

Abstract
Background
Recent efforts have described the evolution of glioblastoma from initial diagnosis to post-treatment recurrence on a genomic and transcriptomic level. However, the evolution of the proteomic landscape is largely unknown.
Methods
Sequential window acquisition of all theoretical fragment ion spectra mass spectrometry (SWATH-MS) was used to characterize the quantitative proteomes of two independent cohorts of paired newly diagnosed and recurrent glioblastomas. Recurrence-associated proteins were validated using immunohistochemistry and further studied in human glioma cell lines, orthotopic xenograft models, and human organotypic brain slice cultures. External spatial transcriptomic, single-cell, and bulk RNA sequencing data were analyzed to gain mechanistic insights.
Results
Although overall proteomic changes were heterogeneous across patients, we identified BCAS1, INF2, and FBXO2 as consistently upregulated proteins at recurrence and validated these using immunohistochemistry. Knockout of FBXO2 in human glioma cells conferred a strong survival benefit in orthotopic xenograft mouse models and reduced invasive growth in organotypic brain slice cultures. In glioblastoma patient samples, FBXO2 expression was enriched in the tumor infiltration zone and FBXO2-positive cancer cells were associated with synaptic signaling processes.
Conclusions
These findings demonstrate a potential role of FBXO2-dependent glioma-microenvironment interactions to promote tumor growth. Furthermore, the published datasets provide a valuable resource for further studies.
Keywords: glioblastoma, microenvironment, PCT-SWATH, proteome
Key Points.
Proteome profiling of two cohorts of initial-recurrent glioblastoma pairs by PCT-SWATH.
FBXO2 knockout improves survival in vivo and reduces invasive growth in brain slice cultures.
FBOX2 is enriched in the infiltration zone and associates with neuronal signaling in FBXO2+ cells.
Importance of the Study.
The formation of incurable recurrent tumors poses a major challenge in the management of glioblastoma patients. Several studies have described the evolution of glioblastoma from initial diagnosis to post-treatment recurrence on the genomic and transcriptomic levels but failed to improve treatment regimens so far. In contrast, patient proteomes are generally understudied although quantitative protein-level information might be more relevant for defining functionally relevant phenotypes and identifying potential drug targets. In this study, we present quantitative proteomic landscapes of paired initial and recurrent glioblastoma samples from two independent patient cohorts using SWATH-MS, which revealed an increased abundance of BCAS1, FBXO2, and INF2 in recurrent tumors. Functional experiments using human knockout glioma cells in vitro, in vivo, and in organotypic brain slice cultures as well as integration of spatial, single-cell, and bulk transcriptome data suggest a potential microenvironment-dependent tumor-promoting role of FBXO2.
Glioblastoma is the most common and most aggressive primary brain tumor in adults.1 Despite multimodal first-line treatment comprising surgery and radiochemotherapy with temozolomide, recurrence is inevitable and the treatment options at recurrence are limited.2 Therefore, there is a need for a better understanding of the molecular changes associated with disease progression. Recent efforts characterized the longitudinal evolution of glioblastoma upon disease progression and upon therapy at the genomic and transcriptomic levels. These studies revealed an overall similar clonal architecture between primary and recurrent tumors and both linear and branched evolution patterns of gene expression.3–5
However, mRNA levels do not accurately predict the abundance of functionally relevant protein levels.6 Accordingly, the evolution of the proteomic landscape of glioblastoma during disease progression remains poorly understood. Technical advances in the last years now allow label-free, reproducible, and precise whole proteome quantification of archived formalin-fixed, paraffin-embedded (FFPE) samples. A recently described method combines pressure cycling technology (PCT) for sample processing and sequential window acquisition of all theoretical fragment ion spectra mass spectrometry (SWATH-MS) to generate precise and comprehensive digital quantitative proteome maps as a resource that can be re-analyzed with different peptide spectral libraries depending on the scientific question.7 Importantly, we have previously demonstrated that this method works comparably well with fresh frozen and FFPE tissue samples.8 In this study, we apply this technology to characterize the evolution of the quantitative proteomic landscapes of glioblastoma in matched pairs of untreated initial and post-treatment recurrent tumor samples from two independent cohorts. Experimental validation of the proteomic data and integration of external bulk, single-cell, and spatial RNA sequencing data reveals a potential involvement of the ubiquitin ligase adaptor protein FBXO2 in regulating invasive growth of glioma cells in a tissue-dependent manner. Furthermore, we provide a valuable resource for further studies on the glioblastoma proteome.
Materials and Methods
A detailed list of key resources and manufacturer information can be found in Supplementary Tables S2–S4.
Data Availability
Protein matrices, clinical information, and analysis code are available in the Supplementary material. Single-cell sequencing data and spatial transcriptomic data are available online.9,10 Proteomics raw data are deposited on ProteomeXchange (https://www.iprox.org/, ID: IPX0003752001).
Cell Lines and Material
Human long-term glioma cell lines and glioma-initiating cells (Supplementary Table S2) were cultured as described.11 NIH-3T3 fibroblasts were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal calf serum and 2 mM l-glutamine. Cell lines were authenticated at the Leibniz Institute DSMZ (Braunschweig, Germany) and regularly tested negative for mycoplasma.
Generation of Knockout Cell Lines
Knockout cell lines were generated by Cas9/sgRNA-ribonucleoprotein complex (RNP) electroporation12 using the Neon electroporation system (Invitrogen, Carlsbad, CA, USA) using two different sgRNA (Lubio Science/Integrated DNA Technologies, Zurich, Switzerland) per target gene in order to create small deletions (Supplementary Table S3). Control cells were prepared identically but electroporated in the absence of targeted gRNA. Single-cell-derived clones were generated for both knockout cells and controls by clonal expansion. Homozygous editing of individual clones was tested by PCR, Sanger sequencing, and immunoblot.
Cell Proliferation Assay
In total, 1500 cells were seeded in triplicates in 100 µL complete medium in 96-well ViewPlates. At each indicated time point, cells were stained with Hoechst 33342, incubated for 3 hours at 37°C/5% CO2 and nuclei were captured with the MuviCyte imaging system (4× objective, 3 fields of view per well). Nuclei were counted using CellProfiler13 and nuclear counts from each field of view within a well were aggregated to derive cell doubling times defined as ln(2)/slope from a log-linear model using R/RStudio.14,15
Spheroid Growth Assay
In total, 1000 cells per well were seeded in 100 µL full medium containing 1:100 Corning Type I collagen in six replicates into cell-repellent 96-well U-bottom plates. After seeding, plates were centrifuged for 5 minutes at 100g, and spheroids were allowed to form for 48 hours, followed by brightfield image acquisition every 24 hours using the MuviCyte imaging system at 4× magnification. Spheroid size was estimated using Matlab/SpheroidSizer.16
Spheroid Invasion Assay
Spheroid invasion assay was performed as described17 using NIH-3T3 conditioned medium as chemoattractant. Cells were allowed to invade for 24-48 hours at 37°C/5% CO2 followed by nuclear staining with Hoechst 33342. Spheroids were imaged with the MuviCyte imaging system (4× objective). Images were contrast-enhanced, converted to binary with ImageJ/Fiji18 and used as input for the automated quantification.17
Clonogenicity Assay
In total, 200 LN-229 cells were seeded in culture-treated 6-well plates, cultivated for 14 days, and stained with crystal violet. Whole-well images were acquired with the MuviCyte imaging system (4× objective) and colonies were quantified in ImageJ/Fiji.
Soft Agar Assay
Six-well suspension plates were pre-coated with 1.5 mL of 0.6% low-melt agarose in complete medium. In total, 2000 cells/1.5 mL 0.3% low-melt agarose in complete medium were seeded into pre-coated wells in triplicates, incubated at 4°C for 30 minutes, and transferred to 37°C/5% CO2. Cells were cultured for 30 days and supplied with 200 µL complete medium twice per week. Cell clusters were stained with MTT reagent for 30 minutes followed by imaging and quantification using ImageJ/Fiji.18
Immunohistochemistry
Tissue microarrays (TMA) were processed as described for deparaffination, antigen retrieval, and staining.19 Antibodies specified in Supplementary Table S2 were used at 0.3 µg/mL (anti-BCAS1), 2.7 µg/mL (anti-FBXO2) or 0.35 µg/mL (anti-INF2). Rabbit IgG isotype control was used at matched concentrations. Microarrays were subsequently incubated in mouse anti-rabbit IgG-HRP (0.53 µg/mL in LowCross-Buffer; anti-FBXO2 staining) or Histofine anti-rabbit immuno-peroxidase polymer (anti-BCAS1/INF2 staining). Automated staining score (H score) estimation was performed using the TMARKER software.20
Animal Experiments
All experiments were performed under the guidelines of the Swiss federal law on animal protection and were approved by the cantonal veterinary office (ZH98/2018). CD1 female nu/nu mice (Janvier, Le Genest-Saint-Isle, France) of 6-12 weeks of age were used in all experiments, and 150 000 LN-229- or 270 000 ZH-161-derived cells were implanted as described.21 For histological confirmation of tumorigenicity, brains from three randomly selected glioma-bearing mice were collected per group, when the first mouse in the whole experiment became symptomatic, respectively.
Patient Samples
Studies were approved by the Institutional Review Board and ethics committee at the University Hospital Zurich (KEK-StV-Nr.19/08), the University Hospital Düsseldorf (study number 2019-641), and the University of Freiburg (protocol 100020/09 and 472/15_160880). Tissues were examined by board-certified neuropathologists to identify the tumor region. For the first cohort, 4-5 tissue punches (1 × 2 mm) were collected across the tumor area. For the independent validation cohort (cohort 2), whole tissue sections were collected and processed.
Organotypic Brain Slice Cultures
A pT4 plasmid backbone containing the zsGreen coding sequence controlled by an EF1a promoter was designed and used to fluorescently label FBXO2-KO cells and as a control for FBXO2 overexpression experiments. The same backbone was used to design an FBXO2 overexpression construct by placing a T2A splicing sequence followed by the FBXO2 coding sequence directly after the zsGreen coding sequence. Both constructs were ordered from GeneScript (GenScript Biotech B.V., Leiden, the Netherlands). mRNA encoding for the SB100X transposase was generated by in vitro transcription. 500 000 cells were transfected with 3 µg of pT4 donor plasmid and 3 µg of SB100X mRNA using the Neon transcription system. After 48 hours, positively transfected cells were selected with 3 µg/mL puromycin. Fluorescently labeled cells were then used for organotypic brain slice cultures, as described in previous work (Supplementary Methods).
PCT‐assisted Sample Preparation and SWATH Mass Spectrometry
Tissue samples were processed using an NEP2320‐45k barocycler.8 1.5 µg of cleaned peptides was analyzed by SWATH‐MS using a SCIEX 6600 TripleTOF mass spectrometer connected to an Eksigent NanoLC 400 system. Raw data were processed and normalized and protein matrices were generated as described using OpenSWATH.8 Protein matrices containing 9307 (cohort 1, 57.6% NA) and 11 233 (cohort 2, 62.8% NA) protein groups (PG), respectively, were imported into RStudio.14 PG detected in less than 40% of samples were filtered out, resulting in 4139 PG/18.7% NA (cohort 1) and 4793 PG/29% NA (cohort 2), respectively. For downstream analyses, protein matrices were log2-transformed.
Downstream Analyses of Protein Matrices
R code used for the analysis of the proteomic datasets is provided in the Supplementary material. Hierarchical clustering was performed on row-wise Z score-transformed data using ComplexHeatmap.22 Gene set enrichment analysis was performed using clusterProfiler.23 For the identification of differentially expressed proteins, protein matrices were filtered for PG detected in both cohorts (N = 2994). PG with at least a 2-fold change in abundance at recurrence across both independent datasets were pre-selected and individually tested for significance using one-sided paired Wilcoxon rank-sum tests (Ha = greater for increased and Ha = less for decreased proteins, respectively). Significant proteins (P < .05) shared between both cohorts were then cross-validated on published RNA sequencing data.5 Interpatient correlations of expressed proteins within initial and recurrent tumors were calculated by bootstrapping (subsample size: 2 × 7, 1000 iterations), and the resulting correlation distributions were compared. Patient survival was analyzed on the basis of external RNA sequencing5 and two independent proteomics datasets24,25 using univariate Cox proportional hazard regression models implemented in survival/survminer.26,27
Analysis of Patient MRI and RNA Microarray Data
Patient MRI imaging and Affy-U133a Array mRNA expression data from 118 IDH wildtype patients were obtained from the TCGA network (https://www.cancer.gov/tcga). Tumors were segmented using the BraTS-toolkit28 and tumor shape parameters were extracted from segmented tumors using pyRadiomics (v3).29
Spatial and Single-Cell Transcriptomics
Single-cell RNA sequencing (scRNA-seq) data9 were batch-corrected and horizontal integration was performed through a mutual nearest neighbor approach.30FBXO2 gene expression across cell states was quantified using the SPATA toolbox.31 Spatial transcriptomic data, including non-malignant samples as well as glioblastoma specimens from newly diagnosed and recurrent tumors,10 were acquired from the Freiburg Spatial GBM Atlas and analyzed with the SPATA toolbox.31 Gene expression at spatial resolution was quantified by Moran’s I statistics. Gene set enrichment analysis was performed using clusterProfiler.23
Statistical Analysis
All data were analyzed with the statistical software R/RStudio.14,15 A detailed list of relevant packages and references is provided in Supplementary Table S2. Statistical tests were performed as indicated in the figure legends. Significance levels were indicated as follows: *P < .05, **P < .01, and ***P < .001.
Results
Study Design and Data Overview
To characterize the quantitative proteomic landscapes of newly diagnosed and recurrent glioblastoma, we applied PCT-SWATH to two independent patient cohorts of matched paired glioblastoma samples obtained at initial diagnosis and recurrence from two different centers (Figure 1A, Supplementary Table S1). Cohort 1 comprised pooled tissue punches covering different tumor regions from 26 matched pairs, whereas cohort 2 comprised one complete tissue section with a tumor content of more than 75% from 16 matched pairs. All tumors had an IDH-wildtype status and all patients received standard of care radiochemotherapy with temozolomide. In total, 8607 PG were detected in both cohorts, and 3001 PG were quantified in at least 40% of the samples and shared between both cohorts (Figure 1B and C).
Fig. 1.

Proteomic profiling of two independent cohorts of matched newly diagnosed and recurrent glioblastoma samples. (A) Scheme of the sample collection with tissue punches from cohort 1 and whole sections from cohort 2 from a different institution, subsequent protein extraction by PCT, and SWATH mass spectrometry. (B) Overview of the number of detected proteins and proportion of samples in which the proteins were quantified. (C) Number of common and unique proteins detected in cohort 1 and cohort 2, respectively.
