

Abstract
Cancers evade immune surveillance, which can be reversed through immune checkpoint therapy in a small subset of cases. Here we report that the MYC oncogene suppresses innate immune surveillance and drives resistance to immunotherapy. In 33 different human cancers, MYC genomic amplification and overexpression increased immune checkpoint expression, predicted non-responsiveness to immune checkpoint blockade, and was associated with both Th2-like immune profile and reduced CD8 T cell infiltration. MYC transcriptionally suppressed innate immunity and MHCI mediated antigen presentation, which in turn impeded T cell response. Combined, but not individual, blockade of PDL1 and CTLA4 could reverse MYC-driven immune suppression by leading to recruitment of pro-inflammatory antigen-presenting macrophages with increased CD40 and MHCII expression. Depletion of macrophages abrogated the anti-neoplastic effects of PDL1 and CTLA4 blockade in MYC-driven hepatocellular carcinoma (HCC). Hence, MYC is a predictor of immune checkpoint responsiveness and a key driver of immune evasion through the suppression of pro-inflammatory macrophages. The immune evasion induced by MYC in HCC can be overcome by combined PDL1 and CTLA4 blockade.
Keywords: MYC, macrophage, immunotherapy, immune checkpoint
INTRODUCTION
The MYC oncogene is one of the most common drivers of human neoplasia, with a majority of human cancers having genetic alterations either in MYC or in one of the members of the proximal MYC network (1). This makes MYC a very desirable therapeutic target. However, no existing therapies directly target MYC. MYC contributes to tumorigenesis both by causing tumor intrinsic effects such as autonomous cellular proliferation, as well as by tumor extrinsic effects such as remodeling the tumor microenvironment via immune evasion (2). MYC inhibits anti-tumor immunity by multiple mechanisms, including impeding CD4+ T cell activation, promoting expression of immune checkpoints like PDL1 and CD47, and blocking natural killer (NK) cell activation (2–7). These immune effects suggest that MYC-driven tumors may be vulnerable to specific combinations of immune therapies.
MYC activation is a major oncogenic event in many types of epithelial tumors including hepatocellular carcinoma (HCC). HCC is an aggressive malignancy with dismal prognosis and the MYC pathway activated in 70% of these tumors (1). Moreover, several studies have established that MYC plays a crucial pathogenic role in HCC progression (8–12). Yet, MYC-driven HCCs remain a therapeutically challenging subset of HCCs. Immune checkpoint inhibitors (ICI) have shown promise in many human cancers but, notably, have failed as first-line monotherapy for HCC (13,14). Recently, the combination of PD1 and CTLA4 blockade has shown promise in HCC. However, only around 20% of patients showed objective response, while around 50% of the patients experienced serious adverse events (15). Understanding the mechanisms by which individual or combined ICIs are effective in subsets of patients could help identify the right patient for the right treatment, and lead to development of further therapies targeting the mechanisms of resistance.
We hypothesize that MYC is a major driver of immune evasion and dictates response to immune checkpoint inhibition in HCC. Here we demonstrate the combination of checkpoint inhibitors PDL1 and CTL4 can overcome MYC immune evasion by restoring macrophage activation via CD40 and MHCII expression, thereby eliciting robust anti-tumor CD8+ T-cell responses.
METHODS
Human pan-cancer TCGA analysis
We used the TumorMap (16) (https://tumormap.ucsc.edu/), a tool that enables grouping the cancer genome atlas (TCGA) samples in a visually accessible way. Tumor Map uses dimensionality reduction methods to condense high-dimensional omics data to a two-dimensional space. In the TumorMap, each node is a sample, and clusters of samples indicate groups with similar genomic alteration events or oncogenic signatures. We used the TCGA pan-cancer data to visualize samples with MYC amplification (17). We visualized the MYC gene signature derived from gene set enrichment analysis of MYC amplification across the TCGA pan-cancer samples (http://www.broadinstitute.org/gsea/msigdb/cards/chr8q24). The Spearman test was used for correlation analysis. Kaplan-Meier analysis was performed for survival analysis. We used the immune signatures and immune deconvolution analysis published in the pan-cancer TCGA immune landscape data (18).
RNA sequencing
RNA sequencing of MYC-HCC was performed at the Beijing Genomics Institute (BGI) using their BGIseq 500 platform single end 150 bp, 20 million reads per sample. MYC-activated tumors (MYC-On, n=3)) and tumors four days after MYC inactivation (MYC-Off, n=3) underwent whole transcriptome sequencing. Gene expression level was quantified by a software package called RSEM. For each RNA sequencing data sample deposited in Gene Expression Omnibus (GEO), we counted the number of identified expressed genes and calculated its proportion to the total gene number in the database. DEseq software was used to perform differential expression analysis. Random forest classifier algorithm was used to derive a 56-gene signature by comparing mouse MYC-HCC On vs. Off, which was used to stratify human HCC in the TCGA cohort. Qlucore Omics Explorer v. 2.2 was used to create heatmaps for visual representation of data.
Ingenuity Pathway Analysis (IPA, Qiagen) was used to perform functional pathway analysis and similarity analysis using “Analysis Match”. To compare the MYC-HCC transcriptional changes to other analyses, ‘Analysis Match’ builds a signature from the highest confidence predictions for MYC-HCC and compares it to a curated list of analyses from public gene expression datasets. A signature in the MYC-HCC analysis was considered to match a signature from another human HCC transcriptome analysis if the predicted activated and inhibited entities strongly overlap.
The CIBERSORT gene expression deconvolution package was used to estimate the immune cell composition in the MYC-HCC (19). The LM22 signature was used as the immune cell gene signature after the careful modification of genes in the signature to their respective mouse orthologs. The settings for the run were: 1000 permutations with quantile normalization disabled. The Student’s t-test was used to infer the statistical significance of the predicted immune cell populations where p<0.05 was considered significant.
The immunotherapy treated MYC-HCC were sequenced using the Nanostring IO360 data using software developed by ROSALIND, Inc. (San Diego, CA, United States) follows the nCounter® Advanced Analysis protocol (20). Housekeeping probes to be used for normalization were selected based on the geNorm algorithm as implemented in the NormqPCR R library (20). Significant p-values (p < 0.05) were adjusted using the Benjamini–Hochberg method of estimating false discovery rates. We used the Meta-Analysis pipeline from ROSALIND to compare the transcriptional changes induced by the combination therapy and the individual monotherapies versus control antibody treated MYC-HCC. ROSALIND Meta-Analysis clusters the data using K-means cluster for value k=2 to 15, then calculates the optimal number of clusters using the PAM method from R package fpc with method ‘clara’ and runs enrichment analysis using hypergeometric tests. Hypergeometric distribution was used to analyze the enrichment of pathways, gene ontology, domain structure, and other ontologies. The topGO R library was used to determine local similarities and dependencies between GO terms to perform Elim pruning correction. Enrichment was calculated relative to a set of background genes relevant for the experiment.
Transgenic Mice
Animals were housed in a pathogen-free environment at Stanford University and all procedures were performed in accordance with Stanford’s Administrative Panel on Laboratory Animal Care (APLAC) protocols and approved by the Institutional Animal Care and Use Committee (IACUC). LAP-tTA/tet-O-MYC transgenic lines were used, as previously described (21). Mice were administered weekly doses of 0.1 mg/mL doxycycline (Sigma) in drinking water during mating and until four weeks of age. At 4 weeks, mice were taken off doxycycline. Mice were screened for tumors via MRI at 2–3 months of age, at which time they developed tumors between 50–150mm3.
Magnetic Resonance Imaging (MRI)
MRI was performed using a 7T small animal MRI (Agilent conversion) with a 40 mm Varian Millipede RF coil (ExtendMR LLC, Milpitas, CA) at the Stanford Small Animal Imaging Facility as previously described (22,23). Briefly, animals were anesthetized with 1–3% isoflurane and placed into the MRI scanner containing a 40 mm Varian Millipede RF coil (ExtendMR LLC, Milpitas, CA). ParaVision (PV6.01) was used to acquire the DICOM images, and tumor volumes were quantified from images using Osirix image processing software (Osirix, UCLA, and Los Angeles, CA). Tumor sizes in mice were monitored weekly, and once before and after the completion of treatments.
Immunotherapy Treatment
Two days following tumor detection, mice were enrolled into one of four treatment groups. Control Rat IgG (BioXCell) and αPDL1 (clone 10 F.9G2, BioXCell) antibodies were given intra peritoneally (100 μg/mouse) every other day, αCTLA4 antibody (clone UC10–4 F10–11, BioXCell) was given i.p. (100 μg/mouse) every three days twice. For the long-term treatment, mice were treated for two weeks, and for the mass cytometry experiment, mice were treated for one week. For the macrophage-depletion experiments, anti-CSF1R antibody (#BE0213, clone AFS98, BioCXell) was administered (400 μg/mouse) thrice a week for two weeks (24,25).
Immunohistochemistry
Tissues were fixed in 10% paraformaldehyde and embedded in paraffin for sectioning. Sections were deparaffinized by incubation in xylene and rehydrated by sequential incubation in 100%, 95%, 80%, 60% ethanol. The sections were incubated with primary antibody overnight at 4C (Supp Table 1) and subsequently incubated with biotinylated anti-mouse (1:300, Vector Lab) or biotinylated anti-rabbit (1:300, Vector Lab) for 30 minutes at room temperature. Sections were incubated for 30 minutes at room temperature in an ABC reagent (1:300, Vectastain ABC kit, Vector Lab). Sections were developed using 3,3’- Diaminobenzidine (DAB) for 30– 60 seconds, counterstained with hematoxylin, and mounted with Permount. The stained sections were scanned on Digital Pathology Slide Scanner (Philips) and quantified on ImageJ and Qupath software (NIH).
Mass-cytometry analysis
Infiltration of immune cells in the tumor was assessed using mass-cytometry (CyTOF). After euthanization, the mice were perfused by injecting PBS into the left ventricle. The dissected tumor was cut into small pieces and processed into a single cell suspension using the Tumor Dissociation Kit (MACS Miltenyi) according to the manufacturer’s instructions. The cell suspension was filtered through a 70 μm cell strainer and treated with the ACK lysis buffer (Thermo Fisher). Approximately 5 *106 cells were stained for CyTOF analysis. In brief, the cells were incubated for 15 min at RT with 1 μM Intercalator-Rh (Fluidigm) diluted in Maxpar Cell Staining Buffer (Fluidigm) for dead cell discrimination. Surface Fc receptors were blocked prior to staining by adding 2.5μg Rat anti-mouse CD16/32 Fc block (BD Pharmingen). Without washing out the blocking antibody, the cells were stained with metal-conjugated antibodies according to the list in Supp Table 2 in a total volume of 100 μl per sample and incubated for 45 min at RT. Antibodies were washed out using the Maxpar Cell Staining Buffer and the cells were resuspended in 125 nM Intercalator-Ir (Fluidigm) diluted in Maxpar Fix and Perm Buffer (Fluidigm) and incubated overnight at 4°C. The intercalator was washed out using the Maxpar Cell Staining Buffer following a washing step in Maxpar water (Fluidigm). The cells were kept as a pellet on ice until analysis. Immediately before analysis, the cells were resuspended to a concentration of approx. 0.5 *106 cells/ml in Maxpar water containing 10% (v/v) EQ Four Element Calibration Beads (Fluidigm). The samples were analyzed on the DVS CyTOF 2 instrument (Fluidigm). Data analysis was performed using FlowJo v10 and Cytosplore (26). The cells were first gated on “Live CD45+ single cells” in FlowJo and then tSNE analysis was performed in Cytosplore. The different immune cell populations were defined based on the expression of the immune markers. Some antibodies were labeled with metal-conjugates in-house using the Maxpar X8 Multimetal Labeling Kit (Fluidigm) according to the manufacturer’s instructions. For each labeling, 100 μg pure antibody was used, and the metal-conjugated antibody was subsequently resuspended in 120 μl of antibody stabilization buffer (Candor Bioscience, Germany). The metal-conjugated antibodies were titrated prior to use for staining of cells.
Data Availability Statement
The RNA sequencing data for this project is publicly available in the GEO database at GSE215010 and GSE215006.
RESULTS
The MYC Oncogene Drives Human Tumorigenesis Associated with Immune Evasion
We evaluated the role of MYC in the immune status of human cancers using the cancer genome atlas (TCGA) pan-cancer studies (n= 11,069 samples, n=33 cancers, Fig 1A). First, MYC was commonly activated by genomic amplification (n=889, 8%), especially in solid tumors like HCC (11%) (Fig 1A; Supp Fig 1A)(27). MYC amplification was strongly associated with MYC gene overexpression (p<0.0001; Supp Fig 1B). Even tumors without MYC amplification showed overexpression of a gene signature associated with MYC amplification (17)(Fig 1A), which also correlated with MYC mRNA expression (p<0.0001, r=0.241). This highlights the ubiquitous nature of MYC activation in cancers. Both MYC amplification and MYC mRNA overexpression were associated with worse overall- and progression-free survival (Fig 1B). Further, MYC-amplified tumors had higher tumor grade (p= 7.0 X 10−8) and belonged to more advanced stages (p=2.7 × 10−7) (Supp Fig 1C–D). Thus, MYC activation by amplification and/or overexpression is a global driver of tumorigenesis in human cancers.
Figure 1. The MYC Oncogene Drives Human Tumorigenesis Associated with Immune Evasion.

A. TumorMap rendering of the Pan-Cancer-Atlas of 33 cancers; tumors are defined by different tissues of origin in the first box. Second box shows the prevalence of MYC genomic amplification. Epithelial tumors with high rates of MYC amplification are annotated. Third box shows the gene expression of a signature derived from MYC amplification. (LAML- Acute Myeloid Leukemia, ACC- Adrenocortical carcinoma, BLCA- Bladder Urothelial Carcinoma, LGG- Brain Lower Grade Glioma, BRCA- Breast invasive carcinoma, CESC- Cervical squamous cell carcinoma and endocervical adenocarcinoma, CHOL- Cholangiocarcinoma, LCML- Chronic Myelogenous Leukemia, COAD- Colon adenocarcinoma, ESCA- Esophageal carcinoma, GBM- Glioblastoma multiforme, HNSC- Head and Neck squamous cell carcinoma, KICH- Kidney Chromophobe, KIRC- Kidney renal clear cell carcinoma, KIRP- Kidney renal papillary cell carcinoma, LIHC- Liver hepatocellular carcinoma, LUAD- Lung adenocarcinoma, LUSC- Lung squamous cell carcinoma, DLBC- Lymphoid Neoplasm Diffuse Large B-cell Lymphoma, MESO- Mesothelioma, OV- Ovarian serous cystadenocarcinoma, PAAD- Pancreatic adenocarcinoma, PCPG- Pheochromocytoma and Paraganglioma, PRAD- Prostate adenocarcinoma, READ- Rectum adenocarcinoma, SARC- Sarcoma, SKCM- Skin Cutaneous Melanoma, STAD- Stomach adenocarcinoma, TGCT- Testicular Germ Cell Tumors, THYM- Thymoma, THCA- Thyroid carcinoma, UCS- Uterine Carcinosarcoma, UCEC- Uterine Corpus Endometrial Carcinoma, UVM- Uveal Melanoma).
B. Overall and progression-free survival in patients with MYC amplification and MYC overexpression (top and bottom quartiles).
C. Correlation of MYC expression with gene expression of immune checkpoints PDL1 and CTLA4 (top) and with gene signature of immune checkpoint blockade (ICB) (Jiang et al. 2018 (28); bottom).
D. Expression of gene signatures associated with T helper cells 1 (Th1), Th17 and Th2 compared between tumors with (1) and without (0) MYC amplification.
E. Expression of gene signatures associated with CD8 T cells, NK cells and monocytes compared between tumors with (1) and without (0) MYC amplification.
F. Immune deconvolution analysis showing comparison of abundance of different immune subsets between tumors with (1) and without (0) MYC amplification.
G. Immunohistochemistry showing CD8 T-cell infiltration in representative HCC tumors from the TCGA cohort with (1, n=20) and without MYC amplification (0, n=8). *p<0.05, ***p<0.0001.
Next, we found that MYC activation influences the immune state of human cancers. First, the expression of MYC strongly correlated with the expression of the immune checkpoints PDL1 and CTLA4 (p=9.4X10−64) (Fig 1C), with higher quartiles of MYC mRNA expression showing higher expression of both PDL1 and CTLA4 (Supp Fig 2A). Second, MYC overexpression was associated with a gene signature predictive of T cell dysfunction and resistance to immune checkpoint blockade (28) (9=3.7X10−91) (Fig 1C). Third, MYC-amplified tumors and MYC-overexpressing tumors had lower expression of anti-tumor Th1 and Th17 signatures and higher expression of pro-tumorigenic Th2 immune signature (p<0.001) (Fig 1D, Supp Fig 2B). Fourth, MYC-amplified tumors had lower infiltration with anti-tumor immune subsets like CD8+ T cells, NK cells, and monocytes (all p<0.001), with no major differences in other subsets, upon immune deconvolution analysis (Fig 1E–F). Finally, we examined if MYC overexpression in HCC was associated with immune evasion since MYC is frequently amplified and overexpressed in HCC (Supp Fig 3A–B) and HCC exhibits limited responsiveness to immune checkpoint blockade (13,29). Indeed, by IHC, we found that MYC amplification was associated with lower CD8+ T cell infiltration in HCC (Fig 1G). Thus, MYC overexpression is common in human cancer and is associated with a poor prognosis. Moreover, MYC-driven tumors are immune evasive and less likely to respond to immune checkpoint blockade.
MYC causes Reversible Suppression of Anti-Tumor Immune Response
To examine how MYC suppresses the immune response, we used a MYC-driven conditional transgenic mouse model of HCC (21). The transcriptional profile of our transgenic mouse model of MYC-HCC was similar to the transcriptome to multiple human HCC cohorts including ten human HCC datasets and the TCGA data (similarity z score 27.2, p value= 1X10−15) (Fig 2A). Specifically, there was an overlap between mouse MYC-HCC transcriptome and the human HCC immune signatures of inactivated 41BB-ve PD1+ T cells in human HCC (30) (similarity z-score 3.2, p value= 1X10−9) and exhausted PD1+ CD8 T cells (31) (similarity z-score 16.7, p= 1X10−21) (Fig 2B). Cross-species analysis by classification of human HCC in the TCGA cohort using a 56-gene orthologous signature derived from the mouse model of MYC-HCC (mMYCOn) found that 16% (n=58/373) of human HCC showed strong enrichment on the mMYCOn gene signature (Fig 2C). We confirmed the mMYCOn. signature did indeed identify MYC-driven human HCCs by its association with MYC molecular pathway activation (NES=2.0, p=0.001) and strong enrichment for MYC gene amplification (p<0.001) (Fig 2D). Human HCC with high expression of mMYCOn signature showed enrichment of a gene program expressed by cancer cells associated with T cell exclusion and resistance to anti-PD1 checkpoint therapy (32), and also showed strong upregulation of CTLA4 (Fig 2E). Thus, the mouse MYC-HCC and MYC-driven human HCC exhibit similar genetic and immune profiles associated with exhausted T cells, and resistance to immune checkpoint monotherapy.
Figure 2: MYC causes Reversible Transcriptional Repression of Antitumor Immune Responses.

A. Heat map shows similarity between MYC-driven murine HCC (MYC-On, n=3 vs. MYC-Off, n=3) and 11 human HCC transcriptome data sets including the TCGA cohort; the top 10 upregulated and downregulated genes with enrichment z-scores across the different data sets are shown. Bar plot at the bottom shows log of p-value of similarity between the individual cohorts and murine MYC-HCC.
B. Heatmap shows z-scores of enrichment of canonical pathways and biological functions in murine MYC-HCC transcriptome and the human HCC immune signatures of exhausted PD1+ CD8 T cells(31) and inactivated 41BB-ve PD1+ T cells in human HCC(30).
C. Gene signature derived from murine MYC HCC used to classify human HCC in the TCGA cohort to identify MYC-driven human HCCs (created using Biorender.com).
D. Human HCCs enriched in mMYCOn signature show MYC molecular pathway activation and strong association with MYC gene amplification.
E. Human HCCs with high expression of mMYCOn signature showed enrichment of a gene program of T cell exclusion and resistance to anti-PD1 checkpoint therapy (32), and upregulation of CTLA4.
F. PCA plot comparing the transcriptional profile of a transgenic model of HCC overexpressing wild-type MYC (MYCOn, n=16), a mutant MYC defective in Miz1 binding (V394D) (MYCVd, n=11) and tumors upon MYC inactivation for a short time point of 16 hours (MYCOff, n=8)(36) to identify MYC/Miz-1 repressed genes.
G. Heatmap shows genes which are reversibly repressed by MYC in the MHC protein complex, complement system and cytokines.
H. Immunohistochemistry shows the abundance of macrophages (F4/80+) by day 1 and CD4 T-cell recruitment by day 4 of MYC inactivation in HCC.
I. Immune deconvolution analysis showing comparison of abundance of different immune subsets in murine MYC HCC upon MYC inactivation.
**p<0.01, ***p<0.0001.
Next, we examined the immune-related transcriptional changes induced upon MYC inactivation in the mouse model of MYC-driven HCC (MYC On (n=3) vs. Off (4 days, n=3) (DE genes 756 upregulated, 1783 downregulated; pAdj<0.05; Supp Table 3). Inactivating MYC in the MYC-HCC resulted in the upregulation of anti-tumor immune pathways including the Il6, Tnfa and Ifng pathways, and activation of inflammatory biological processes like phagosome formation, production of ROS by macrophages, and phagocyte migration (Supp Fig 4). This led us to hypothesize that MYC transcriptionally represses macrophage-mediated anti-tumor immunity.
To test this hypothesis, we further analyzed transcriptional data from a transgenic model of HCC expressing a mutant MYC defective in Miz1 binding (V394D)(33), since interaction with Miz1 is a well characterized mechanism of transcriptional repression by MYC (34,35). We compared gene expression between HCC with wild-type MYC overexpression (MYCOn, n=16), HCC with MYC-V394D mutant (MYCVd, n=11) and tumors upon MYC inactivation for a short time point of 16 hours (MYCOff, n=8)(36) (Fig 2F). Genes that were overexpressed in tumors expressing MYC Vd mutant vs. MYC-WT HCC, and also upregulated upon short-term MYC inactivation were identified as the ones specifically transcriptionally repressed by the MYC/Miz1 interaction (n=1028, pAdj<0.05, Fig 2F, Supp Table 4). Pathway analysis of the MYC/Miz1 repressed gene program revealed enrichment of several critical immune-related pathways particularly involving antigen presentation, MHC I protein complex and T cell mediated cytotoxicity (Supp Fig 5A–B). Multiple genes related to antigen presentation, particularly belonging to the MHCI protein complex pathway like Cd74, B2m and MHC-I genes were found to be strongly transcriptionally repressed by the MYC-Miz1 complex (Fig 2G). Even short-term MYC inactivation was noted to activate the innate immune response in a Miz1 dependent manner with upregulation of multiple components of the complement pathway (C2, C3, C4a, C4b, C8, C9) and cytokines which recruit innate immune cells Il1b, Ccl15, Cxcl10 and Cxcl16 (Fig 2G). Thus, we show that MYC interacts with Miz1 to transcriptionally repress genes and programs specifically essential to invoke anti-tumor innate immunity.
To confirm activation of anti-tumor immune responses upon MYC inactivation, we performed temporal analysis of immune changes in the tumor microenvironment. We found that MYC inactivation was associated with early macrophage recruitment by day one, especially in the peritumoral regions (Fig 2H). This was followed by subsequent infiltration of CD4T cells by day four (Fig 2H). These findings are consistent with our earlier work showing that CD4+ T-cells play a causal role in tumor regression upon oncogene inactivation (37). Additionally, immune deconvolution analysis of transcriptional changes noted at day four of MYC inactivation demonstrated recruitment of pro-inflammatory M1-macrophages and CD4+ T cells, along with a decrease in the proportion of pro-tumorigenic immune cells like regulatory T (Treg) cells (Fig 2I). Thus, the mouse MYC-HCC and human HCC exhibit activation of gene programs of immune evasion. Further, in the mouse model, MYC inactivation reverses immune evasion by activating innate and adaptive anti-tumor immunity.
Combined Anti- PDL1 and Anti-CTLA4 overcome MYC induced immune evasion
We evaluated if inhibiting the immune checkpoints PDL1 and/or CTLA4 could overcome MYC-induced immune evasion (Fig 3A). In mouse MYC-HCC, only mice treated with both anti-PDL1 and anti-CTLA4, but not either alone, showed significantly delayed progression on three-dimensional tumor volume assessment by MRI compared to mice treated with control antibody (mean tumor volume [858.31 μm3, SEM = 53.60] vs. [1897.67 μm3, SEM = 258.62]; p = 0.004)) (Fig 3B–C). Mice treated with combined PDL1 and CTLA4 blockade (n=5) had smaller and fewer liver tumors than mice treated with control antibody (n=5) ([858.31 μm3, SEM = 53.60] vs. [1897.67 μm3, SEM = 258.62]; p = 0.004) (Fig 3D–E). However, there were no differences in final tumor volumes of mice treated with anti-PDL1 (n=5)(1235.81 μm3, SEM = 288.87) or anti-CTLA4 alone (n=5) (1922.64 μm3, SEM = 509.45) compared to IgG treated mice (p = 0.126; p = 0.966 respectively) (Fig 3D–E). The combination therapy led to a decrease in the proliferative index of the tumor as measured by phospho-histone 3 staining without affecting MYC expression levels in the tumor cells (Supp Fig 6), and also led to increased cell death as shown by cleaved caspase 3 staining (Supp Fig 7). Thus, only combined, but not individual, inhibition of PDL1 and CTLA4, impeded tumor progression in MYC-HCC.
Figure 3. Combined Anti- PDL1 and Anti-CTLA4 delays tumor progression in MYC-HCC.

A. Experimental scheme of MYC-HCC treatment with IgG control (n=5), PDL1 antibody (n=5), CTLA4 antibody (n=5), or their combination (n=5) (created using Biorender.com).
B. Weekly MRI showing tumor progression in representative MYC-HCC mice in the 4 treatment groups (n=5 each group).
C. Quantification of volumetric tumor measurement using MRI of MYC-HCC mice in the 4 treatment groups (n=5 each group).
D. End-of-treatment gross appearance, histology of representative MYC-HCC mice in the 4 treatment groups (n=5 each group).
E. Quantification of liver tumor burden at end-of-treatment of MYC-HCC mice in the 4 treatment groups (n=5 each group).
**p<0.01, ***p<0.0001
Treatment with combination therapy did not result in weight loss or hepatotoxicity in mice (Fig 4A). End-of-treatment analysis revealed slight lymphopenia with individual or combined treatments. Treatment with anti-CTLA4 alone and in combination with anti-PDL1 resulted in lower peripheral blood monocyte count (Fig 4B). Histological evaluation of liver, colon, and lungs by a blinded pathologist showed no areas of severe autoimmune organ damage with the combination therapy even though immunohistochemical analysis revealed higher levels of CD8+ T cell infiltration with combination therapy (Fig 4C). Thus, the combination of anti-PDL1 and anti-CTLA4 shows synergistic efficacy in overcoming MYC-induced immune evasion without severe toxicity.
Figure 4. Safety profile of combined Anti- PDL1 and Anti-CTLA in MYC-HCC.
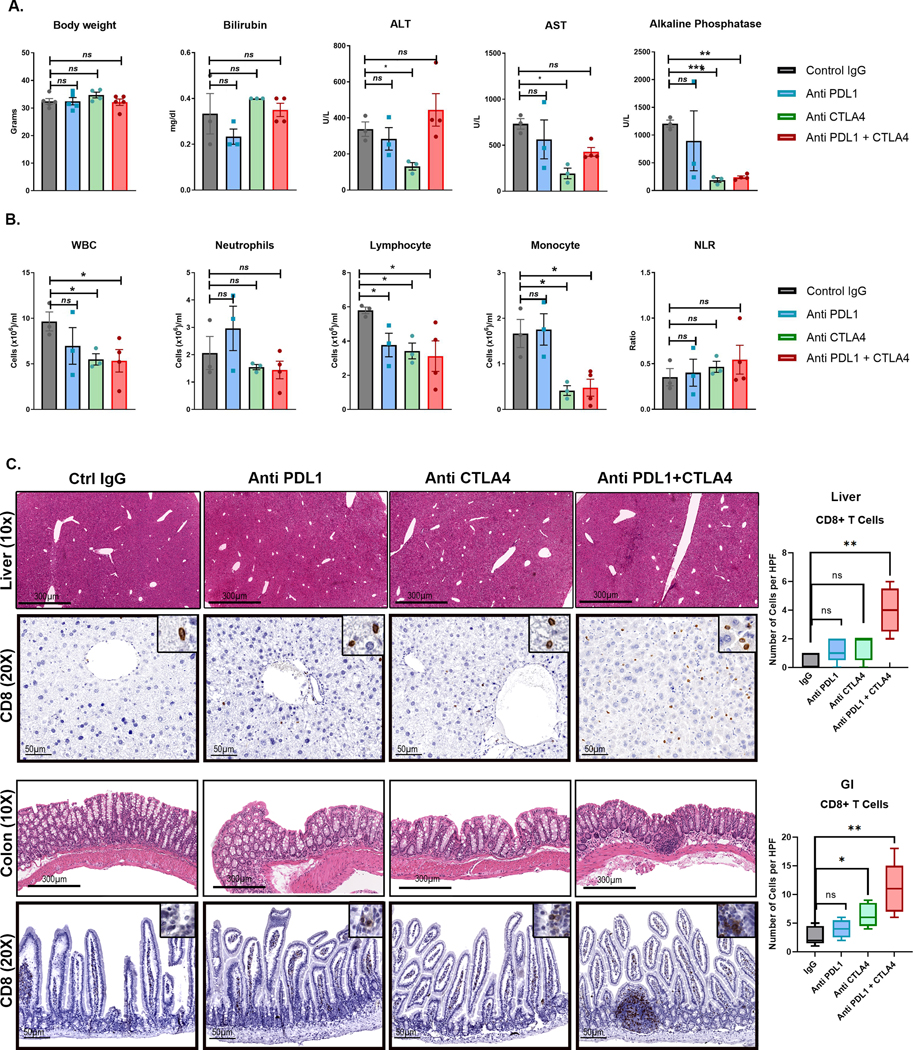
A. Body weight and liver tests at end-of-treatment of MYC-HCC bearing mice treated with either IgG control (n=5) or PDL1 antibody (n=5) or CTLA4 antibody (n=5) or their combination (n=5).
B. Peripheral white cell counts at end-of-treatment of MYC-HCC bearing mice treated with either IgG control (n=5) or PDL1 antibody (n=5) or CTLA4 antibody (n=5) or their combination (n=5).
C. Histology and CD8 T cell infiltration in normal surrounding liver and colon tissue of mice treated with the 4 treatment groups. *p<0.05, **p<0.01.
Dual targeting of PDL1 and CTLA4 Restores Macrophage-Mediated Anti-tumor immunity
Next, we evaluated how dual targeting of anti-PDL1 and CTLA4 remodeled the immune microenvironment of the tumors. IHC analysis showed that the combination of PDL1 and CTLA4 blockade elicited recruitment of a higher number of CD4+ T cells (p=0.005) and CD8+ T cells (p<0.0001) than individual monotherapies (Fig 5A). Interestingly, we observed significantly increased macrophage infiltration in tumors treated with the combination of PDL1 and CTLA4 compared to IgG treated mice (p=0.005) but not in mice treated with anti- PDL1 (p=0.31) or anti-CTLA4 (p=0.06) as monotherapy (Fig 5A). Despite an overall increase in macrophage infiltration, the proportion of immunosuppressive PDL1+ macrophages were lower in mice treated with combined anti-PDL1 and anti CTLA4 compared to mice treated with IgG control antibody (p=0.001) (Supp Fig 8). Thus, dual targeting of PDL1 and CTLA4 elicits a more robust T cell immune response and macrophage infiltration than either monotherapy.
Figure 5. Dual targeting of PDL1 and CTLA4 Restores Macrophage-Mediated Anti-tumor immunity.
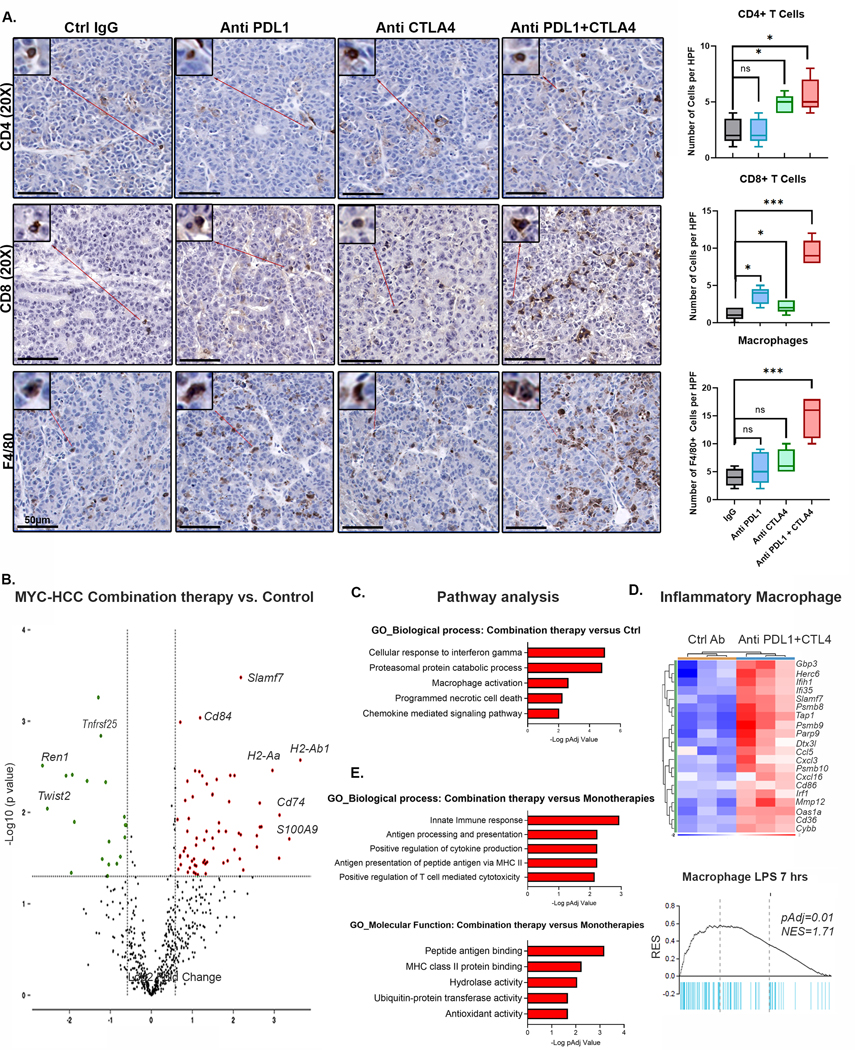
A. Immunohistochemistry for CD4+ T cells, CD8+ T cells and F4/80+ macrophages in MYC-HCC bearing mice treated with IgG control (n=5), PDL1 antibody (n=5), CTLA4 antibody (n=5), or their combination (n=5). Boxplots show quantification of cell counts.
B. Violin plot showing differentially expressed genes between MYC-HCC treated with combination therapy (n=3) versus control IgG (n=3). Macrophage-related genes are upregulated with treatment with anti PDL1+CTLA4.
C. Pathway analysis of differentially expressed genes between MYC-HCC treated with combination therapy (n=3) versus control IgG (n=3).
D. Enrichment of inflammatory signature of macrophages stimulated with lipopolysaccharide (LPS) in tumors treated with combination therapy (n=3) versus control IgG (n=3).
E. Pathways analysis of meta-analysis comparing the transcriptional changes induced by the combination therapy (n=3) to those induced by anti PDL1 (n=3) or CTLA4 monotherapies (n=3).
To identify mechanisms by which combination therapy is effective in MYC-HCC we evaluated the transcriptomic differences between the tumors treated with the four different therapies using a targeted immune-oncology RNAseq panel. We identified 87 genes which were differentially expressed (DEG) between tumors treated with the anti PDL1+CTLA4 combination therapy versus control antibody (pAdj<0.05) (Fig 5B) with upregulation of multiple genes in the innate immune response including Cd84, Cd74, S100A9 and upregulation of MHC-II genes (Fig 5B). Pathway analysis of the DEG revealed that apart from activation of the interferon gamma pathway, as expected (Fig 5C), a top pathway upregulated in tumors treated with anti PDL1+CTLA4 was macrophage activation, with enrichment of a signature associated with inflammatory macrophages stimulated with lipopolysaccharide (LPS)(38)(Fig 5D).
Next, we performed a meta-analysis comparing the transcriptional changes induced by the combination therapy to those induced by anti PDL1 or CTLA4 monotherapies (Supp Fig 9A). The pathways that were preferentially upregulated in the MYC-HCC treated with combination therapy included innate immune response, antigen processing and presentation and MHC class II pathway (Fig 5E, Supp Fig 9B). Crucial genes in the antigen presentation pathway were preferentially upregulated in tumors treated with combination therapy including B2m, H2-Aa, H2-Ab1, Cd74 and Tap1. Moreover, multiple macrophage-related genes like Mmp13, Atf3 and monocyte chemo attractants like Ccl5 and Ccl8 were differentially upregulated in tumors treated with combination therapy (Supp Fig 9C). Thus, the combination therapy leads to differential activation of inflammatory macrophages and upregulation of the antigen presentation pathway which is transcriptionally repressed by MYC.
Combined PD-L1 and CTLA-4 Inhibition Elicits Early Repolarization of Myeloid Cells
To determine the sequence of macrophage and T cell activation with immune checkpoint inhibition, we evaluated immune responses at an earlier time point of 1 week after treatment with PDL1 and/or CTLA4 inhibitors. We used high-dimensional single-cell profiling of the tumor immune infiltrates with mass cytometry (CyTOF) (Fig 6A). After dimensionality reduction of the high-order parametric data, twenty-seven unique immune subsets were identified (Fig 6B and Supp Fig 10). The most significant changes in the mice treated with combined anti-PDL1 and anti-CTLA4, compared to the individual monotherapies, were found in the myeloid compartment. The frequency of pro-inflammatory macrophages (Ly6CHigh/CCR2High) was significantly increased, and immunosuppressive macrophages (PDL1+/Ly6CLow/CCR2Low) was significantly decreased in the tumors treated with both anti-PDL1 and anti-CTLA4, and not either alone, compared to mice treated with IgG control (Fig 6C–6D). We further confirmed this on immunofluorescent staining which showed higher M1-like CCR2+ macrophages at the outer edges of the tumors (Supp Fig 11) with depletion of intratumoral M2-like CD206+ macrophages (Supp Fig 12)
Figure 6. Mass cytometry reveals unique early immune response to dual targeting of PDL1 and CTLA4.

A. Experimental scheme of MYC-HCC treatment with IgG control (n=3), PDL1 antibody (n=3), CTLA4 antibody (n=2), or their combination (n=4) for 1 week, followed by mass cytometry analysis (created using Biorender.com).
B. tSNE plot grouped by major immune subsets identified in the tumor immune microenvironment.
C. tSNE plot showing the visual representation of myeloid marker expression between MYC-HCC treated with IgG control (n=3), PDL1 antibody (n=3), CTLA4 antibody (n=2), or their combination (n=4)
D. Boxplots shows quantification of CCR2+/Ly6High M1-Like Macrophages, Ly6C expression in the macrophages, abundance of PDL1+/Ly6CLow/CCR2Low M2-Like Macrophages and myeloid derived suppressor cells (MDSCs) in MYC-HCC treated with IgG control (n=3), PDL1 antibody (n=3), CTLA4 antibody (n=2), or their combination (n=4) respectively.
E. Boxplots show quantification of CD40 expression in different myeloid subsets in MYC-HCC treated with IgG control (n=3), PDL1 antibody (n=3), CTLA4 antibody (n=2), or their combination (n=4) respectively.
F. Boxplots shows quantification of MHCII expression in CD39+ and PDL1+ M2-Like Macrophages in MYC-HCC treated with IgG control (n=3), PDL1 antibody (n=3), CTLA4 antibody (n=2), or their combination (n=4) respectively.
CD40 expression on myeloid cells is known to activate the tumoricidal activities of macrophages (39,40). The combination therapy, but not either anti PDL1 or anti CTLA4 alone, significantly increased CD40 expression on multiple myeloid cells including inflammatory macrophages (Ly6CHigh/CCR2High), conventional dendritic cells (CD11c+/CD11b-/CD103+), myeloid dendritic cells (CD11c+/CD11b+/CD103-) and MDSCs (Fig 6C–6E). On the other hand, immunosuppressive macrophage subsets like the PDL1+ TAMs and CD39+ TAMs showed significantly increased expression of MHC II with combination anti-PDL1 and anti-CTLA4, but not with the individual monotherapies (Fig 6C–6F). We did find a trend towards increased effector CD8+ T cells (LyCHigh/PD1Low) with combination therapy but no other significant differences in the proportion of T cell subpopulations at this early stage of treatment (Supp Fig 13A–B). There were no significant changes in other innate immune cells like NK cells, eosinophils, or neutrophils (Supp Fig 13A–B). Thus, we show that combined blockade of both PDL1 and CTLA4, and not either alone, elicits an early immune response with the expansion of the pro-inflammatory myeloid compartments and repolarization to an anti-tumor profile with upregulation of CD40 and MHC-II.
Macrophages are Essential for the anti-tumor efficacy of Immune Checkpoint Therapy
To evaluate the causal role of macrophages, we depleted macrophages prior to treatment with combined PDL1 and CTLA4 blockade (Fig 7A). We used the anti-CSF1R antibodies to deplete macrophages (24,25), after first confirming that the CSF1R antibody effectively depleted macrophages, without impacting tumor progression in MYC-driven HCC (Supp Fig 14A–C).
Figure 7. Macrophages are Essential for the anti-tumor efficacy of Combined Immune Checkpoint Therapy in MYC-HCC.

A. Experimental scheme of macrophage depletion in MYC-HCC followed by treatment with either IgG control (n=4) or dual PDL1 and CTLA4 antibodies (n=3) (created using Biorender.com).
B. Quantification of tumor burden at end-of-treatment with either IgG control (n=4) or dual PDL1 and CTLA4 antibodies (n=3) in macrophage-depleted MYC-HCC mice.
C. End-of-treatment gross appearance, histology and immunofluorescence for CD8 T cells in representative macrophage-depleted MYC-HCC-bearing mice treated with IgG control (n=4) or dual PDL1 and CTLA4 antibodies (n=3).
D. Quantitation of CD8T cell infiltration in macrophage-depleted MYC-HCC-bearing mice treated with IgG control (n=4) or dual PDL1 and CTLA4 antibodies (n=3).
E. Figurative representation of the mechanism of efficacy of combined PDL1 and CTLA4 therapy in MYC-HCC. MYC-driven cancers are immune evasive with MHCI repression and PDL1 overexpression on cancer cells and macrophages which in turn lead to T cell exhaustion. Treatment with CTLA4 and PDL1 inhibitors leads to repolarization of macrophages to the M1-like phenotype with increased expression of CD40 and MHCII, which leads to enhanced antigen presentation and robust T cell activation resulting in delayed tumor progression (created using Biorender.com).
There was no difference in liver tumor burden between macrophage-depleted mice treated with combination PDL1 and CTLA4 inhibition (n=3) or control antibody (n=4) (liver/body ratio 0.25 vs. 0.17, p=0.23) (Fig 7B). Further, we confirmed that the combined blockade of PDL1 and CTLA4 was not able to elicit antitumor CD8 T cell response in macrophage-depleted mice (Fig 7C–D). Together, these data show that macrophage depletion blocks the anti-tumor efficacy of combined PDL1 and CTLA4 inhibition in MYC-HCC. Thus, our data demonstrate that combined PDL1 and CTLA4 blockade requires the activation of macrophages to overcome MYC-induced immune evasion (Fig 7E).
DISCUSSION
We show that MYC is involved in immune evasion during HCC tumorigenesis which can be overcome through combined blockade of immune checkpoints PDL1 and CTLA4. A pan-cancer analysis of 33 human tumors reveals that MYC is not only commonly amplified and overexpressed, but it is also associated with increased immune checkpoint expression, immunotherapy resistance, a Th2-like immune profile, and reduced anti-tumor CD8 T-cell infiltration. We demonstrate that MYC transcriptionally represses MHC class I genes thus impeding antigen presentation in a murine model of MYC-driven HCC. Notably, MYC immune evasion can be abrogated through combined immune checkpoint blockade with anti-PDL1 and CTLA4, which was associated with the causally required activation of macrophages to a pro-inflammatory phenotype with enhanced antigen presentation. The restoration of immune responsiveness was associated with increased expression of CD40 and MHCII on antigen-presenting cells (APCs) and robust anti-tumor T cell responses (Fig 7E). We conclude that MYC is a key driver of immune evasiveness in human cancer, blocking both innate and adaptive antitumor immune responses. Further, we identify MYC-driven HCC as a specific subset of cancers that are unlikely to respond to monotherapy with immune checkpoint inhibitors and need combination immunotherapy to achieve tumor remission.
The MYC oncogene has been shown to play a pivotal role in remodeling the tumor microenvironment (4,7,37,41,42). In transgenic mouse models of MYC-driven pancreatic and lung cancer, inactivation of MYC led to rapid efflux of M2-like CD206+ macrophages with recruitment of T cells (41,42). However, in a different mouse model of pancreatic adenocarcinoma MYC inactivation did not influence macrophage infiltration, but instead reprogrammed macrophages to secrete Cxcl13, leading to recruitment of B and NK cells (43). We show here that MYC reversibly transcriptionally repressed several crucial genes involved in antigen presentation including MHC class I genes thus handicapping both innate and adaptive immunity. MYC inactivation de-repressed MHC I expression and also led to the secretion of monocyte chemoattractants like IL1β (44,45) and CXCL10 (46) associated with rapid recruitment of M1-like macrophages to the peritumoral areas and subsequent recruitment of CD4 T cells. In this context, we show that combined inhibition of PDL1 and CTLA4 in MYC-HCC led to loss of M2-like CD206+ macrophages, recruitment of CCR2+ inflammatory macrophages and increased expression of CD40 and MHC II on multiple pro-inflammatory myeloid cells, thus relieving the MYC-induced block on antigen presentation and resulting in enhanced T cell cytotoxicity.
Inhibition of immune checkpoints like PDL1 or CTLA4 has shown promising efficacy in many cancers (47,48). However, less than a third of patients respond to ICI monotherapy, making it critical to understand mechanisms of resistance to immunotherapy. While the role of MYC in driving immune evasion has been demonstrated by previous studies, (4,5,41,49,50), its functional relevance in modulating response to immune checkpoint inhibition is not understood. Our study identifies MYC oncogene amplification or overexpression as a major driver of tumor-intrinsic resistance to immune checkpoint inhibition. We demonstrate that MYC-driven HCC exhibit primary resistance to monotherapy with PDL1 or CTLA4 blockade, implying that additional aspects of the immune response are modulated by MYC. These results imply that MYC-amplification or MYC-overexpression can potentially serve as a biomarker to prioritize patients to be considered for combination immunotherapies, rather than individual checkpoint inhibitors.
Our results identify a unique mechanistic basis for the synergistic efficacy of combined PDL1 and CTLA4 blockade, beyond just additive effects of their individual mechanisms. We suggest a model whereby the combination of anti-PDL1 and CTLA4 restores immune responsiveness by the activation of pro-inflammatory CCR2+ macrophages, which we show are causally required, as macrophage depletion abrogated the anti-tumor efficacy of PDL1 and CTLA4 blockade (Figure 7E). These macrophages appear to be involved in the subsequent recruitment of adaptive immune cells including CD4+ T cells and cytotoxic CD8+ T cells. We suggest that the upregulation of at least two ligands on APCs, CD40, and MHCII, is involved in the mechanism of activating adaptive immunity. CD40 expression is known to license APCs to activate cytotoxic T cells and invigorate the tumoricidal activity of macrophages (51). Increased MHCII expression enables efficient antigen presentation by APCs to CD4+ T cells (52). The upregulation of both CD40 and MHCII on APCs, seen uniquely with combined PDL1 and CTLA4 blockade and not with the monotherapies, could be a key mechanism that contributes to the anti-tumor efficacy of the combination.
Finally, no existing therapies that directly target the MYC oncogene exist. Several strategies to indirectly target MYC are currently under evaluation (53,54), and our data suggest that such MYC inhibitors will potentially synergize with immunotherapy. In the existing treatment paradigm, our results show that combined PDL1 and CTLA4 inhibition may be effective in restoring macrophage-mediated antitumor immune response in MYC-driven human HCC.
Supplementary Material
Statement of Significance.
Macrophage-mediated immune evasion is a therapeutic vulnerability of MYC-driven cancers, which has implications for prioritizing MYC-driven hepatocellular carcinoma for combination immunotherapy.
Acknowledgements
RD- National Institutes of Health (NIH) grant CA222676 from the National Cancer Institute
(NCI), American College of Gastroenterology Junior Faculty Career Development Grant.
DF- National Institutes of Health (NIH) grant CA208735 and CA253180 from the National
Cancer Institute (NCI).
Pauline Chu- Helped with mouse histology services.
Rachana Baskar for help with immunohistochemistry.
Human HCC TCGA samples were obtained from the Mayo Clinic Hepatobiliary SPORE support (Award Number P50 CA210964) from the National Cancer Institute.
Biorender was used to create graphical representations.
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Schaub FX, Dhankani V, Berger AC, Trivedi M, Richardson AB, Shaw R, et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst [Internet]. 2018;6:282–300.e2. Available from: 10.1016/j.cels.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dhanasekaran R, Deutzmann A, Mahauad-Fernandez WD, Hansen AS, Gouw AM, Felsher DW. The MYC oncogene - the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol [Internet]. 2021; Available from: 10.1038/s41571-021-00549-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4 T Cells Contribute to the Remodeling of the Microenvironment Required for Sustained Tumor Regression upon Oncogene Inactivation [Internet]. Cancer Cell. 2010. page 696. Available from: 10.1016/j.ccr.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science [Internet]. 2016;352:227–31. Available from: 10.1126/science.aac9935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med [Internet]. 2019;25:301–11. Available from: 10.1038/s41591-018-0321-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MYC-Mediated Translation of PD-L1 Promotes Liver Cancer Immune Escape. Cancer Discov [Internet]. 2019;9:317. Available from: 10.1158/2159-8290.CD-RW2019-011 [DOI]
- 7.Swaminathan S, Hansen AS, Heftdal LD, Dhanasekaran R, Deutzmann A, Fernandez WDM, et al. MYC functions as a switch for natural killer cell-mediated immune surveillance of lymphoid malignancies. Nat Commun [Internet]. 2020;11:2860. Available from: 10.1038/s41467-020-16447-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xin B, Yamamoto M, Fujii K, Ooshio T, Chen X, Okada Y, et al. Critical role of Myc activation in mouse hepatocarcinogenesis induced by the activation of AKT and RAS pathways. Oncogene [Internet]. 2017;36:5087–97. Available from: 10.1038/onc.2017.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, Jin R, Zhang X, Lv F, Liu L, Liu D, et al. Oncogenic activation of glypican-3 by c-Myc in human hepatocellular carcinoma. Hepatology [Internet]. 2012;56:1380–90. Available from: 10.1002/hep.25891 [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Wang P, Xu M, Song X, Wu H, Evert M, et al. Distinct functions of transforming growth factor-β signaling in c-MYC driven hepatocellular carcinoma initiation and progression. Cell Death Dis [Internet]. 2021;12:200. Available from: 10.1038/s41419-021-03488-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wangensteen KJ, Wang YJ, Dou Z, Wang AW, Mosleh-Shirazi E, Horlbeck MA, et al. Combinatorial genetics in liver repopulation and carcinogenesis with a in vivo CRISPR activation platform. Hepatology [Internet]. 2018;68:663–76. Available from: 10.1002/hep.29626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhanasekaran R, Baylot V, Kim M, Kuruvilla S, Bellovin DI, Adeniji N, et al. and cooperate to drive metastasis by eliciting crosstalk between cancer and innate immunity. Elife [Internet]. 2020;9. Available from: 10.7554/eLife.50731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yau T, Park JW, Finn RS, Cheng A-L, Mathurin P, Edeline J, et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC) [Internet]. Annals of Oncology. 2019. page v874–5. Available from: 10.1093/annonc/mdz394.029 [DOI] [Google Scholar]
- 14.Finn RS, Ryoo B-Y, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J Clin Oncol [Internet]. 2020;38:193–202. Available from: 10.1200/JCO.19.01307 [DOI] [PubMed] [Google Scholar]
- 15.Abou-Alfa GK, Lau G, Kudo M, Chan SL, Kelley RK, Furuse J, et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evidence [Internet]. Massachusetts Medical Society; 2022;1. Available from: 10.1056/EVIDoa2100070 [DOI] [PubMed] [Google Scholar]
- 16.UCSC TumorMap [Internet]. [cited 2022 Oct 12]. Available from: https://tumormap.ucsc.edu/
- 17.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell [Internet]. 2018;173:291–304.e6. Available from: 10.1016/j.cell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, et al. The Immune Landscape of Cancer. Immunity [Internet]. 2019;51:411–2. Available from: 10.1016/j.immuni.2019.08.004 [DOI] [PubMed] [Google Scholar]
- 19.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods [Internet]. 2015;12:453–7. Available from: 10.1038/nmeth.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perkins JR, Dawes JM, McMahon SB, Bennett DLH, Orengo C, Kohl M. ReadqPCR and NormqPCR: R packages for the reading, quality checking and normalisation of RT-qPCR quantification cycle (Cq) data. BMC Genomics [Internet]. 2012;13:296. Available from: 10.1186/1471-2164-13-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature [Internet]. 2004;431:1112–7. Available from: 10.1038/nature03043 [DOI] [PubMed] [Google Scholar]
- 22.Lai I, Swaminathan S, Baylot V, Mosley A, Dhanasekaran R, Gabay M, et al. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J Immunother Cancer [Internet]. 2018;6:125. Available from: 10.1186/s40425-018-0431-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dhanasekaran R, Park J, Yevtodiyenko A, Bellovin DI, Adam SJ, Kd AR, et al. MYC ASO Impedes Tumorigenesis and Elicits Oncogene Addiction in Autochthonous Transgenic Mouse Models of HCC and RCC. Mol Ther Nucleic Acids [Internet]. 2020;21:850–9. Available from: 10.1016/j.omtn.2020.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDonald KPA, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation [Internet]. Blood. 2010. page 3955–63. Available from: 10.1182/blood-2010-02-266296 [DOI] [PubMed] [Google Scholar]
- 25.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature [Internet]. 2017;545:495–9. Available from: 10.1038/nature22396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Höllt T, Pezzotti N, van Unen V, Koning F, Eisemann E, Lelieveldt B, et al. Cytosplore: Interactive Immune Cell Phenotyping for Large Single-Cell Datasets [Internet]. Computer Graphics Forum. 2016. page 171–80. Available from: 10.1111/cgf.12893 [DOI] [Google Scholar]
- 27.Newton Y, Novak AM, Swatloski T, McColl DC, Chopra S, Graim K, et al. TumorMap: Exploring the Molecular Similarities of Cancer Samples in an Interactive Portal [Internet]. Cancer Research. 2017. page e111–4. Available from: 10.1158/0008-5472.can-17-0580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med [Internet]. 2018;24:1550–8. Available from: 10.1038/s41591-018-0136-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finn RS, Ryoo B-Y, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J Clin Oncol [Internet]. 2020;38:193–202. Available from: 10.1200/JCO.19.01307 [DOI] [PubMed] [Google Scholar]
- 30.Kim H-D, Park S, Jeong S, Lee YJ, Lee H, Kim CG, et al. 4–1BB Delineates Distinct Activation Status of Exhausted Tumor-Infiltrating CD8 T Cells in Hepatocellular Carcinoma. Hepatology [Internet]. 2020;71:955–71. Available from: 10.1002/hep.30881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim H-D, Song G-W, Park S, Jung MK, Kim MH, Kang HJ, et al. Association Between Expression Level of PD1 by Tumor-Infiltrating CD8 T Cells and Features of Hepatocellular Carcinoma. Gastroenterology [Internet]. 2018;155:1936–50.e17. Available from: 10.1053/j.gastro.2018.08.030 [DOI] [PubMed] [Google Scholar]
- 32.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell [Internet]. 2018;175:984–97.e24. Available from: 10.1016/j.cell.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herold S, Wanzel M, Beuger V, Frohme C, Beul D, Hillukkala T, et al. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell [Internet]. 2002;10:509–21. Available from: 10.1016/s1097-2765(02)00633-0 [DOI] [PubMed] [Google Scholar]
- 34.Peukert K. An alternative pathway for gene regulation by Myc [Internet]. The EMBO Journal. 1997. page 5672–86. Available from: 10.1093/emboj/16.18.5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herkert B, Eilers M. Transcriptional repression: the dark side of myc. Genes Cancer [Internet]. 2010;1:580–6. Available from: 10.1177/1947601910379012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kress TR, Pellanda P, Pellegrinet L, Bianchi V, Nicoli P, Doni M, et al. Identification of MYC-Dependent Transcriptional Programs in Oncogene-Addicted Liver Tumors. Cancer Res [Internet]. 2016;76:3463–72. Available from: 10.1158/0008-5472.CAN-16-0316 [DOI] [PubMed] [Google Scholar]
- 37.Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation [Internet]. Cancer Cell. 2010. page 485–98. Available from: 10.1016/j.ccr.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bordano V, Kinsella GK, Cannito S, Dianzani C, Gigliotti CL, Stephens JC, et al. G protein-coupled receptor 21 in macrophages: An in vitro study. Eur J Pharmacol [Internet]. 2022;926:175018. Available from: 10.1016/j.ejphar.2022.175018 [DOI] [PubMed] [Google Scholar]
- 39.Vonderheide RH, