Å, Supporting Figure S7).
The Mpro:13 complex structure unambiguously shows that the nucleophilic thiolate of Cys145 covalently reacts with the terminal alkyne group of 13 at the more electrophilic internal alkyne position (distance of the Cys145 S-atom to the internal C-atom of the vinyl group: 1.8 Å; Figure 4a). The groups at the P1-4 substrate-equivalent positions of 13 occupy the same Mpro substrate binding pockets as observed for 1, i.e., the cycloglutamine group occupies the S1 pocket, the bicyclic leucine isostere occupies the hydrophobic S2 pocket, the tert-butyl group is solvent-exposed, and the trifluoroacetamide occupies the S4 pocket, and are positioned to interact with Mpro via similar H-bonding interactions as observed for 1 (Supporting Figure S8). In contrast with the Mpro:1 complex structure, in the alkyne 13 structure, the terminal olefin C-atom of the vinyl thioether formed by the reaction with Cys145 is not positioned in the oxyanion hole (distances of the terminal olefin C-atom to the Cys145 and Gly143 main chain N-atoms: 3.6 and 3.8 Å, respectively) compared to the thioimidate nitrogen in 1 (distances of the thioimidate N-atom to the Cys145 and Gly143 main chain N-atoms: 3.1 and 3.3 Å, respectively; PDB ID: 7R7H(32)), indicating the absence of H-bonding-type interactions of the intermediate vinyl anion with the oxyanion hole in the Mpro:13 complex (Figure 4). Although care should be taken in correlating the precise mechanism in solution with crystal structures, this observation suggests that the proposed vinyl anion intermediate is protonated, potentially by one of the oxyanion hole NH groups or by water. Note that two SARS-CoV-2 Mpro complex structures in which the nucleophilic thiolate of Cys145 has reacted with activated bromo alkynes of unspecific protease inhibitors have been reported; in these structures, the terminal olefin C-atom of the vinyl thioether formed with Cys145 is positioned in the oxyanion hole (Supporting Figure S9).53 The apparently different binding mode of 13 compared to 1 with respect to the oxyanion hole is of interest, given the differences in relative potencies for some of the nitriles and alkynes versus WT and Ser144Ala Mpro, as described above.
Figure 4.

Crystallographic evidence that the alkyne of the nirmatrelvir derivative 13 reacts covalently with the nucleophilic thiolate of Mpro Cys145. Color code: SARS-CoV-2 Mpro, gray; carbon backbone of 13 in complex with Mpro is in orange; oxygen, red; nitrogen, blue; sulfur, yellow; and fluorine, light blue. (a) Reaction of Mpro with alkyne 13; (b) representative OMIT electron density map (mFo–DFc) contoured to 3σ around Cys145 and 13 in complex with Mpro (PDB ID: 8B2T); (c) superimposition of a view from the Mpro:13 complex structure (PDB ID: 8B2T) with the reported Mpro:nirmatrelvir (1) complex structure (pale yellow: Mpro; green: carbon backbone of 1 in complex with Mpro; PDB ID: 7TE0(22)) showing the interaction of 1 but not 13 with residues forming the oxyanion hole, i.e., Cys145 and Gly143.
Evidence That Nirmatrelvir Alkyne Derivative 13 Is an Irreversible Covalent Mpro Inhibitor
It has been reported that the reaction of nirmatrelvir (1) with the nucleophilic thiolate of SARS-CoV-2 Mpro Cys145 is reversible,1 in accord with the reversible reaction of other small-molecule nitrile inhibitors with nucleophilic cysteine or serine residues32,44,77 and a mechanism in which the negatively charged thioimide electron pair occupies the Mpro oxyanion hole (Figure 1). By contrast, it appears feasible that the initially formed vinyl anion (by the reaction of the nucleophilic thiolate of Mpro with the alkyne group of 13) is protonated as it is oriented away from the oxyanion hole in the Mpro:13 complex structure (Figure 4), suggesting that the reaction of 13 with Mpro may be irreversible. To investigate this, we initially analyzed the covalent reaction of Mpro with synthetic alkyne and nitrile inhibitors (i.e., 1, 13–15, 18–21, 23, 27a, 27b, and 28) at a fixed concentration using protein-observed SPE-MS. The results revealed that all of the synthesized nitrile and alkyne inhibitors react covalently and once with Mpro, likely with the nucleophilic active site Cys145, as supported by crystallographic analysis (Figure 4). Their apparent reaction rates with Mpro differed, with nirmatrelvir (1) and its alkyne derivative 13 appearing to react fastest with Mpro (Supporting Figures S10–S21).
To assess the reversibility of the covalent reaction of Mpro with nirmatrelvir (1) and alkyne 13, we then separately incubated Mpro with an excess of 1 or 13, until apparent complete formation of the corresponding covalent Mpro adducts was observed by SPE-MS (Figure 5a). Unreacted 1 and 13 were removed from the mixtures, and a 10-fold excess of the CF3-bearing alkyne 28 was added to the covalent complexes of Mpro with 1 and 13; competition for binding to Mpro was then monitored by SPE-MS as a function of time (Figure 5). Alkyne 28 was used because it is a potent Mpro inhibitor, which covalently reacts with Cys145 (Table 3 and Figure 5a) and because its mass differs substantially from the masses of 1 and 13 (by ∼67 Da), thus enabling the differentiation of the Mpro complex with 28 from those with 1 or 13, by SPE-MS. The results revealed that, while the covalent Mpro adduct with nirmatrelvir (1) slowly exchanges with 28 over time reaching ∼20% exchange 4 h post incubation (Figure 5b,c), the covalent Mpro adduct with 13 did not exchange with 28, even after prolonged incubation (>48 h). Thus, the covalent reaction of Mpro with nitriles such as 1 is reversible, whereas the covalent reaction of Mpro with alkynes such as 13 is irreversible or at least substantially less reversible than the reaction with nitriles.
Figure 5.

Nirmatrelvir alkyne derivatives inhibit SARS-CoV-2 Mpro via covalent reaction with Cys145. (a) Reaction of Mpro (bottom) with nirmatrelvir (1, center) and its alkyne derivative 13 (top); SPE-MS analysis indicates near-quantitative reaction of 1 or 13 with Mpro (∼500 Da mass shifts). (b) Addition of a 10-fold excess of alkyne 28 to the covalent complexes of Mpro with 1 or 13, followed by 30 min incubation; SPE-MS analysis of the mixtures reveals formation of a covalent complexes of Mpro with 28 only for the Mpro complex with 1 (∼66 Da mass shift, center), but not with 13 (top), as compared with unreacted Mpro (bottom). (c) SPE-MS analysis of the mixtures of the covalent complexes of Mpro with 1 or 13 with 28 indicates that 28 reacts slowly with the Mpro complex with 1 (∼66 Da mass shift, center), but not with 13 (top), as compared with unreacted Mpro (bottom). Mpro assays were performed using SPE-MS as described in the Experimental Section employing SARS-CoV-2 Mpro (2.0 μM) in buffer (20 mM HEPES, pH 7.5).43,62
Discussion
Alkynes are established functional groups in active pharmaceutical ingredients (APIs) of approved human therapeutics (Figure 6a) and molecules under clinical investigation, though they can induce cytotoxicity and they can be metabolized via reaction with CYP450 monooxygenases.63 Nonactivated terminal alkynes are present in APIs, e.g., in the dihydrofolate reductase inhibitor pralatrexate (29),78 in the monoamine oxidase-B inhibitors erlotinib (30), selegiline, and rasagiline which are used to alleviate symptoms of Parkinson’s disease,79 and in the steroidal drugs danazol, gestrinone, and 17α-ethynylestradiol (31), the latter of which is widely used as a contraceptive.63 C-Terminal derivatized alkynes occur in APIs of drugs, e.g., in the DPP4 inhibitor linagliptin,80 ponatinib (32, used to treat acute myeloid leukemia),81 the antifungal therapeutic terbinafine (33),82,83 the HIV reverse transcriptase inhibitor efavirenz (34),84 the HIV therapeutic lenacapavir,85 the steroid mifepristone used for medical abortions,63 and in the antibody-drug conjugates gemtuzumab ozogamicin and inotuzumab ozogamicin (used to treat leukemia).86
Figure 6.
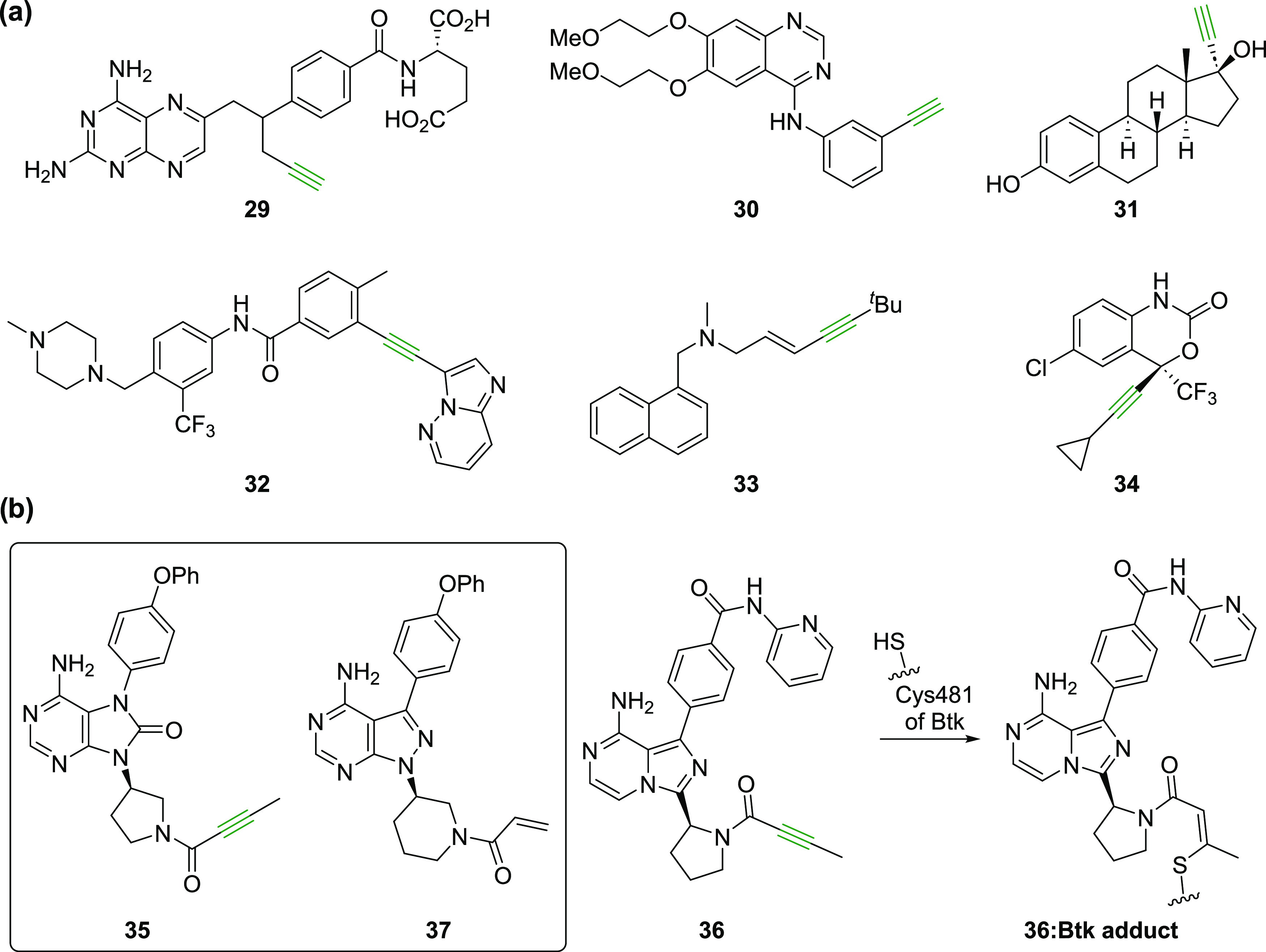
Alkynes are important functional groups in human therapeutics. (a) Representative human therapeutics that contain alkynes (in green); (b) covalent reaction of acalabrutinib (36)87 with Btk Cys481.
Bruton’s tyrosine kinase (Btk) inhibitors tirabrutinib (35) and acalabrutinib (36)87 (used to treat inter alia chronic lymphocytic leukemia and lymphoma, respectively) contain an yneamide group, the electrophilic alkyne of which covalently reacts with the nucleophilic thiolate of Btk Cys481 resulting in efficient inhibition (Figure 6b),87,88 further highlighting the use of alkynes as clinically useful warheads. These two Btk inhibitors were designed based on the structure of the clinically used compound ibrutinib (37),89,90 which, however, employs an acrylamide group rather than an yneamide group for covalent reactions with Btk Cys481 (Figure 6b).88 It has been proposed that off-target Michael reactions of ibrutinib and related acrylamide-bearing Btk inhibitors with other nucleophilic cysteine residues are in part responsible for side effects of ibrutinib and that the corresponding yneamides are less reactive electrophiles with better pharmacokinetic properties.87,91,92
Propargyl amides have also been used as (latent) electrophilic warheads for activity-based probes of deubiquitinases, including of SARS-CoV-2 PLpro, which covalently reacts via its active site cysteine Cys111 with terminal alkynes.51,54 However, nonactivated terminal alkynes have, to our knowledge, not been used as electrophilic warheads in SARS-CoV-2 Mpro inhibitors. Mpro inhibitors containing terminal alkynes, which are not covalently modified,93 and Mpro inhibitors with activated alkynes, i.e., yneamides20 and bromo alkynes,53 have been reported; however, their inhibition mechanisms have not been investigated in detail. We chose to investigate whether nirmatrelvir derivatives with an alkyne electrophilic warhead inhibit SARS-CoV-2 Mpro by reaction with Cys145 because (i) nirmatrelvir (1) is optimized for tight binding to the Mpro active site; hence, its scaffold should favor the reaction with terminal alkynes which are normally less reactive with nucleophiles than nitriles (template effect);1 (ii) nirmatrelvir alkyne derivatives may show a different reactivity profile with potential future nirmatrelvir-resistant Mpro variants;94 (iii) nirmatrelvir alkyne derivatives may show an altered selectivity profile with regard to inhibiting human cysteine proteases; and (iv) nirmatrelvir alkyne derivatives may manifest improved cellular properties, including with respect to penetration and efflux, in the latter case potentially avoiding the need for coadministration with an efflux inhibitor, as done in cellular studies with 1. Note that unlike GC376 (3), which also efficiently inhibits human cathepsins in vitro,32,951 is reported to be a selective Mpro inhibitor, which does not inhibit human proteases;21 however, this observation may be refined as human cathepsins may catalyze hydrolysis with similar substrate recognition sites as Mpro.96
Considering that an efficient reaction of alkynes with thiols is proposed to be largely proximity-driven (template effect),48,50 it is remarkable that alkyne derivatives of structurally simple scaffolds like the one of GC376 (3) undergo relatively efficient reaction with Mpro Cys145, implying a broader utility of appropriately complexed nonactivated alkynes to covalently react with nucleophilic cysteine residues and potentially proteins with other nucleophilic residues. Note that a cysteine to serine Btk variant has been reported to be resistant towards treatment with common covalent Btk inhibitors such as ibrutinib, implying that different preorganized binding modes are required for the reaction of alkynes with different types of nucleophiles.97 This proposal is consistent with studies showing that the substitution of nucleophilic Cys to Ser, or vice versa, in nucleophilic enzymes normally leads to loss of activity, including in the case of Mpro.98,99
Our studies reveal that both activated and non-activated alkynes are suitable irreversibly reacting warheads to covalently inhibit isolated Mpro and SARS-CoV-2 progression in infected cells (Tables 1–3). Considering that alkynes are versatile functional groups which can easily be modified, including at a late stage using mild protocols which include, but are not limited to, trifluoromethylation and Sonogashira couplings as described here (Scheme 3), the studies suggest that there is potential to further optimize nirmatrelvir and related Mpro inhibitors, e.g., by introducing substituents on the alkyne, which bind tightly in the Mpro S′ sites and/or modulate pharmacodynamic and/or pharmacokinetic properties. The irreversible nature of the reaction of the alkynes compared to the analogous nitriles means that the alkynes should be useful to inform on the selectivity of the analogous nitriles in terms of reactivity in cells, using activity-based profiling-type approaches.100
Importantly, our results show that the CF3-capped alkyne 28 inhibits SARS-CoV-2 progression in infected VeroE6 cells ∼5-fold more efficiently than the terminal alkyne 13 (EC50 ∼ 5.1 μM, Table 3, entry iv), highlighting the potential of appropriately capped alkynes for potent inhibition of isolated SARS-CoV-2 Mpro and SARS-CoV-2 progression in cells (and by analogy other nucleophilic cysteine enzymes). Likely, alkynes 13 and 28 can be optimized to achieve similar or better inhibitory activity in cells than nirmatrelvir (1) and GC376 (3).
Crystallographic analysis reveals that the terminal methylene group of the vinyl thioether resulting from the reaction of 13 with Cys145 is not located in the oxyanion hole, which is formed by the NH groups of Gly143, Ser144, and Cys145, contrasting with the thioimidate nitrogen in the complex formed by 1, which is located in the oxyanion hole (Figure 4). This observation is of interest given that Ser144Ala Mpro is implicated in nirmatrelvir resistance and because of the differences in relative potencies observed for some of the nitriles and alkynes versus WT and Ser144Ala Mpro. Although further work is required, it may be that optimized derivatives of irreversibly binding alkyne-based inhibitors of WT Mpro and/or Mpro active site variants can be developed that are more efficient than those bearing reversibly binding nitrile or other electrophilic warheads.
Importantly, weobserved that the covalent reaction of Mpro with nirmatrelvir alkyne 13 is irreversible or at least substantially less reversible than its covalent reaction with nirmatrelvir (1) (Figure 5). This result contrasts with the reported reversibility of the covalent reaction of Mpro with 1 (and other nitrile-bearing small-molecule inhibitors with nucleophilic serine or cysteine proteins32,44,77) and with the observed conformation of its thioimidate in the Mpro:1 complex, which suggests that the negative charge at the thioimidate N-atom is stabilized by interactions with the oxyanion hole, potentially hampering the protonation of the N-atom.1,24
The covalent reaction of the Cys145 thiolate with the terminal alkyne of 13 likely results in the initial formation of a vinyl anion (Figure 4a); vinyl anions are typically more basic than thioimidate anions, likely resulting in protonation of the vinyl anion by an NH in the oxyanion hole, by a nearby water, or by an Mpro residue with an acidic proton. This process is likely irreversible as the basicity of an oxyanion hole may be insufficient to deprotonate a non-activated alkene which are per se not very acidic, in accord with our MS data (Figure 5) and the observation that the vinyl group in the Mpro:13 complex is oriented away from the oxyanion hole (Figure 4). Note that the Mpro:13 complex structure was obtained using data from multiple (12) microcrystals at the recently constructed VMXm microfocus/nanofocus beamline at the Diamond Light Source synchrotron, a method that should have general utility.75,76
The inhibition results show that the electrophilic nitrile group is preferred over the isoelectronic alkyne group for inhibition of isolated Mpro in a manner independent of the inhibitor scaffold, at least with the evaluated scaffolds under the tested conditions (Tables 1–3). Importantly, however, they imply that the increased potency for the nitrile over the alkyne may be substantially less in the cellular context (Table 1). It should be possible to increase the potency of the alkyne Mpro inhibitors by optimizing the P1-4 substrate-equivalent groups as well as the terminal alkyne substituent. Further SAR studies are required to validate our observations, but the results highlight the importance of optimizing potency in cells as well as against isolated Mpro. The relative difference between the inhibition results with isolated Mpro and in cells may in part reflect the irreversible reaction of alkyne inhibitors with Mpro.
Conclusions
The results reveal the therapeutic potential for the covalent inhibition of Mpro and other nucleophilic cysteine proteases by alkynes, which, in contrast to more electrophilic nitriles such as in nirmatrelvir (1), react irreversibly and can be functionalized at the alkyne terminal position, properties that can be used to optimize inhibition, including with respect to drug-resistant Mpro variants, selectivity/safety, and pharmacokinetics.
Experimental Section
The syntheses and characterizations of the SARS-CoV-2 Mpro inhibitors used in this work are disclosed in the associated Supporting Information. All compounds are ≥95% pure by NMR and HPLC analysis unless stated otherwise; NMR spectra and HPLC traces are shown for all lead compounds in the associated Supporting Information.
SARS-CoV-2 Mpro Inhibition Assays
Solid-phase extraction coupled to mass spectrometry (SPE-MS) SARS-CoV-2 Mpro inhibition assays were performed in buffer (20 mM HEPES, pH 7.5, 50 mM NaCl) at 20 °C as reported, using a freshly thawed aliquot of recombinant SARS-CoV-2 Mpro (multiple freeze–thaw cycles were avoided). The Mpro sequence was based on the Wuhan-Hu-1 genome101 (National Center for Biotechnology Information (NCBI) reference sequence: NC_045512.2). Mpro Mpro was prepared according to established procedures,43 and a 37-mer oligopeptide (ALNDFSNSGSDVLYQPPQTSITSAVLQ/SGFRKMAFPS-NH2), which was based on the on the sequence of the N-terminal SARS-CoV-2 Mpro self-cleavage site and which was synthesized as a C-terminal amide and purified by GL Biochem (Shanghai) Ltd. (Shanghai, China), was used as a substrate (2.0 μM).62 Note that SPE-MS assays were performed using a lower protein concentration than that reported (0.05 μM rather than 0.15 μM) and in the presence of the N-terminally acetylated C-terminal product peptide (Ac-SGFRKMAFPS-NH2) as an internal standard to account for inhibitor-induced suppression of ionization of the product peptide.
In detail, solutions of the inhibitors (100% DMSO) were dry-dispensed across 384-well polypropylene assay plates (Greiner) in an approximately threefold and 11-point dilution series (100 μM top concentration) using an ECHO 550 acoustic dispenser (Labcyte). DMSO and formic acid were used as negative and positive inhibition controls, respectively. The final DMSO concentration was kept constant at 0.5%v/v throughout all experiments (using the DMSO backfill option of the acoustic dispenser). Each reaction was performed in technical duplicates in adjacent wells of the assay plates, and assays were performed in at least two independent duplicates.
An enzyme mixture (25 μL/well), containing freshly thawed SARS-CoV-2 Mpro (0.1 μM) in buffer (20 mM HEPES, pH 7.5, 50 mM NaCl), was dispensed across the inhibitor-containing 384-well assay plates with a multidrop dispenser (Thermo Fisher Scientific) at 20 °C under an ambient atmosphere. The plates were subsequently centrifuged (1000 rpm, 5 s) and incubated for 15 min at 20 °C. A substrate mixture (25 μL/well), containing ALNDFSNSGSDVLYQPPQTSITSAVLQ/SGFRKMAFPS-NH2 (4.0 μM) and Ac-SGFRKMAFPS-NH2 (0.8 μM) in buffer (20 mM HEPES, pH 7.5, 50 mM NaCl), was added using the multidrop dispenser. The plates were centrifuged (1000 rpm, 5 s), and after incubating for 30 min, the reaction was stopped by addition of 10%v/v aqueous formic acid (5 μL/well). The plates were then centrifuged (1000 rpm, 30 s) and analyzed by MS.
Note that Ser144Ala Mpro inhibition assays were performed similarly to those for WT Mpro, however, using double the concentration of Ser144Ala Mpro (0.1 μM final assay concentration) compared to WT Mpro (0.05 μM final assay concentration). The Ser144Ala mutation was introduced into the plasmid DNA encoding for WT SARS-CoV-2 Mpro using standard protocols, and recombinant Ser144Ala Mpro was produced and purified as described for WT Mpro.43
MS analyses were performed using a RapidFire RF 365 high-throughput sampling robot (Agilent) attached to an iFunnel Agilent 6550 accurate mass quadrupole time-of-flight (Q-TOF) mass spectrometer operated in the positive ionization mode as previously reported.43,62 For data analysis, the m/z +1 charge states of the C-terminal product peptide (SGFRKMAFPS-NH2) and the N-terminally acetylated C-terminal product peptide (Ac-SGFRKMAFPS-NH2) were used to extract and integrate ion chromatogram data using RapidFire Integrator software (Agilent). Data were exported into Microsoft Excel and used to calculate the product concentration using the following equation: product concentration = 0.4 μM × (integral C-terminal product peptide)/(integral N-terminally acetylated C-terminal product peptide). Normalized dose–response curves (using the formic acid and DMSO controls) were obtained from the raw data by nonlinear regression (GraphPad Prism 5) and used to determine IC50 values.
Protein-Observed Mpro Assays
Assays were performed as described using SPE-MS.62 Solutions of the inhibitors (100% DMSO) were dry-dispensed across 384-well polypropylene assay plates (Greiner) using an ECHO 550 acoustic dispenser (Labcyte). DMSO was used as a negative control. An enzyme mixture (50 μL/well), containing Mpro (2.0 μM) in buffer (20 mM HEPES, pH 7.5), was dispensed across the inhibitor-containing 384-well assay plates with a multidrop dispenser (Thermo Fisher Scientific). The reaction mixture was incubated for the indicated time at 20 °C under an ambient atmosphere prior to analysis by SPE-MS. MS analyses were performed using a RapidFire RF 365 high-throughput sampling robot (Agilent) attached to an iFunnel Agilent 6550 accurate mass Q-TOF mass spectrometer using a C4 cartridge and the same parameters as described.43,62
Cell Viability Assays
Inhibitor toxicity was assayed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay kit (Abcam – Ab211091) as per the manufacturers’ recommendation. Compounds were dispensed into 96-well plates using an ECHO 550 acoustic dispenser (Labcyte) to give a final concentration range of 100–0.11 μM. All assays were carried out in technical duplicates. VeroE6 cells (4.5 × 104) were added to each well, and the 96-well plate was then incubated for 24 h at 37 °C in a 5% CO2 atmosphere. Following incubation, the media were removed, the MTT reagent was added, and the cells were incubated for 3 h at 37 °C. Subsequently, all media were removed from the cells, the MTT solvent was added to each well, and the plate was incubated on a shaker at rt for 15 min protected from light. The absorbance was measured at 600 nm. OD600 values of the different concentrations of compound were divided by the OD600 value of the negative control to calculate the percentage cytotoxicity; the cytotoxicity curves were plotted using GraphPad Prism.
Cell-Based Antiviral Assays
Compounds were dispensed into 96-well plates using an ECHO 550 acoustic dispenser (Labcyte) to give a final concentration range of 100–0.11 μM. Compounds were preincubated with 50 μL of SARS-CoV-2 virus (Victoria strain–100 FFU) and 50 μL of VeroE6 cells (9 × 105/mL) in separate 96-well plates. Following incubation for 1 h at 37 °C in a 5% CO2 atmosphere, the virus was added to the well containing compound-treated cells. The virus was allowed to infect the cells for 2 h at 37 °C in a 5% CO2 atmosphere, followed by the addition of 100 μL of compound-adulterated carboxymethyl cellulose (1.5%) to each well. Subsequently, the plates were incubated for a further 20 h at 37 °C in a 5% CO2 atmosphere. All assays were carried out in technical duplicates.
Cells were washed with 200 μL of DPBS and then fixed with paraformaldehyde 4%v/v (100 μL/well) for 30 min at rt. Cells were permeabilized with TritonX100 (1% in PBS) and then stained for SARS-CoV-2 nucleoprotein using a human monoclonal antibody (FB9B102). Bound antibodies were detected following incubation with a goat anti-human IgG HRP conjugate (Sigma, U.K.) and following TrueBlue Peroxidase substrate (Insight Biotechnology, U.K.) addition imaged using an ELISPOT reader. The half-maximal effective concentration (EC50) was defined as the concentration of the compound that reduced the Foci forming unit (FFU) by 50% compared to the control wells.
Crystallization
A frozen SARS-CoV-2 Mpro solution was thawed and diluted to 6 mg/mL (using 20 mM HEPES, pH 7.5, 50 mM NaCl). Alkyne 13 was added to the protein solution to a final concentration of 10 mM; the mixture was incubated for 6 h at ambient temperature prior to dispensing plates. The drop composition was 0.15 μL of protein ligand solution, 0.3 μL of 11%v/v PEG 4000, 0.1 M MES, pH 6.5, and 0.05 μL of Mpro crystal seed stock. The Mpro crystal seed stock was prepared by crushing Mpro crystals with a pipette tip, suspending them in 30% PEG 4000, 5%v/v DMSO, 0.1 M MES, pH 6.5, and vortexing for 30 s with approximately 10 glass beads (1.0 mm diameter, BioSpec products). The reservoir solution contained: 11%v/v PEG 4K, 5%v/v DMSO, 0.1 M MES, pH 6.5. Note that DMSO was added to the reservoir solution; but in the crystallization drop, DMSO is provided by the solution of the ligand incubated with the protein. Crystals were grown using the sitting drop vapor diffusion method at 20 °C and appeared within 24 h. Crystal morphology was needle-like with a diameter of approximately 1 μm.
Data Collection and Structure Determination
Crystals were applied to an electron microscopy grid as described.75 In brief, standard holey carbon grids (Quantifoil, Cu 200 mesh, R2/2) were glow-discharged for 30 s at 15 mA. Crystals in a drop were resuspended in 10 μL of reservoir solution, and 3 μL of the resulting suspension was applied to the grid surface in a Leica EM GP2 plunge-freezing device at >80% humidity. Then, 2 μL of reservoir solution was dispensed onto the rear face of the grid. The grid was blotted from the back for 10 s prior to plunging into liquified ethane.
Diffraction data were collected at 100 K and a wavelength of 0.5813 Å at the VMXm beamline at Diamond Light Source from 24 crystals. 40° sweeps were collected from each crystal using 0.1° oscillations. The data were collected on an Eiger 9M CdTe detector. Data were processed using Dials103 via xia2.multiplex;76,104 data from 12 crystals were present in the final merged dataset. The datasets were phased using Molrep105 and the Mpro apo structure (PDB ID: 6YB7). Ligand restraints were generated using AceDRG;106 96.0% of the residues are in the favored regions of the Ramachandran plot, 3.3% in the allowed region, and 0.7% in high-energy conformations (two residues). Crystal structures were manually rebuilt in Coot and refined using Refmac107 and PDB_Redo (Supporting Table S4).108
The crystal structure data for the SARS-CoV-2 Mpro:13 complex structure have been deposited in the protein data bank (PDB) with accession code 8B2T. Additionally, data of the following reported crystal structure have been used: 7TE0.22
Acknowledgments
The investigators acknowledge the philanthropic support of the donors to the University of Oxford’s COVID-19 Research Response Fund and King Abdulaziz University, Saudi Arabia, for funding. This research was funded in part by the Wellcome Trust (106244/Z/14/Z). For the purpose of open access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. The authors thank the Cancer Research UK (C8717/A18245) and the Biotechnology and Biological Sciences Research Council (BB/J003018/1 and BB/R000344/1) for funding. L.D. acknowledges financial support from the EU ERAMUS+ program. T.R.M. was supported by the BBSRC (BB/M011224/1). M.W.C., S.M.L., and D.N. were funded by the Oak Foundation (0010608, Platform for Screening COVID-19 against new Antibodies and Drugs). The authors acknowledge the Diamond Light Source for the award of beamtime through the COVID-19 dedicated call (proposal ID MX27088) and thank the Diamond MX group for their support and expertise.
Glossary
Abbreviations Used
- API
active pharmaceutical ingredient
- COVID-19
coronavirus disease 2019
- Btk
Bruton’s tyrosine kinase
- DUB
deubiquitinase
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- Mpro
main protease
- PLpro
papain-like protease
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01627.
Author Contributions
⊥ L.B., L.D., and Y.Z. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A. E.; Baker S. C.; Baric R. S.; de Groot R. J.; Drosten C.; Gulyaeva A. A.; Haagmans B. L.; Lauber C.; Leontovich A. M.; Neuman B. W.; Penzar D.; Perlman S.; Poon L. L. M.; Samborskiy D. V.; Sidorov I. A.; Sola I.; Ziebuhr J. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannalire R.; Cerchia C.; Beccari A. R.; Di Leva F. S.; Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem. 2022, 65, 2716–2746. 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Shyr Z.; Lo D. C.; Zheng W. Viral proteases as targets for coronavirus disease 2019 drug development. J. Pharmacol. Exp. Ther. 2021, 378, 166–172. 10.1124/jpet.121.000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Yang J. A review of the latest research on Mpro targeting SARS-COV inhibitors. RSC Med. Chem. 2021, 12, 1026–1036. 10.1039/D1MD00066G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citarella A.; Scala A.; Piperno A.; Micale N. SARS-CoV-2 Mpro: a potential target for peptidomimetics and small-molecule inhibitors. Biomolecules 2021, 11, 607. 10.3390/biom11040607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K.; Wang R.; Chen J.; Tepe J. J.; Huang F.; Wei G.-W. Perspectives on SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021, 64, 16922–16955. 10.1021/acs.jmedchem.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anirudhan V.; Lee H.; Cheng H.; Cooper L.; Rong L. Targeting SARS-CoV-2 viral proteases as a therapeutic strategy to treat COVID-19. J. Med. Virol. 2021, 93, 2722–2734. 10.1002/jmv.26814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E.; Li G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; Duan Y.; Yu J.; Wang L.; Yang K.; Liu F.; Jiang R.; Yang X.; You T.; Liu X.; Yang X.; Bai F.; Liu H.; Liu X.; Guddat L. W.; Xu W.; Xiao G.; Qin C.; Shi Z.; Jiang H.; Rao Z.; Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. T.; Yang Q.; Gribenko A.; Perrin B. S. Jr.; Zhu Y.; Cardin R.; Liberator P. A.; Anderson A. S.; Hao L. Genetic surveillance of SARS-CoV-2 Mpro reveals high sequence and structural conservation prior to the introduction of protease inhibitor paxlovid. mBio 2022, 13, e00869-22. 10.1128/mbio.00869-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greasley S. E.; Noell S.; Plotnikova O.; Ferre R.; Liu W.; Bolanos B.; Fennell K.; Nicki J.; Craig T.; Zhu Y.; Stewart A. E.; Steppan C. M. Structural basis for the in vitro efficacy of nirmatrelvir against SARS-CoV-2 variants. J. Biol. Chem. 2022, 298, 101972. 10.1016/j.jbc.2022.101972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangeel L.; Chiu W.; De Jonghe S.; Maes P.; Slechten B.; Raymenants J.; André E.; Leyssen P.; Neyts J.; Jochmans D. Remdesivir, molnupiravir and nirmatrelvir remain active against SARS-CoV-2 omicron and other variants of concern. Antiviral Res. 2022, 198, 105252. 10.1016/j.antiviral.2022.105252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco M. D.; Hu Y.; Gongora M. V.; Meilleur F.; Kemp M. T.; Zhang X.; Wang J.; Chen Y. The P132H mutation in the main protease of omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell Res. 2022, 32, 498–500. 10.1038/s41422-022-00640-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich S.; Ekanayake K. B.; Otting G.; Nitsche C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629. 10.1016/j.bmcl.2022.128629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uraki R.; Kiso M.; Iida S.; Imai M.; Takashita E.; Kuroda M.; Halfmann P. J.; Loeber S.; Maemura T.; Yamayoshi S.; Fujisaki S.; Wang Z.; Ito M.; Ujie M.; Iwatsuki-Horimoto K.; Furusawa Y.; Wright R.; Chong Z.; Ozono S.; Yasuhara A.; Ueki H.; Sakai-Tagawa Y.; Li R.; Liu Y.; Larson D.; Koga M.; Tsutsumi T.; Adachi E.; Saito M.; Yamamoto S.; Hagihara M.; Mitamura K.; Sato T.; Hojo M.; Hattori S.-i.; Maeda K.; Valdez R.; Bennett-Baker P.; Chu Z.; Davis D.; Kowalski-Dobson T.; Eckard A.; Gherasim C.; Gremel W.; Lindsey K.; Manthei D.; Meyers A.; Moya J. Z.; Rico A.; Stoneman E.; Blanc V.; Sneeringer S.; Warsinske L.; Okuda M.; Murakami J.; Duong C.; Godbole S.; Douek D. C.; Maeda K.; Watanabe S.; Gordon A.; Ohmagari N.; Yotsuyanagi H.; Diamond M. S.; Hasegawa H.; Mitsuya H.; Suzuki T.; Kawaoka Y. Characterization and antiviral susceptibility of SARS-CoV-2 omicron BA.2. Nature 2022, 607, 119–127. 10.1038/s41586-022-04856-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Lin C.; Zhou X.; Zhong F.; Zeng P.; Yang Y.; Zhang Y.; Yu B.; Fan X.; McCormick P. J.; Fu R.; Fu Y.; Jiang H.; Zhang J. Structural basis of the main proteases of coronavirus bound to drug candidate PF-07321332. J. Virol. 2022, 96, e02013-21. 10.1128/jvi.02013-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankadara S.; Dawson M. D.; Fong J. Y.; Oh Q. Y.; Ang Q. A.; Liu B.; Chang H. Y.; Koh J.; Koh X.; Tan Q. W.; Joy J.; Chia C. S. B. A warhead substitution study on the coronavirus main protease inhibitor nirmatrelvir. ACS Med. Chem. Lett. 2022, 13, 1345–1350. 10.1021/acsmedchemlett.2c00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stille J. K.; Tjutrins J.; Wang G.; Venegas F. A.; Hennecker C.; Rueda A. M.; Sharon I.; Blaine N.; Miron C. E.; Pinus S.; Labarre A.; Plescia J.; Patrascu M. B.; Zhang X.; Wahba A. S.; Vlaho D.; Huot M. J.; Schmeing T. M.; Mittermaier A. K.; Moitessier N. Design, synthesis and in vitro evaluation of novel SARS-CoV-2 3CLpro covalent inhibitors. Eur. J. Med. Chem. 2022, 229, 114046. 10.1016/j.ejmech.2021.114046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duveau D. Y.; Thomas C. J. The remarkable selectivity of nirmatrelvir. ACS Pharmacol. Transl. Sci. 2022, 5, 445–447. 10.1021/acsptsci.2c00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. S.; Leeuwon S. Z.; Xu S.; Liu W. R. Evolutionary and structural insights about potential SARS-CoV-2 evasion of nirmatrelvir. J. Med. Chem. 2022, 65, 8686–8698. 10.1021/acs.jmedchem.2c00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoh Y.; Uehara S.; Nakahara K.; Nobori H.; Yamatsu Y.; Yamamoto S.; Maruyama Y.; Taoda Y.; Kasamatsu K.; Suto T.; Kouki K.; Nakahashi A.; Kawashima S.; Sanaki T.; Toba S.; Uemura K.; Mizutare T.; Ando S.; Sasaki M.; Orba Y.; Sawa H.; Sato A.; Sato T.; Kato T.; Tachibana Y. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 2022, 65, 6499–6512. 10.1021/acs.jmedchem.2c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D. W.; Li H.; Phillips G.; Weiss K. L.; Zhang Q.; Arnould M. A.; Jonsson C. B.; Surendranathan S.; Parvathareddy J.; Blakeley M. P.; Coates L.; Louis J. M.; Bonnesen P. V.; Kovalevsky A. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 2022, 13, 2268. 10.1038/s41467-022-29915-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Hurst B.; Townsend J. A.; Hu Y.; Szeto T.; Zhang X.; Tarbet B.; Marty M. T.; Chen Y.; Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L.; Ye F.; Feng Y.; Yu F.; Wang Q.; Wu Y.; Zhao C.; Sun H.; Huang B.; Niu P.; Song H.; Shi Y.; Li X.; Tan W.; Qi J.; Gao G. F. Both boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J.; Li Y.-S.; Zeng R.; Liu F.-L.; Luo R.-H.; Huang C.; Wang Y.-F.; Zhang J.; Quan B.; Shen C.; Mao X.; Liu X.; Sun W.; Yang W.; Ni X.; Wang K.; Xu L.; Duan Z.-L.; Zou Q.-C.; Zhang H.-L.; Qu W.; Long Y.-H.-P.; Li M.-H.; Yang R.-C.; Liu X.; You J.; Zhou Y.; Yao R.; Li W.-P.; Liu J.-M.; Chen P.; Liu Y.; Lin G.-F.; Yang X.; Zou J.; Li L.; Hu Y.; Lu G.-W.; Li W.-M.; Wei Y.-Q.; Zheng Y.-T.; Lei J.; Yang S. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W.; Jochmans D.; Xie H.; Yang H.; Li J.; Su H.; Chang D.; Wang J.; Peng J.; Zhu L.; Nian Y.; Hilgenfeld R.; Jiang H.; Chen K.; Zhang L.; Xu Y.; Neyts J.; Liu H. Design, synthesis, and biological evaluation of peptidomimetic aldehydes as broad-spectrum inhibitors against enterovirus and SARS-CoV-2. J. Med. Chem. 2022, 65, 2794–2808. 10.1021/acs.jmedchem.0c02258. [DOI] [PubMed] [Google Scholar]
- Yang K. S.; Ma X. R.; Ma Y.; Alugubelli Y. R.; Scott D. A.; Vatansever E. C.; Drelich A. K.; Sankaran B.; Geng Z. Z.; Blankenship L. R.; Ward H. E.; Sheng Y. J.; Hsu J. C.; Kratch K. C.; Zhao B.; Hayatshahi H. S.; Liu J.; Li P.; Fierke C. A.; Tseng C.-T. K.; Xu S.; Liu W. R. A quick route to multiple highly potent SARS-CoV-2 main protease inhibitors. ChemMedChem 2021, 16, 942–948. 10.1002/cmdc.202000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampalla C. S.; Kim Y.; Bickmeier N.; Rathnayake A. D.; Nguyen H. N.; Zheng J.; Kashipathy M. M.; Baird M. A.; Battaile K. P.; Lovell S.; Perlman S.; Chang K.-O.; Groutas W. C. Structure-guided design of conformationally constrained cyclohexane inhibitors of severe acute respiratory syndrome coronavirus-2 3CL protease. J. Med. Chem. 2021, 64, 10047–10058. 10.1021/acs.jmedchem.1c00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Lovell S.; Tiew K.-C.; Mandadapu S. R.; Alliston K. R.; Battaile K. P.; Groutas W. C.; Chang K.-O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B.; Belovodskiy A.; Hena M.; Kandadai A. S.; Joyce M. A.; Saffran H. A.; Shields J. A.; Khan M. B.; Arutyunova E.; Lu J.; Bajwa S. K.; Hockman D.; Fischer C.; Lamer T.; Vuong W.; van Belkum M. J.; Gu Z.; Lin F.; Du Y.; Xu J.; Rahim M.; Young H. S.; Vederas J. C.; Tyrrell D. L.; Lemieux M. J.; Nieman J. A. Peptidomimetic α-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J. Med. Chem. 2022, 65, 2905–2925. 10.1021/acs.jmedchem.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R. L.; Kania R. S.; Brothers M. A.; Davies J. F.; Ferre R. A.; Gajiwala K. S.; He M.; Hogan R. J.; Kozminski K.; Li L. Y.; Lockner J. W.; Lou J.; Marra M. T.; Mitchell L. J. Jr.; Murray B. W.; Nieman J. A.; Noell S.; Planken S. P.; Rowe T.; Ryan K.; Smith G. J. III; Solowiej J. E.; Steppan C. M.; Taggart B. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boras B.; Jones R. M.; Anson B. J.; Arenson D.; Aschenbrenner L.; Bakowski M. A.; Beutler N.; Binder J.; Chen E.; Eng H.; Hammond H.; Hammond J.; Haupt R. E.; Hoffman R.; Kadar E. P.; Kania R.; Kimoto E.; Kirkpatrick M. G.; Lanyon L.; Lendy E. K.; Lillis J. R.; Logue J.; Luthra S. A.; Ma C.; Mason S. W.; McGrath M. E.; Noell S.; Obach R. S.; O’Brien M. N.; O’Connor R.; Ogilvie K.; Owen D.; Pettersson M.; Reese M. R.; Rogers T. F.; Rosales R.; Rossulek M. I.; Sathish J. G.; Shirai N.; Steppan C.; Ticehurst M.; Updyke L. W.; Weston S.; Zhu Y.; White K. M.; García-Sastre A.; Wang J.; Chatterjee A. K.; Mesecar A. D.; Frieman M. B.; Anderson A. S.; Allerton C. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021, 12, 6055. 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redhead M. A.; Owen C. D.; Brewitz L.; Collette A. H.; Lukacik P.; Strain-Damerell C.; Robinson S. W.; Collins P. M.; Schäfer P.; Swindells M.; Radoux C. J.; Hopkins I. N.; Fearon D.; Douangamath A.; von Delft F.; Malla T. R.; Vangeel L.; Vercruysse T.; Thibaut J.; Leyssen P.; Nguyen T.-T.; Hull M.; Tumber A.; Hallett D. J.; Schofield C. J.; Stuart D. I.; Hopkins A. L.; Walsh M. A. Bispecific repurposed medicines targeting the viral and immunological arms of COVID-19. Sci. Rep. 2021, 11, 13208. 10.1038/s41598-021-92416-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.-Y.; Chen C.; Su J.; Li J.-Q.; Jiang Z.; Gao H.; Chigan J.-Z.; Ding H.-H.; Zhai L.; Yang K.-W. Ebsulfur and ebselen as highly potent scaffolds for the development of potential SARS-CoV-2 antivirals. Bioorg. Chem. 2021, 112, 104889. 10.1016/j.bioorg.2021.104889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thun-Hohenstein S. T. D.; Suits T. F.; Malla T. R.; Tumber A.; Brewitz L.; Salah E.; Choudhry H.; Schofield C. J. Structure-activity studies reveal scope for optimisation of ebselen-type inhibition of SARS-CoV-2 main protease. ChemMedChem 2022, 17, e202100582. 10.1002/cmdc.202100582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno S.; Kobayashi K.; Senda M.; Funai Y.; Seki Y.; Tamai I.; Schäkel L.; Sakata K.; Pillaiyar T.; Taguchi A.; Taniguchi A.; Gütschow M.; Müller C. E.; Takeuchi K.; Hirohama M.; Kawaguchi A.; Kojima M.; Senda T.; Shirasaka Y.; Kamitani W.; Hayashi Y. 3CL protease inhibitors with an electrophilic arylketone moiety as anti-SARS-CoV-2 agents. J. Med. Chem. 2022, 65, 2926–2939. 10.1021/acs.jmedchem.1c00665. [DOI] [PubMed] [Google Scholar]
- Chamakuri S.; Lu S.; Ucisik M. N.; Bohren K. M.; Chen Y.-C.; Du H.-C.; Faver J. C.; Jimmidi R.; Li F.; Li J.-Y.; Nyshadham P.; Palmer S. S.; Pollet J.; Qin X.; Ronca S. E.; Sankaran B.; Sharma K. L.; Tan Z.; Versteeg L.; Yu Z.; Matzuk M. M.; Palzkill T.; Young D. W. DNA-encoded chemistry technology yields expedient access to SARS-CoV-2 Mpro inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2111172118. 10.1073/pnas.2111172118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Xu M.; Chen C. Z.; Guo H.; Shen M.; Hu X.; Shinn P.; Klumpp-Thomas C.; Michael S. G.; Zheng W. Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016. 10.1021/acsptsci.0c00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Raghavaiah J.; Shahabi D.; Yadav M.; Anson B. J.; Lendy E. K.; Hattori S.-i.; Higashi-Kuwata N.; Mitsuya H.; Mesecar A. D. Indole chloropyridinyl ester-derived SARS-CoV-2 3CLpro inhibitors: enzyme inhibition, antiviral efficacy, structure–activity relationship, and X-ray structural studies. J. Med. Chem. 2021, 64, 14702–14714. 10.1021/acs.jmedchem.1c01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Xia Z.; Sacco M. D.; Hu Y.; Townsend J. A.; Meng X.; Choza J.; Tan H.; Jang J.; Gongora M. V.; Zhang X.; Zhang F.; Xiang Y.; Marty M. T.; Chen Y.; Wang J. Discovery of di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 2021, 143, 20697–20709. 10.1021/jacs.1c08060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malla T. R.; Tumber A.; John T.; Brewitz L.; Strain-Damerell C.; Owen C. D.; Lukacik P.; Chan H. T. H.; Maheswaran P.; Salah E.; Duarte F.; Yang H.; Rao Z.; Walsh M. A.; Schofield C. J. Mass spectrometry reveals potential of β-lactams as SARS-CoV-2 Mpro inhibitors. Chem. Commun. 2021, 57, 1430–1433. 10.1039/D0CC06870E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villhauer E. B.; Brinkman J. A.; Naderi G. B.; Burkey B. F.; Dunning B. E.; Prasad K.; Mangold B. L.; Russell M. E.; Hughes T. E. 1-[[(3-Hydroxy-1-adamantyl)amino]acetyl]-2-cyano-(S)-pyrrolidine: a potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J. Med. Chem. 2003, 46, 2774–2789. 10.1021/jm030091l. [DOI] [PubMed] [Google Scholar]
- Brogi S.; Ibba R.; Rossi S.; Butini S.; Calderone V.; Gemma S.; Campiani G. Covalent reversible inhibitors of cysteine proteases containing the nitrile warhead: recent advancement in the field of viral and parasitic diseases. Molecules 2022, 27, 2561. 10.3390/molecules27082561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuck C.-P.; Chen C.; Ke Z.; Wan D. C.-C.; Chow H.-F.; Wong K.-B. Design, synthesis and crystallographic analysis of nitrile-based broad-spectrum peptidomimetic inhibitors for coronavirus 3C-like proteases. Eur. J. Med. Chem. 2013, 59, 1–6. 10.1016/j.ejmech.2012.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B.; Arutyunova A.; Bashir Khan M.; Lu J.; Joyce M. A.; Saffran H. A.; Shields J. A.; Srinivas Kandadai A.; Belovodskiy A.; Hena M.; Vuong W.; Lamer T.; Young H. S.; Vederas J. C.; Tyrrell D. L.; Lemieux M. J.; Nieman J. A. Peptidomimetic nitrile warheads as SARS-CoV-2 3CL protease inhibitors. RSC Med. Chem. 2021, 12, 1722–1730. 10.1039/D1MD00247C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenbach J.; Lemke C.; Pillaiyar T.; Schäkel L.; Al Hamwi G.; Diett M.; Gedschold R.; Geiger N.; Lopez V.; Mirza S.; Namasivayam V.; Schiedel A. C.; Sylvester K.; Thimm D.; Vielmuth C.; Vu L. P.; Zyulina M.; Bodem J.; Gütschow M.; Müller C. E. Targeting the main protease of SARS-CoV-2: from the establishment of high throughput screening to the design of tailored inhibitors. Angew. Chem., Int. Ed. 2021, 60, 10423–10429. 10.1002/anie.202016961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mons E.; Jansen I. D. C.; Loboda J.; van Doodewaerd B. R.; Hermans J.; Verdoes M.; van Boeckel C. A. A.; van Veelen P. A.; Turk B.; Turk D.; Ovaa H. The alkyne moiety as a latent electrophile in irreversible covalent small molecule inhibitors of cathepsin K. J. Am. Chem. Soc. 2019, 141, 3507–3514. 10.1021/jacs.8b11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekkebus R.; van Kasteren S. I.; Kulathu Y.; Scholten A.; Berlin I.; Geurink P. P.; de Jong A.; Goerdayal S.; Neefjes J.; Heck A. J. R.; Komander D.; Ovaa H. On terminal alkynes that can react with active-site cysteine nucleophiles in proteases. J. Am. Chem. Soc. 2013, 135, 2867–2870. 10.1021/ja309802n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S.; Weikart N. D.; Linne U.; Mootz H. D. Covalent inhibition of SUMO and ubiquitin-specific cysteine proteases by an in situ thiol–alkyne addition. Bioorg. Med. Chem. 2013, 21, 2511–2517. 10.1016/j.bmc.2013.02.039. [DOI] [PubMed] [Google Scholar]
- Shin D.; Mukherjee R.; Grewe D.; Bojkova D.; Baek K.; Bhattacharya A.; Schulz L.; Widera M.; Mehdipour A. R.; Tascher G.; Geurink P. P.; Wilhelm A.; van der Heden van Noort G. J.; Ovaa H.; Müller S.; Knobeloch K.-P.; Rajalingam K.; Schulman B. A.; Cinatl J.; Hummer G.; Ciesek S.; Dikic I. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mons E.; Kim R. Q.; van Doodewaerd B. R.; van Veelen P. A.; Mulder M. P. C.; Ovaa H. Exploring the versatility of the covalent thiol–alkyne reaction with substituted propargyl warheads: a deciding role for the cysteine protease. J. Am. Chem. Soc. 2021, 143, 6423–6433. 10.1021/jacs.0c10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douangamath A.; Fearon D.; Gehrtz P.; Krojer T.; Lukacik P.; Owen C. D.; Resnick E.; Strain-Damerell C.; Aimon A.; Ábrányi-Balogh P.; Brandão-Neto J.; Carbery A.; Davison G.; Dias A.; Downes T. D.; Dunnett L.; Fairhead M.; Firth J. D.; Jones S. P.; Keeley A.; Keserü G. M.; Klein H. F.; Martin M. P.; Noble M. E. M.; O’Brien P.; Powell A.; Reddi R. N.; Skyner R.; Snee M.; Waring M. J.; Wild C.; London N.; von Delft F.; Walsh M. A. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm T.; Ebert G.; Calleja D. J.; Allison C. C.; Richardson L. W.; Bernardini J. P.; Lu B. G. C.; Kuchel N. W.; Grohmann C.; Shibata Y.; Gan Z. Y.; Cooney J. P.; Doerflinger M.; Au A. E.; Blackmore T. R.; van der Heden van Noort G. J.; Geurink P. P.; Ovaa H.; Newman J.; Riboldi-Tunnicliffe A.; Czabotar P. E.; Mitchell J. P.; Feltham R.; Lechtenberg B. C.; Lowes K. N.; Dewson G.; Pellegrini M.; Lessene G.; Komander D. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Faham A.; Albericio F. COMU: a third generation of uronium-type coupling reagents. J. Pept. Sci. 2010, 16, 6–9. 10.1002/psc.1204. [DOI] [PubMed] [Google Scholar]
- Nahm S.; Weinreb S. M. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. 10.1016/S0040-4039(01)91316-4. [DOI] [Google Scholar]
- Lin D.; Qian W.; Hilgenfeld R.; Jiang H.; Chen K.; Liu H. Improved synthesis of rupintrivir. Sci. China Chem. 2012, 55, 1101–1107. 10.1007/s11426-011-4478-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou K.; Xu B.; Ma C.; Yang X.; Zou X.; Lü Y.; Xu P. Novel CADD-based peptidyl vinyl ester derivatives as potential proteasome inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2198–2202. 10.1016/j.bmcl.2007.12.077. [DOI] [PubMed] [Google Scholar]
- Ohira S. Methanolysis of dimethyl (1-diazo-2-oxopropyl) phosphonate: generation of dimethyl (diazomethyl) phosphonate and reaction with carbonyl compounds. Synth. Commun. 1989, 19, 561–564. 10.1080/00397918908050700. [DOI] [Google Scholar]
- Müller S.; Liepold B.; Roth G. J.; Bestmann H. J. An improved one-pot procedure for the synthesis of alkynes from aldehydes. Synlett 1996, 1996, 521–522. 10.1055/s-1996-5474. [DOI] [Google Scholar]
- Dess D. B.; Martin J. C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. 10.1021/jo00170a070. [DOI] [Google Scholar]
- Malla T. R.; Brewitz L.; Muntean D.-G.; Aslam H.; Owen C. D.; Salah E.; Tumber A.; Lukacik P.; Strain-Damerell C.; Mikolajek H.; Walsh M. A.; Schofield C. J. Penicillin derivatives inhibit the SARS-CoV-2 main protease by reaction with its nucleophilic cysteine. J. Med. Chem. 2022, 65, 7682–7696. 10.1021/acs.jmedchem.1c02214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talele T. T. Acetylene group, friend or foe in medicinal chemistry. J. Med. Chem. 2020, 63, 5625–5663. 10.1021/acs.jmedchem.9b01617. [DOI] [PubMed] [Google Scholar]
- Sacco M. D.; Ma C.; Lagarias P.; Gao A.; Townsend J. A.; Meng X.; Dube P.; Zhang X.; Hu Y.; Kitamura N.; Hurst B.; Tarbet B.; Marty M. T.; Kolocouris A.; Xiang Y.; Chen Y.; Wang J. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020, 6, eabe0751. 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel-Andreasen J.; Wellhöfer I.; Wich K.; Olsen C. A. Backbone-fluorinated 1,2,3-triazole-containing dipeptide surrogates. J. Org. Chem. 2017, 82, 11613–11619. 10.1021/acs.joc.7b01744. [DOI] [PubMed] [Google Scholar]
- Zhang J.-H.; Chung T. D. Y.; Oldenburg K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screening 1999, 4, 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Sonogashira K.; Tohda Y.; Hagihara N. A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. 10.1016/S0040-4039(00)91094-3. [DOI] [Google Scholar]
- Chinchilla R.; Nájera C. The Sonogashira reaction: a booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- Tresse C.; Guissart C.; Schweizer S.; Bouhoute Y.; Chany A.-C.; Goddard M.-L.; Blanchard N.; Evano G. Practical methods for the synthesis of trifluoromethylated alkynes: oxidative trifluoromethylation of copper acetylides and alkynes. Adv. Synth. Catal. 2014, 356, 2051–2060. 10.1002/adsc.201400057. [DOI] [Google Scholar]
- Bott G.; Field L. D.; Sternhell S. Steric effects. A study of a rationally designed system. J. Am. Chem. Soc. 1980, 102, 5618–5626. 10.1021/ja00537a036. [DOI] [Google Scholar]
- Carcenac Y.; Diter P.; Wakselman C.; Tordeux M. Experimental determination of the conformational free energies (A values) of fluorinated substituents in cyclohexane by dynamic 19F NMR spectroscopy. Part 1. Description of the method for the trifluoromethyl group. New J. Chem. 2006, 30, 442–446. 10.1039/b516636e. [DOI] [Google Scholar]
- Hu Y.; Lewandowski E. M.; Tan H.; Zhang X.; Morgan R. T.; Zhang X.; Jacobs L. M. C.; Butler S. G.; Gongora M. V.; Choy J.; Deng X.; Chen Y.; Wang J.. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. bioRxiv 2022, 10.1101/2022.06.28.497978. [DOI] [PMC free article] [PubMed]
- Moghadasi S. A.; Heilmann E.; Khalil A. M.; Nnabuife C.; Kearns F. L.; Ye C.; Moraes S. N.; Costacurta F.; Esler M. A.; Aihara H.; von Laer D.; Martinez-Sobrido L.; Palzkill T.; Amaro R. E.; Harris R. S.. Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. bioRxiv 2022, 10.1101/2022.08.07.503099. [DOI] [PMC free article] [PubMed]
- Brewitz L.; Kamps J. J. A. G.; Lukacik P.; Strain-Damerell C.; Zhao Y.; Tumber A.; Malla T. R.; Orville A. M.; Walsh M. A.; Schofield C. J. Mass spectrometric assays reveal discrepancies in inhibition profiles for the SARS-CoV-2 papain-like protease. ChemMedChem 2022, 17, e202200016. 10.1002/cmdc.202200016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawshaw A. D.; Beale E. V.; Warren A. J.; Stallwood A.; Duller G.; Trincao J.; Evans G. A sample preparation pipeline for microcrystals at the VMXm beamline. J. Vis. Exp. 2021, e62306. 10.3791/62306. [DOI] [PubMed] [Google Scholar]
- Gildea R. J.; Beilsten-Edmands J.; Axford D.; Horrell S.; Aller P.; Sandy J.; Sanchez-Weatherby J.; Owen C. D.; Lukacik P.; Strain-Damerell C.; Owen R. L.; Walsh M. A.; Winter G. xia2.multiplex: a multi-crystal data-analysis pipeline. Acta Crystallogr., Sect. D: Struct. Biol. 2022, 78, 752–769. 10.1107/S2059798322004399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon J. B.; Coleman R. S.; Hanzlik R. P. Reversible covalent inhibition of papain by a peptide nitrile. Carbon-13 NMR evidence for a thioimidate ester adduct. J. Am. Chem. Soc. 1986, 108, 1350–1351. 10.1021/ja00266a066. [DOI] [Google Scholar]
- DeGraw J. I.; Colwell W. T.; Piper J. R.; Sirotnak F. M. Synthesis and antitumor activity of 10-propargyl-10-deazaaminopterin. J. Med. Chem. 1993, 36, 2228–2231. 10.1021/jm00067a020. [DOI] [PubMed] [Google Scholar]
- Oldfield V.; Keating G. M.; Perry C. M. Rasagiline: a review of its use in the management of Parkinson’s disease. Drugs 2007, 67, 1725–1747. 10.2165/00003495-200767120-00006. [DOI] [PubMed] [Google Scholar]
- Eckhardt M.; Langkopf E.; Mark M.; Tadayyon M.; Thomas L.; Nar H.; Pfrengle W.; Guth B.; Lotz R.; Sieger P.; Fuchs H.; Himmelsbach F. 8-(3-(R)-Aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2007, 50, 6450–6453. 10.1021/jm701280z. [DOI] [PubMed] [Google Scholar]
- Huang W.-S.; Metcalf C. A.; Sundaramoorthi R.; Wang Y.; Zou D.; Thomas R. M.; Zhu X.; Cai L.; Wen D.; Liu S.; Romero J.; Qi J.; Chen I.; Banda G.; Lentini S. P.; Das S.; Xu Q.; Keats J.; Wang F.; Wardwell S.; Ning Y.; Snodgrass J. T.; Broudy M. I.; Russian K.; Zhou T.; Commodore L.; Narasimhan N. I.; Mohemmad Q. K.; Iuliucci J.; Rivera V. M.; Dalgarno D. C.; Sawyer T. K.; Clackson T.; Shakespeare W. C. Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J. Med. Chem. 2010, 53, 4701–4719. 10.1021/jm100395q. [DOI] [PubMed] [Google Scholar]
- Stuetz A.; Petranyi G. Synthesis and antifungal activity of (E)-N-(6,6-dimethyl-2-hepten-4-ynyl)-N-methyl-1-naphthalenemethanamine (SF 86-327) and related allylamine derivatives with enhanced oral activity. J. Med. Chem. 1984, 27, 1539–1543. 10.1021/jm00378a003. [DOI] [PubMed] [Google Scholar]
- Stütz A. Allylamine derivatives—a new class of active substances in antifungal chemotherapy. Angew. Chem. Int. Ed. 1987, 26, 320–328. 10.1002/anie.198703201. [DOI] [Google Scholar]
- Young S. D.; Britcher S. F.; Tran L. O.; Payne L. S.; Lumma W. C.; Lyle T. A.; Huff J. R.; Anderson P. S.; Olsen D. B.; Carroll S. S. L-743, 726 (DMP-266): a novel, highly potent nonnucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 1995, 39, 2602–2605. 10.1128/AAC.39.12.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link J. O.; Rhee M. S.; Tse W. C.; Zheng J.; Somoza J. R.; Rowe W.; Begley R.; Chiu A.; Mulato A.; Hansen D.; Singer E.; Tsai L. K.; Bam R. A.; Chou C.-H.; Canales E.; Brizgys G.; Zhang J. R.; Li J.; Graupe M.; Morganelli P.; Liu Q.; Wu Q.; Halcomb R. L.; Saito R. D.; Schroeder S. D.; Lazerwith S. E.; Bondy S.; Jin D.; Hung M.; Novikov N.; Liu X.; Villaseñor A. G.; Cannizzaro C. E.; Hu E. Y.; Anderson R. L.; Appleby T. C.; Lu B.; Mwangi J.; Liclican A.; Niedziela-Majka A.; Papalia G. A.; Wong M. H.; Leavitt S. A.; Xu Y.; Koditek D.; Stepan G. J.; Yu H.; Pagratis N.; Clancy S.; Ahmadyar S.; Cai T. Z.; Sellers S.; Wolckenhauer S. A.; Ling J.; Callebaut C.; Margot N.; Ram R. R.; Liu Y.-P.; Hyland R.; Sinclair G. I.; Ruane P. J.; Crofoot G. E.; McDonald C. K.; Brainard D. M.; Lad L.; Swaminathan S.; Sundquist W. I.; Sakowicz R.; Chester A. E.; Lee W. E.; Daar E. S.; Yant S. R.; Cihlar T. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. 10.1038/s41586-020-2443-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricart A. D. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. 10.1158/1078-0432.CCR-11-0486. [DOI] [PubMed] [Google Scholar]
- Barf T.; Covey T.; Izumi R.; van de Kar B.; Gulrajani M.; van Lith B.; van Hoek M.; de Zwart E.; Mittag D.; Demont D.; Verkaik S.; Krantz F.; Pearson P. G.; Ulrich R.; Kaptein A. Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252. 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
- Liclican A.; Serafini L.; Xing W.; Czerwieniec G.; Steiner B.; Wang T.; Brendza K. M.; Lutz J. D.; Keegan K. S.; Ray A. S.; Schultz B. E.; Sakowicz R.; Feng J. Y. Biochemical characterization of tirabrutinib and other irreversible inhibitors of Bruton’s tyrosine kinase reveals differences in on - and off - target inhibition. Biochim. Biophys. Acta, Gen. Subj. 2020, 1864, 129531. 10.1016/j.bbagen.2020.129531. [DOI] [PubMed] [Google Scholar]
- Pan Z.; Scheerens H.; Li S.-J.; Schultz B. E.; Sprengeler P. A.; Burrill L. C.; Mendonca R. V.; Sweeney M. D.; Scott K. C. K.; Grothaus P. G.; Jeffery D. A.; Spoerke J. M.; Honigberg L. A.; Young P. R.; Dalrymple S. A.; Palmer J. T. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Honigberg L. A.; Smith A. M.; Sirisawad M.; Verner E.; Loury D.; Chang B.; Li S.; Pan Z.; Thamm D. H.; Miller R. A.; Buggy J. J. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13075–13080. 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter H. S.; Rule S. A.; Dyer M. J. S.; Karlin L.; Jones C.; Cazin B.; Quittet P.; Shah N.; Hutchinson C. V.; Honda H.; Duffy K.; Birkett J.; Jamieson V.; Courtenay-Luck N.; Yoshizawa T.; Sharpe J.; Ohno T.; Abe S.; Nishimura A.; Cartron G.; Morschhauser F.; Fegan C.; Salles G. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood 2016, 127, 411–419. 10.1182/blood-2015-08-664086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson S. H.; Liu Q.; Beaudoin Bertrand M.; Batt D. G.; Li L.; Pattoli M. A.; Skala S.; Cheng L.; Obermeier M. T.; Moore R.; Yang Z.; Vickery R.; Elzinga P. A.; Discenza L.; D’Arienzo C.; Gillooly K. M.; Taylor T. L.; Pulicicchio C.; Zhang Y.; Heimrich E.; McIntyre K. W.; Ruan Q.; Westhouse R. A.; Catlett I. M.; Zheng N.; Chaudhry C.; Dai J.; Galella M. A.; Tebben A. J.; Pokross M.; Li J.; Zhao R.; Smith D.; Rampulla R.; Allentoff A.; Wallace M. A.; Mathur A.; Salter-Cid L.; Macor J. E.; Carter P. H.; Fura A.; Burke J. R.; Tino J. A. Discovery of branebrutinib (BMS-986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton’s tyrosine kinase (BTK). J. Med. Chem. 2019, 62, 3228–3250. 10.1021/acs.jmedchem.9b00167. [DOI] [PubMed] [Google Scholar]
- Plassche M. A. T.; Barniol-Xicota M.; Verhelst S. H. L. Peptidyl acyloxymethyl ketones as activity-based probes for the main protease of SARS-CoV-2. ChemBioChem 2020, 21, 3383–3388. 10.1002/cbic.202000371. [DOI] [PubMed] [Google Scholar]
- Mótyán J. A.; Mahdi M.; Hoffka G.; Tőzsér J. Potential resistance of SARS-CoV-2 main protease (Mpro) against protease inhibitors: lessons learned from HIV-1 protease. Int. J. Mol. Sci. 2022, 23, 3507. 10.3390/ijms23073507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandyck K.; Abdelnabi R.; Gupta K.; Jochmans D.; Jekle A.; Deval J.; Misner D.; Bardiot D.; Foo C. S.; Liu C.; Ren S.; Beigelman L.; Blatt L. M.; Boland S.; Vangeel L.; Dejonghe S.; Chaltin P.; Marchand A.; Serebryany V.; Stoycheva A.; Chanda S.; Symons J. A.; Raboisson P.; Neyts J. ALG-097111, a potent and selective SARS-CoV-2 3-chymotrypsin-like cysteine protease inhibitor exhibits in vivo efficacy in a syrian hamster model. Biochem. Biophys. Res. Commun. 2021, 555, 134–139. 10.1016/j.bbrc.2021.03.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biniossek M. L.; Nägler D. K.; Becker-Pauly C.; Schilling O. Proteomic identification of protease cleavage sites characterizes prime and non-prime specificity of cysteine cathepsins B, L, and S. J. Proteome Res. 2011, 10, 5363–5373. 10.1021/pr200621z. [DOI] [PubMed] [Google Scholar]
- Woyach J. A.; Furman R. R.; Liu T.-M.; Ozer H. G.; Zapatka M.; Ruppert A. S.; Xue L.; Li D. H.-H.; Steggerda S. M.; Versele M.; Dave S. S.; Zhang J.; Yilmaz A. S.; Jaglowski S. M.; Blum K. A.; Lozanski A.; Lozanski G.; James D. F.; Barrientos J. C.; Lichter P.; Stilgenbauer S.; Buggy J. J.; Chang B. Y.; Johnson A. J.; Byrd J. C. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Wei P.; Fan K.; Liu Y.; Lai L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 2004, 43, 4568–4574. 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- Pillay D.; Boulangé A. F.; Coetzer T. H. T. Expression, purification and characterisation of two variant cysteine peptidases from Trypanosoma congolense with active site substitutions. Protein Expression Purif. 2010, 74, 264–271. 10.1016/j.pep.2010.06.021. [DOI] [PubMed] [Google Scholar]
- Berger A. B.; Vitorino P. M.; Bogyo M. Activity-based protein profiling: applications to biomarker discovery, in vivo imaging and drug discovery. Am. J. PharmacoGenomics 2004, 4, 371–381. 10.2165/00129785-200404060-00004. [DOI] [PubMed] [Google Scholar]
- Wu F.; Zhao S.; Yu B.; Chen Y.-M.; Wang W.; Song Z.-G.; Hu Y.; Tao Z.-W.; Tian J.-H.; Pei Y.-Y.; Yuan M.-L.; Zhang Y.-L.; Dai F.-H.; Liu Y.; Wang Q.-M.; Zheng J.-J.; Xu L.; Holmes E. C.; Zhang Y.-Z. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K.-Y. A.; Tan T. K.; Chen T.-H.; Huang C.-G.; Harvey R.; Hussain S.; Chen C.-P.; Harding A.; Gilbert-Jaramillo J.; Liu X.; Knight M.; Schimanski L.; Shih S.-R.; Lin Y.-C.; Cheng C.-Y.; Cheng S.-H.; Huang Y.-C.; Lin T.-Y.; Jan J.-T.; Ma C.; James W.; Daniels R. S.; McCauley J. W.; Rijal P.; Townsend A. R. Breadth and function of antibody response to acute SARS-CoV-2 infection in humans. PLoS Pathog. 2021, 17, e1009352. 10.1371/journal.ppat.1009352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.; Beilsten-Edmands J.; Devenish N.; Gerstel M.; Gildea R. J.; McDonagh D.; Pascal E.; Waterman D. G.; Williams B. H.; Evans G. DIALS as a toolkit. Protein Sci. 2022, 31, 232–250. 10.1002/pro.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.; Lobley C. M. C.; Prince S. M. Decision making in xia2. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, D69, 1260–1273. 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, D66, 22–25. 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Long F.; Nicholls R. A.; Emsley P.; Gražulis S.; Merkys A.; Vaitkus A.; Murshudov G. N. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2017, 73, 112–122. 10.1107/S2059798317000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Murshudov G. N.; Papiz M. Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003, 374, 300–321. [DOI] [PubMed] [Google Scholar]
- Joosten R. P.; Joosten K.; Murshudov G. N.; Perrakis A. PDB_REDO: constructive validation, more than just looking for errors. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 484–496. 10.1107/S0907444911054515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.