The Proteomic Landscape of Newly Diagnosed and Recurrent Glioblastoma Is Overall Similar
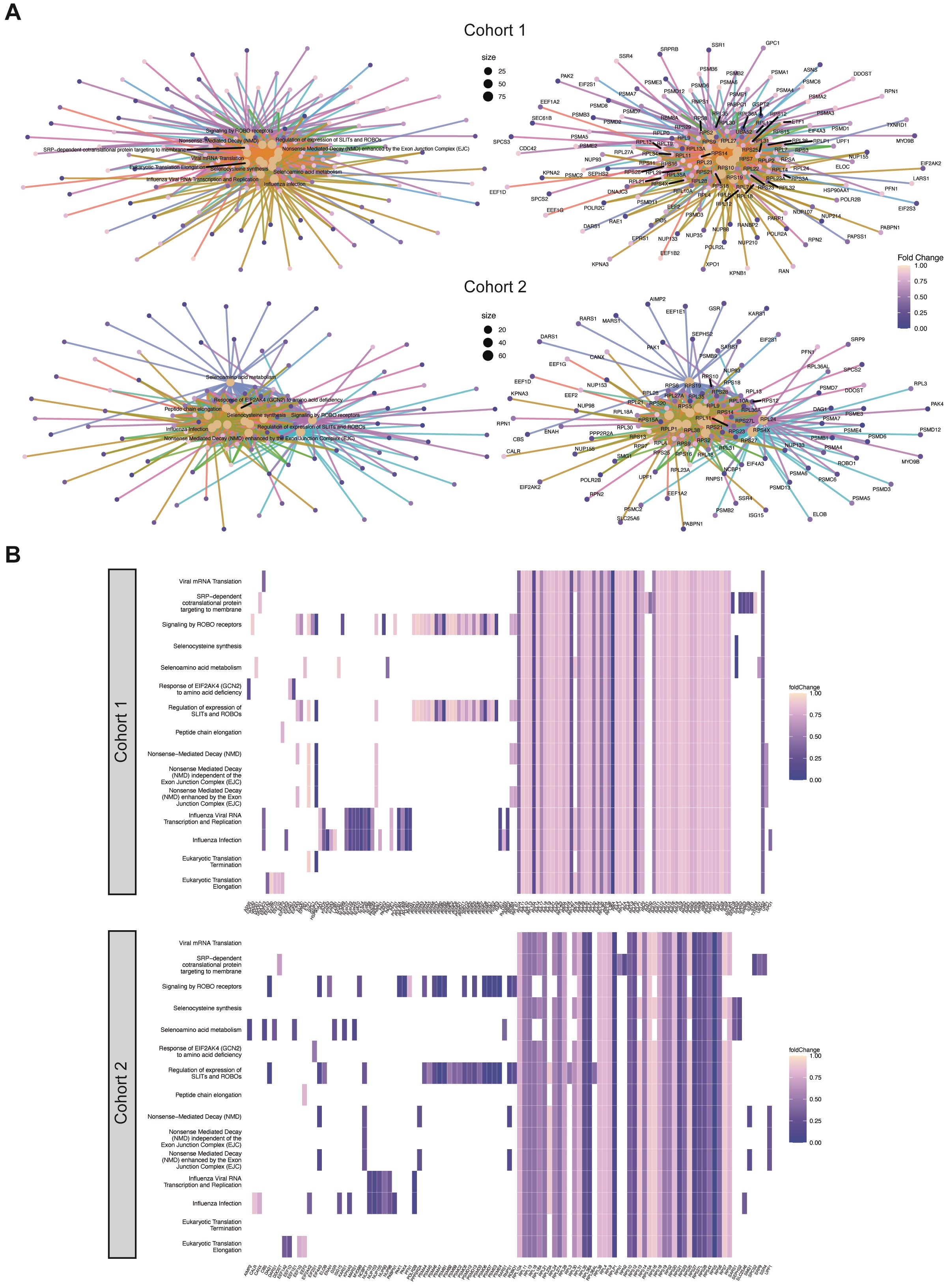
At the global proteome level, initial tumors did not separate uniformly from recurrent tumors in principal component analyses of either cohort (Figure 2A). Furthermore, average protein levels were highly correlated between newly diagnosed and recurrent samples, suggesting a high overall similarity of the overall quantitative proteomes between matched newly diagnosed and recurrent samples (Figure 2B). Unsupervised hierarchical clustering did not reveal clustering of other clinically relevant subgroups as defined by MGMT promoter methylation status, progression-free or overall survival based on the global proteome (Figure 2C). However, correlations of protein levels between patients were significantly lower in recurrent tumors compared to newly diagnosed tumors suggesting an overall higher heterogeneity of the disease at recurrence (Figure 2D). Comparative gene set enrichment analysis between initial and recurrent tumors revealed 15 significantly enriched Reactome pathways shared by both cohorts, all of which are associated with proteins that are less abundant at recurrence (Figure 2E, Supplementary Figure 1). Several of these pathways are involved in modifications of translation elongation, which is a frequently altered process in cancer.32
Fig. 2.

Quantitative proteomic landscapes of newly diagnosed and recurrent glioblastoma are similar. (A) Principal component analysis based on global proteomic data for cohort 1 (left) and cohort 2 (right). (B) Pearson correlation between initial and recurrent glioblastoma samples based on averaged protein abundance. (C) Hierarchical clustering based on the 500 most variable proteins for cohort 1 (top) and 2 (bottom). MGMT, methylation promoter methylation status (U, unmethylated, M, methylated, n.d., not determined). OS, overall survival; PFS, progression-free survival. Sex: f, female; m, male. 450k: DNA methylation class annotation. (D) Pearson correlations of different patient samples based on the global proteome calculated individually within initial and recurrent samples. P values were derived from a two-sided Welch test. (E) Significantly enriched Reactome pathways identified by gene set enrichment analysis. Only the least redundant pathways shared between both cohorts are shown (top: cohort 1, bottom: cohort 2; set size: 50-150; colors: FDR-adjusted P values; dot size: number of detected proteins per gene set, x-axis: enrichment ratio).
BCAS1, FBXO2, and INF2 Are More Abundant at Recurrence
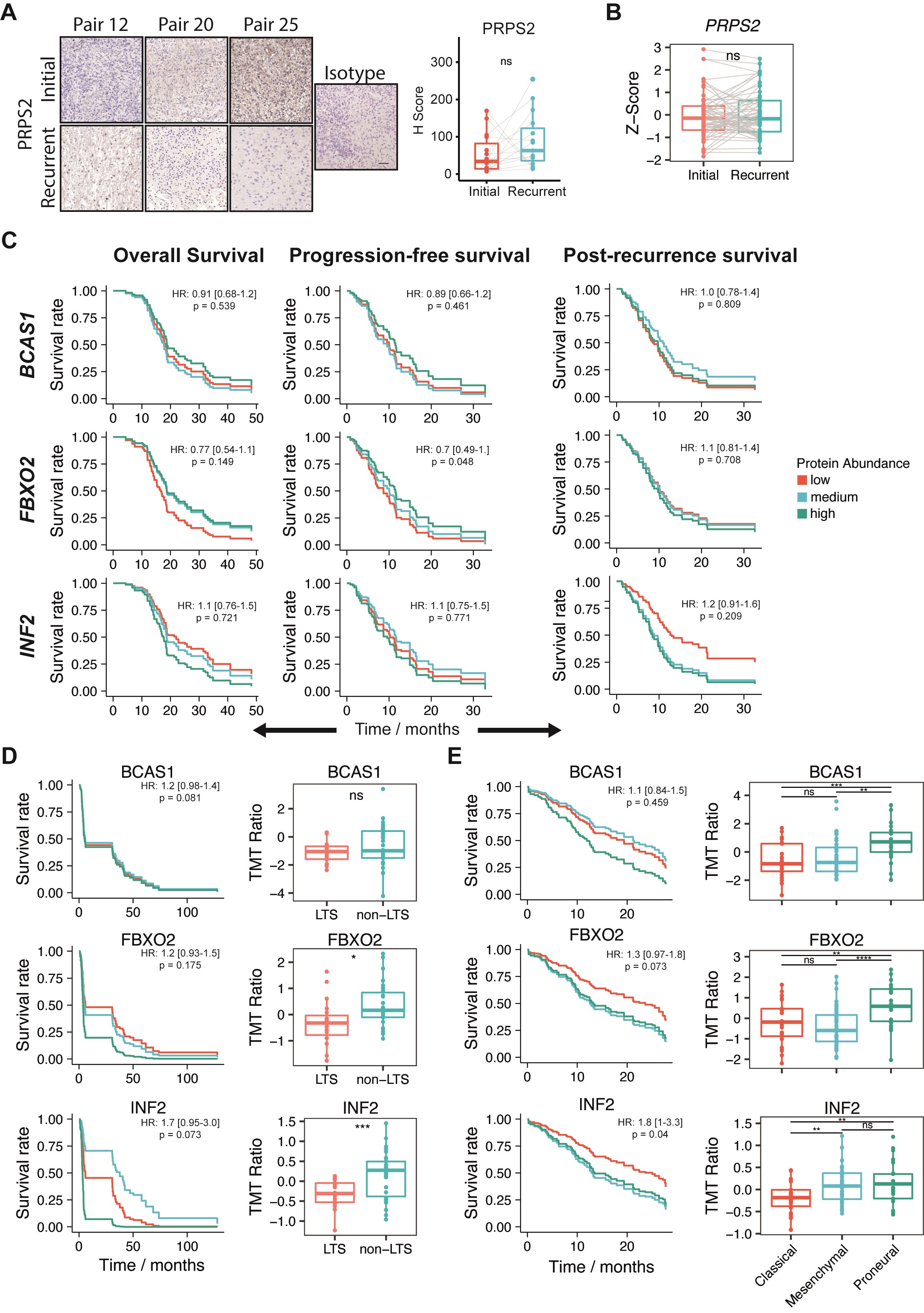
To identify individual proteins that showed quantitative differences from initial diagnosis to recurrence, we investigated longitudinally differentially expressed proteins with a 2-fold change or higher across both cohorts (Supplementary Note). This revealed 75 proteins with higher and 242 proteins with lower abundance at recurrence, of which 5 and 21, respectively, were statistically significant after follow-up statistical inference (Figure 3A). Subsequently, we focused on the 5 upregulated proteins CaMK2, BCAS1, FBXO2, INF2, and PRPS2 as their inhibition might be therapeutically exploited for recurrent glioblastoma. Calcium-calmodulin-dependent protein kinase II (CaMK2), including the beta (CAMK2B) and gamma (CAMK2G) isoforms, has been previously studied in recurrent glioblastoma, with high expression linked to poor prognosis.33,34 In contrast, INF235 and PRPS236 have only been scarcely investigated and BCAS1 and FBXO2 have not been investigated in glioblastoma. Increased abundance of BCAS1, FBXO2, and INF2, but not PRPS2, in recurrent tumors, was confirmed by immunohistochemical staining of a TMA comprising 20 initial-recurrent glioblastoma pairs (Figure 3C, Supplementary Figure 2A) and on independent RNA sequencing data5 (Figure 3D). Univariate Cox regression models of published proteomics24,25 and RNA sequencing data5 showed an opposing trend for an association of FBXO2 protein or transcript level with survival, whereas there was a consistent trend of INF2 expression with inferior survival on both protein and transcript levels (Figure 3E and F, Supplementary Figure 2C–E). In addition, both FBXO2 and INF2 abundance was significantly lower in long-term surviving (≥3-year OS) glioblastoma patients. Furthermore, protein abundances were significantly different among different transcriptional subtypes of glioblastoma37,38 (Figure 3E and F, Supplementary Figure 2D and E).
Fig. 3.

Differentially expressed proteins in matched initial-recurrent glioblastoma samples across both cohorts and external validation. (A) Scatterplot representing differential protein abundance in both cohorts. Only significantly up- (red) and downregulated (blue) proteins are labeled with names (one-sided, paired Wilcoxon rank-sum test; Ha = higher for upregulated proteins, Ha = lower for downregulated proteins). Fold change cutoffs are indicated by red dashed lines. (B) Protein abundance distributions of BCAS1, FBXO2, and INF2 in both cohorts. (C) Representative images and quantification of matched initial-recurrent glioblastoma sections stained for BCAS1 (15 sample pairs), FBXO2 (19 sample pairs), and INF2 (15 sample pairs). Scale bar = 100 µm. (D) Expression of indicated genes in external RNA sequencing data of matched initial-recurrent tumors. Significance in (C) and (D) according to a one-sided Welch test (Ha = greater at recurrence). Gray lines in B–D indicate sample pairs. (E) Association between OS and FBXO2 abundance in external proteomics data derived from long-term (LTS; OS ≥3 years) and non-long-term (non-LTS) surviving patients.24 Left: FBXO2 abundance in LTS and non-LTS patients. Right: Predicted survival based on Cox proportional hazard regression for three protein abundance quantiles (0%-33%: low; 33%-66%: medium, >66%: high). (F) Association between OS and FBXO2 in proteomics data from the CPTAC consortium.25 Left: FBXO2 protein abundance across different transcriptional subtypes. Right: Predicted survival as described in E. Statistical significances in (E) and (F) are based on unpaired Welch test.
FBXO2 Knockout Increases Survival in Orthotopic Xenograft Mouse Models and Reduces Glioma Cell Growth in Human Organotypic Brain Slice Cultures
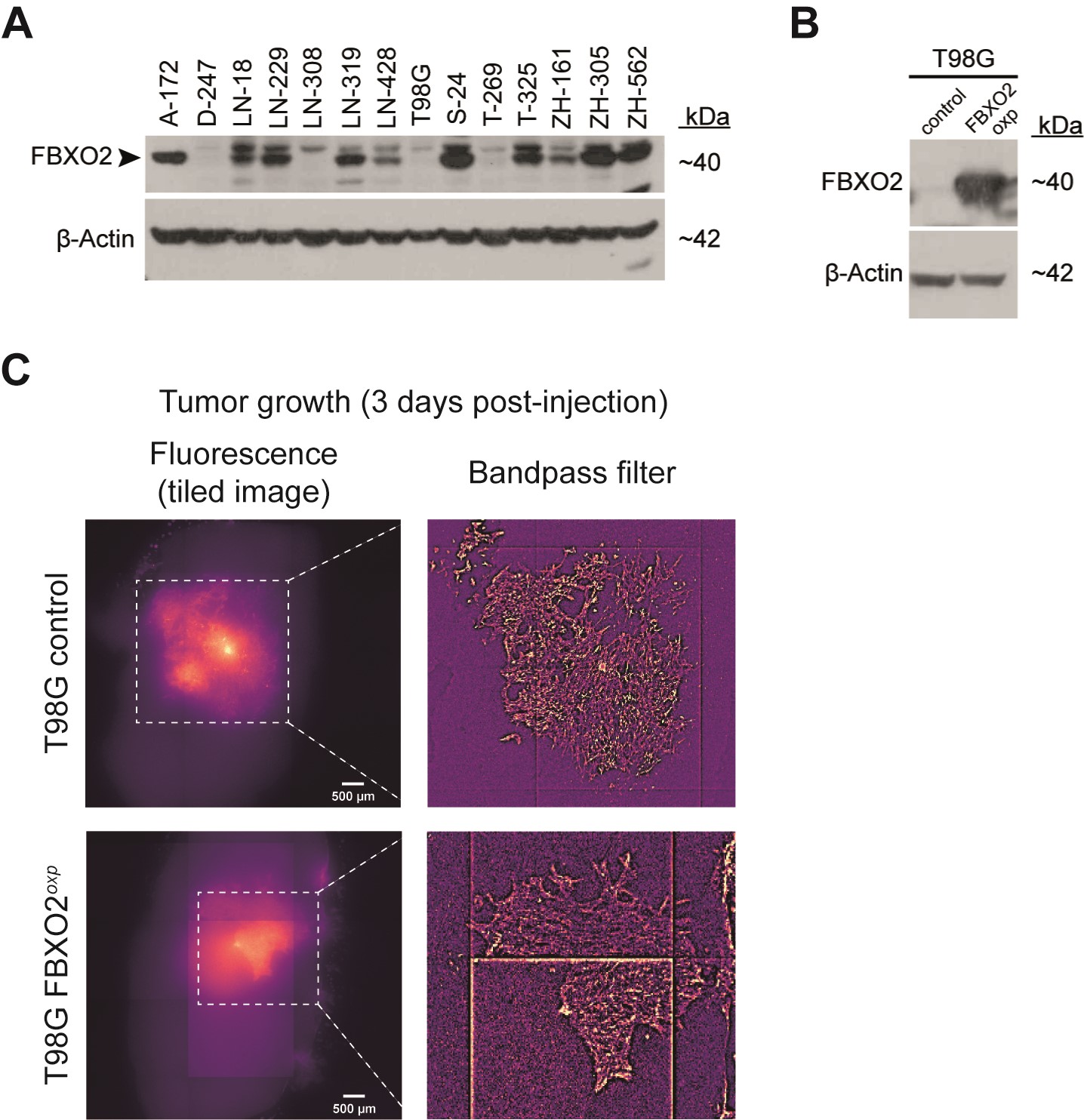
To investigate the functional roles of BCAS1, FBXO2, and INF2 in glioblastoma, we used CRISPR/Cas9 to generate individual knockout cell clones from the human glioma cell line LN-229 and the glioma-initiating cell line ZH-161 (Figure 4A and B, Supplementary Figure 3). Notably, protein-level confirmation of gene knockout was not possible for BCAS1 due to poor specificity of available antibodies. In both glioma cell lines, knockout of BCAS1, FBXO2, and INF2 did not lead to a consistent phenotype in growth and clonogenicity assays in vitro (Figure 4C and D, Supplementary Figure 4) but led to an increased migratory capacity in vitro (Figure 4E, Supplementary Figure 5). In contrast to the in vitro situation, tumor cell growth in vivo depends on complex interactions with the surrounding extracellular matrix (ECM), cell types, metabolites, and growth factors. In orthotopic xenograft models, FBXO2 knockout cells led to improved survival and an increased fraction of long-term surviving mice compared to the control cells in both cell line models (Figure 5A). Histological assessment of mouse brains at the onset of neurological symptoms confirmed the presence of tumors and indicated reduced tumor growth in FBXO2-KO cells (Figure 5A). In contrast, the survival effects upon implantation of BCAS1- or INF2-knockout glioma cells were less consistent between the individual clones and cell lines. To confirm the role of FBXO2 for the integration and growth of glioma cells in a environment recapitulating human microenvironment, we injected LN-229 FBXO2 knockout cells or control cells into human organotypic brain slices39 (Figure 5B). LN-229 control cells exhibited a more infiltrative behavior characterized by the extensive spreading of glioma cells whereas LN-229 FBXO2 knockout cells displayed a spot-like growth pattern, resulting in an overall reduced infiltrated area. Overexpression of FBXO2 in a FBXO2-negative cell line model did not reveal striking morphological differences (Supplementary Figure 6), which might suggest that not FBXO2 itself determines the different growth patterns but rather cellular changes associated with its physiological expression or loss. We further investigated the association of FBXO2 with infiltrative tumor growth using MRI and matched transcriptomic data from the TCGA database. This revealed that tumors with high FBXO2 expression, had a reduced tumor sphericity compared to tumors with low FBXO2 expression, which suggests a more diffuse tumor infiltration of tumors with high FBXO2 expression (Figure 5C).
Fig. 4.

CRISPR/Cas9-mediated knockout of BCAS1, FBXO2, and INF2 in human glioma cell lines and subsequent phenotypic characterization. (A) Illustration of the knockout strategy using a CRISPR/Cas9-RNP approach. (B) Validation of editing in LN-229- or ZH-161-derived knockout or control cells as indicated on DNA level by PCR (left) and protein level by immunoblot (right: FBXO2 and INF2 only). (C) Cell growth of knockout and control cell lines assessed either by microscopic cell tracking and cell doubling time estimation for LN-229-derived clones (left) or spheroid growth assay for ZH-161-derived clones (right). (D) Clonogenic cell growth assessed by crystal violet for LN-229-derived clones or soft agar assay for ZH-161-derived clones. Top panel shows representative images. Lower panel shows the corresponding quantification (N = 3 replicates per condition). Each assay was performed at least three times with similar results. (E) Quantification of migration distances as assessed by spheroid invasion. Boxplots show pooled results of N = 5 (LN-229) or N = 4 (ZH-161) replicate wells, respectively. Significances in (C), (D), and (E) according to Bonferroni-adjusted pairwise t-tests compared to control cells.
Fig. 5.

FBXO2 knockout in human glioma cell lines confers a survival benefit in glioma-bearing mice and affects growth patterns in human brain slice cultures and glioblastoma patients. (A) Left: Survival of nude mice after orthotopic implantation of LN-229- or ZH-161-derived control or knockout clones. Statistical significances were assessed by pairwise log-rank tests FDR-corrected P values are indicated. LTS, long-term surviving mice. Vertical lines indicate censored events. Right: Representative images of tumors stained by hematoxylin-eosin from brains isolated at indicated time points. Dashed lines indicate tumor borders. Black arrowheads indicate clusters of tumor cells near the implantation site in LN-229 FBXO2-KO clones. Scale bar = 100 µm. (B) Left: Experimental workflow for human organotypic brain slice culture experiments. Right: Representative images of brain slice cultures 3 days after implantation of fluorescently labeled control or knockout clones and microscopy-based quantification of tumor growth areas using a Wilcoxon rank-sum test. (C) Association between FBXO2 abundance and tumor sphericity based on RNA microarray and MRI data from the TCGA network (n = 118 cases). Representative MRI images (FLAIR) of low- and high-sphericity tumors are shown with segmented tumor regions as indicated. P values were derived from pairwise Welch test.
FBXO2 Expression in Glioma Cells Is Associated With Developmental Transcription Programs and Pathways Modulating Synaptic Activity and Interaction With the Environment
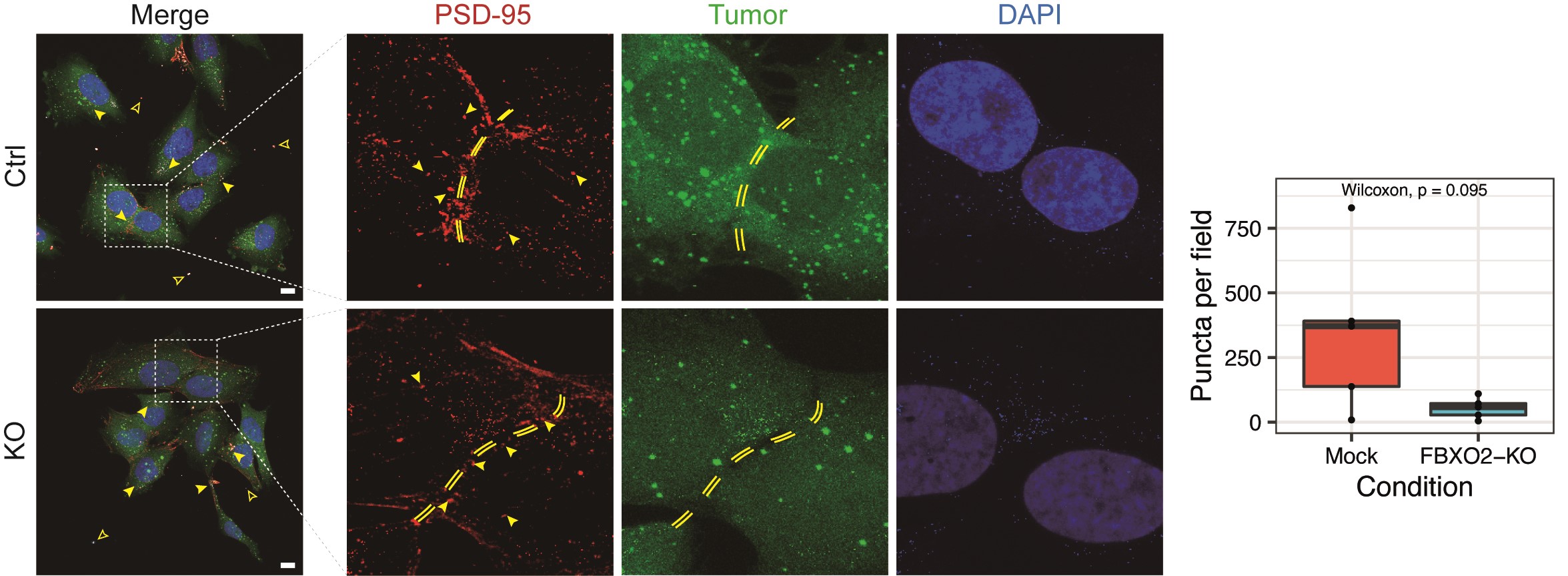
External scRNA-seq data9 as well as spatial transcriptomics data from external glioblastoma samples10 were analyzed to identify glioma cell states that are associated with FBXO2 expression and the spatial distribution of FBXO2-expressing cancer cells. FBXO2-expressing glioma cells were associated with tumor regions exhibiting a reactive immune signature and neurodevelopmental transcription programs (Figure 6A), whereas BCAS1 and INF2 were more broadly expressed across different tumor regions (Supplementary Figure 7A). Incorporation of known gene expression signatures9,10,40 confirmed a strong spatial correlation between FBXO2-enriched tumor regions and neurodevelopmental, NPC/OPC-like, and proneural gene signatures (Figure 6B), thereby confirming increased FBXO2 protein abundance observed in proneural tumors (Figure 3F). In contrast, MES-like signatures and FBXO2 were negatively correlated in a spatial context (Figure 6B, Supplementary Figure 7B). In spatial transcriptomic data from healthy brain tissue, FBXO2 expression was restricted to layer 1 (L1) of the neocortex (Figure 6C), which is a niche for neural progenitor cells.41 Importantly, FBXO2 expression was strongly enriched within the infiltration zone and also upregulated at recurrence (Figure 6D and E), confirming proteomics and bulk RNA-seq data (Figure 3B and D). In FBXO2+ glioma cells, pathways associated with synaptic activity, cell-cell interaction, transmembrane transport, vesicle secretion, and phospholipase C signaling were significantly enriched (Supplementary Figure 7C). These associations were also confirmed by meta-pathway analysis combining both bulk and scRNA-seq data (Figure 6F) and by reduced formation of PSD-95 puncta in the vicinity of FBXO2-KO glioma cells in organotypic brain slice cultures (Figure 6G, Supplementary Figure 8), suggesting a role of FBXO2 in glioma cells in mediating interactions with the surrounding CNS tissue.
Fig. 6.

Single-cell, bulk RNA sequencing, and spatial transcriptomics reveal cell states, locations, and pathways associated with FBXO2 expression in glioma cells. (A) Transcriptional profiles of spatially separated tumor regions. Each vertical line is a tumor region and transcriptional profiles of different regions are indicated as radial glia, reactive immune, OPC, neural development, and reactive hypoxia (upper panel). Glioma cells were identified by copy number variation (CNV) and FBXO2, BCAS1, and INF2 expression in glioma cells was mapped to different tumor zones (lower panel). (B) Spatial correlation between FBXO2 expression and different single-cell, spatial, and bulk expression signatures. (C–E) FBXO2 expression in spatial transcriptomic data from healthy brain cortex (C), glioblastoma tissue samples (D), and initial-recurrent glioblastoma tissue (E). Expression level of FBXO2 in different tumor regions is color-coded by normalized gene expression. (F) Meta-pathway enrichment analysis based on external glioblastoma single-cell and bulk RNA sequencing data. (G) Effect of FBXO2 knockout on PSD-95-positive post-synaptic puncta formation in the vicinity of glioma cells in organotypic brain slice cultures. Filled and empty arrowheads represent positive and negative puncta, respectively. Quantification of puncta by Wilcoxon rank-sum test. Scale bar = 25 µm.
Discussion
Despite multimodal treatment efforts, there is an unmet need to better characterize factors that are associated with disease progression of glioblastoma. Recent studies investigated longitudinal genomic3,4 and transcriptomic5 changes in glioblastoma, whereas only one study so far characterized initial-recurrent tissue pairs on the proteome level.42 This study revealed stochastic patient-individual longitudinal protein changes and poor separation of initial and recurrent tumors but is limited by low sample size and limited protein coverage. We extend these findings using PCT-SWATH-MS of FFPE samples from two independently assembled, processed, and acquired patient cohorts. Despite the overall similar global proteomic landscapes of matched initial and recurrent glioblastomas, we identified shared proteins that were down- or upregulated at recurrence in both cohorts (Figure 3). FBXO2, a brain-enriched ubiquitin ligase substrate adaptor,43 was highly upregulated at recurrence across both independent cohorts and is also significantly less abundant in long-term surviving glioblastoma patients (Figure 3). This protein has not been investigated in the context of glioblastoma and its role in physiological and pathophysiological processes is poorly understood. Recent experimental studies have demonstrated an oncogenic role of FBXO2 in other cancer types.44,45 In addition, retrospective studies have demonstrated an association between FBXO2 expression and metastatic rate in colorectal and gastric cancer.46FBXO2 knockout in two human glioma models did not reveal a consistent phenotype in vitro (Figure 4) but led to a strong survival increase in vivo and impaired growth and infiltration in human organotypic brain slice cultures (Figure 5). Furthermore, our data provide early evidence for reduced post-synaptic puncta formation in the vicinity of FBXO2 knockout cells, suggesting a role of FBXO2 in shaping the interaction of glioma cells with their microenvironment. Recent studies have demonstrated that FBXO2 may modulate synaptic connectivity47 and neurodegeneration,48 which potentially could be exploited by glioma cells to facilitate integration into the microenvironment. Additionally, analyses of spatial transcriptomic, single-cell, and bulk RNA sequencing data demonstrate enrichment of synaptic signaling, cell-cell interaction, and neurodevelopmental pathways in FBXO2-expressing glioma cells and reveal spatially enriched FBXO2 expression at the infiltration margin of tumors. These findings are consistent with recent transcriptomics data showing increased neuronal signaling in recurrent tumors.49 Overall, this study indicates a potential role of FBXO2 in promoting tumor growth in vivo. However, additional efforts will be required to understand the underlying molecular mechanisms for potential therapeutic targeting.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the Mass Spectrometry & Metabolomics Core Facility at Westlake University for peptide fractionation and Westlake University Supercomputer Center for assistance in data storage and computation.
Contributor Information
Marcel Buehler, Department of Neurology and Clinical Neuroscience Center, University Hospital Zurich and University of Zurich, Zurich, Switzerland.
Xiao Yi, Key Laboratory of Structural Biology of Zhejiang Province, School of Life Sciences, Westlake University, Hangzhou, Zhejiang, China; Westlake Intelligent Biomarker Discovery Lab, Westlake Laboratory of Life Sciences and Biomedicine, Hangzhou, Zhejiang, China; Westlake Omics Biotechnology Co., Ltd., Hangzhou, Zhejiang, China.
Weigang Ge, Key Laboratory of Structural Biology of Zhejiang Province, School of Life Sciences, Westlake University, Hangzhou, Zhejiang, China; Westlake Intelligent Biomarker Discovery Lab, Westlake Laboratory of Life Sciences and Biomedicine, Hangzhou, Zhejiang, China; Westlake Omics Biotechnology Co., Ltd., Hangzhou, Zhejiang, China.
Peter Blattmann, Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, Zurich, Switzerland.
Elisabeth Rushing, Department of Neuropathology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Guido Reifenberger, Department of Neuropathology, Heinrich Heine University, Duesseldorf, Germany; German Cancer Consortium, partner site Essen/Düsseldorf, Duesseldorf, Germany.
Joerg Felsberg, Department of Neuropathology, Heinrich Heine University, Duesseldorf, Germany; German Cancer Consortium, partner site Essen/Düsseldorf, Duesseldorf, Germany.
Charles Yeh, Department of Biology, Institute of Molecular Health Sciences, ETH Zürich, Zürich, Switzerland.
Jacob E Corn, Department of Biology, Institute of Molecular Health Sciences, ETH Zürich, Zürich, Switzerland.
Luca Regli, Department of Neurosurgery, Clinical Neuroscience Center, University Hospital Zurich and University of Zurich, Zürich, Switzerland.
Junyi Zhang, Microenvironment and Immunology Research Laboratory, Department of Neurosurgery, Medical Center, University of Freiburg, Germany; German Cancer Consortium (DKTK), partner site Freiburg, Freiburg, Germany; Translational Neuro-Oncology Research Group, Medical Center, University of Freiburg, Freiburg, Germany.
Ann Cloos, Microenvironment and Immunology Research Laboratory, Department of Neurosurgery, Medical Center, University of Freiburg, Germany; German Cancer Consortium (DKTK), partner site Freiburg, Freiburg, Germany; Translational Neuro-Oncology Research Group, Medical Center, University of Freiburg, Freiburg, Germany.
Vidhya M Ravi, Microenvironment and Immunology Research Laboratory, Department of Neurosurgery, Medical Center, University of Freiburg, Germany; German Cancer Consortium (DKTK), partner site Freiburg, Freiburg, Germany; Translational Neuro-Oncology Research Group, Medical Center, University of Freiburg, Freiburg, Germany; Freiburg Institute for Advanced Studies (FRIAS), University of Freiburg, Freiburg, Germany.
Benedikt Wiestler, Department of Neuroradiology, Klinikum rechts der Isar, Technical University Munich, Munich, Germany.
Dieter Henrik Heiland, Microenvironment and Immunology Research Laboratory, Department of Neurosurgery, Medical Center, University of Freiburg, Germany; German Cancer Consortium (DKTK), partner site Freiburg, Freiburg, Germany.
Ruedi Aebersold, Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, Zurich, Switzerland.
Michael Weller, Department of Neurology and Clinical Neuroscience Center, University Hospital Zurich and University of Zurich, Zurich, Switzerland.
Tiannan Guo, Key Laboratory of Structural Biology of Zhejiang Province, School of Life Sciences, Westlake University, Hangzhou, Zhejiang, China; Westlake Intelligent Biomarker Discovery Lab, Westlake Laboratory of Life Sciences and Biomedicine, Hangzhou, Zhejiang, China.
Tobias Weiss, Department of Neurology and Clinical Neuroscience Center, University Hospital Zurich and University of Zurich, Zurich, Switzerland; Key Laboratory of Structural Biology of Zhejiang Province, School of Life Sciences, Westlake University, Hangzhou, Zhejiang, China.
Funding
This work was supported by the Forschungskredit of the University of Zurich (FK-18-054 to T.W.); Betty and David Koetser Foundation for Brain Research (T.W.); Sophienstiftung (T.W.); Promedica Foundation (T.W.); Helmut Horten Stiftung (T.W.); National Key R&D Program of China (2020YFE0202200); National Natural Science Foundation of China (No. 81972492); and Chinese National Science Fund for Young Scholars (No. 21904107).
Conflict of interest statement. T.G. is a shareholder of Westlake Omics Inc. W.G. and X.Y. are currently employees of Westlake Omics Inc. The other authors declare no conflicts of interest.
Authorship statement. Collection of samples: E.R., J.F., G.R., L.R. Experimental design, execution, data analysis, and manuscript writing: T.W., M.B., M.W., R.A., and T.G. Brain slice cultures and transcriptomic data analysis: D.H.H., V.M.R., A.C., and J.Z. MRI analysis: B.W. Mass spectrometry measurements, data analysis, and interpretation: P.B., X.Y., and W.G. CRISPR/Cas9 knockout experiments: J.E.C. and C.Y.
References
- 1. Wen PY, Weller M, Lee EQ, et al. . Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020;22(8):1073–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weller M, van den Bent M, Preusser M, et al. . EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021;18(3):170–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barthel FP, Johnson KC, Varn FS, et al. . Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576(7785):112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Körber V, Yang J, Barah P, et al. . Evolutionary trajectories of IDHWT glioblastomas reveal a common path of early tumorigenesis instigated years ahead of initial diagnosis. Cancer Cell. 2019;35(4):692–704.e12. [DOI] [PubMed] [Google Scholar]
- 5. Wang J, Cazzato E, Ladewig E, et al. . Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48(7):768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buccitelli C, Selbach M. mRNAs, proteins and the emerging principles of gene expression control. Nat Rev Genet. 2020;21(10):630–644. [DOI] [PubMed] [Google Scholar]
- 7. Guo T, Kouvonen P, Koh CC, et al. . Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat Med. 2015;21(4):407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu Y, Weiss T, Zhang Q, et al. . High-throughput proteomic analysis of FFPE tissue samples facilitates tumor stratification. Mol Oncol. 2019;13(11):2305–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neftel C, Laffy J, Filbin MG, et al. . An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178(4):835–849.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ravi VM, Will P, Kueckelhaus J, et al. . Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell. 2022;40(6):639–655.e13 doi: 10.1101/2021.02.16.431475. PMID: 35700707. [DOI] [PubMed] [Google Scholar]
- 11. Le Rhun E, Achenbach C, Lohmann B, et al. . Profound, durable and MGMT‐independent sensitivity of glioblastoma cells to cyclin‐dependent kinase inhibition. Int J Cancer. 2019;145(1):242–253. [DOI] [PubMed] [Google Scholar]
- 12. Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34(3):339–344. [DOI] [PubMed] [Google Scholar]
- 13. McQuin C, Goodman A, Chernyshev V, et al. . CellProfiler 3.0: next-generation image processing for biology. PLoS Biol. 2018;16(7):e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. RStudio Team. RStudio: Integrated Development Environment for R. Boston, MA: RStudio, PBC; 2020. [Google Scholar]
- 15. R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria; 2021. https://www.R-project.org/. [Google Scholar]
- 16. Chen W, Wong C, Vosburgh E, et al. . High-throughput image analysis of tumor spheroids: a user-friendly software application to measure the size of spheroids automatically and accurately. J Vis Exp. 2014;89(89):51639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumar KS, Pillong M, Kunze J, et al. . Computer-assisted quantification of motile and invasive capabilities of cancer cells. Sci Rep. 2015;5(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schindelin J, Arganda-Carreras I, Frise E, et al. . Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weiss T, Schneider H, Silginer M, et al. . NKG2D-dependent antitumor effects of chemotherapy and radiotherapy against glioblastoma. Clin Cancer Res. 2018;24(4):882–895. [DOI] [PubMed] [Google Scholar]
- 20. Wild P, Rupp N, Buhmann J, et al. . TMARKER: a free software toolkit for histopathological cell counting and staining estimation. J Pathol Inform. 2013;4(2):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. von Achenbach C, Weller M, Kaulich K, et al. . Synergistic growth inhibition mediated by dual PI3K/mTOR pathway targeting and genetic or direct pharmacological AKT inhibition in human glioblastoma models. J Neurochem. 2020;153(4):510–524. [DOI] [PubMed] [Google Scholar]
- 22. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–2849. [DOI] [PubMed] [Google Scholar]
- 23. Wu T, Hu E, Xu S, et al. . clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yanovich-Arad G, Ofek P, Yeini E, et al. . Proteogenomics of glioblastoma associates molecular patterns with survival. Cell Rep. 2021;34(9):108787. [DOI] [PubMed] [Google Scholar]
- 25. Wang LB, Karpova A, Gritsenko MA, et al. . Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell. 2021;39(4):509–528.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Therneau TM. A Package for Survival Analysis in R. 2022. https://cran.r-project.org/package=survival. Accessed May 15, 2022. [Google Scholar]
- 27. Kassambara A, Kosinski M, Biecek P.. survminer: Drawing Survival Curves Using “ggplot2”. 2021. https://cloud.r-project.org/package=survminer. [Google Scholar]
- 28. Kofler F, Berger C, Waldmannstetter D, et al. . BraTS Toolkit: translating BraTS brain tumor segmentation algorithms into clinical and scientific practice. Front Neurosci. 2020;14:125. doi: 10.3389/fnins.2020.00125 PMID: 32410929; PMCID: PMC7201293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Griethuysen JJM, Fedorov A, Parmar C, et al. . Computational radiomics system to decode the radiographic phenotype. Cancer Res. 2017;77(21):e104–e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ravi VM, Neidert N, Will P, et al. . T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun. 2022;13(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kueckelhaus J, von Ehr J, Ravi VM, et al. . Inferring spatially transient gene expression pattern from spatial transcriptomic studies. bioRxiv, doi: 10.1101/2020.10.20.346544, October 21 2020, preprint: not peer reviewed. [DOI] [Google Scholar]
- 32. Fabbri L, Chakraborty A, Robert C, Vagner S. The plasticity of mRNA translation during cancer progression and therapy resistance. Nat Rev Cancer. 2021;21(9):558–577. [DOI] [PubMed] [Google Scholar]
- 33. Van den Boom J, Wolter M, Kuick R, et al. . Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163(3):1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y, Xu J, Zhu X. A 63 signature genes prediction system is effective for glioblastoma prognosis. Int J Mol Med. 2018;41(4):2070–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heuser VD, Kiviniemi A, Lehtinen L, et al. . Multiple formin proteins participate in glioblastoma migration. BMC Cancer. 2020;20(1):710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li C, Yan Z, Cao X, Zhang X, Yang L. Phosphoribosylpyrophosphate synthetase 1 knockdown suppresses tumor formation of glioma CD133+ cells through upregulating cell apoptosis. J Mol Neurosci. 2016;60(2):145–156. [DOI] [PubMed] [Google Scholar]
- 37. Verhaak RGW, Hoadley KA, Purdom E, et al. . Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Q, Hu B, Hu X, et al. . Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32(1):42–56.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ravi VM, Joseph K, Wurm J, et al. . Human organotypic brain slice culture: a novel framework for environmental research in neuro-oncology. Life Sci Alliance. 2019;2(4):e201900305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Richards LM, Whitley OKN, MacLeod G, et al. . Gradient of developmental and injury response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat Cancer. 2021;2(2):157–173. [DOI] [PubMed] [Google Scholar]
- 41. Ohira K, Furuta T, Hioki H, et al. . Ischemia-induced neurogenesis of neocortical layer 1 progenitor cells. Nat Neurosci. 2010;13(2):173–179. [DOI] [PubMed] [Google Scholar]
- 42. Dekker LJM, Kannegieter NM, Haerkens F, et al. . Multiomics profiling of paired primary and recurrent glioblastoma patient tissues. Neurooncol Adv. 2020;2(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14(6):369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao X, Guo W, Zou L, Hu B. FBXO2 modulates STAT3 signaling to regulate proliferation and tumorigenicity of osteosarcoma cells. Cancer Cell Int. 2020;20(1):245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Che X, Jian F, Wang Y, et al. . FBXO2 promotes proliferation of endometrial cancer by ubiquitin-mediated degradation of FBN1 in the regulation of the cell cycle and the autophagy pathway. Front Cell Dev Biol. 2020;8:843. doi: 10.3389/fcell.2020.00843. PMID: 32984335; PMCID: PMC7487413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu S, Wang T, Guan ZR, et al. . FBXO2, a novel marker for metastasis in human gastric cancer. Biochem Biophys Res Commun. 2018;495(3):2158–2164. [DOI] [PubMed] [Google Scholar]
- 47. Atkin G, Moore S, Lu Y, et al. . Loss of F-box only protein 2 (Fbxo2) disrupts levels and localization of select NMDA receptor subunits, and promotes aberrant synaptic connectivity. J Neurosci. 2015;35(15):6165–6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu EA, Schultz ML, Mochida C, et al. . Fbxo2 mediates clearance of damaged lysosomes and modifies neurodegeneration in the Niemann-Pick C brain. JCI Insight. 2020;5(20):e136676. doi: 10.1172/jci.insight.136676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Varn FS, Johnson KC, Martinek J, et al. . Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell. 2022;185(12):2184–2199.e16. doi: 10.1016/j.cell.2022.04.038. Epub 2022 May 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Protein matrices, clinical information, and analysis code are available in the Supplementary material. Single-cell sequencing data and spatial transcriptomic data are available online.9,10 Proteomics raw data are deposited on ProteomeXchange (https://www.iprox.org/, ID: IPX0003752001).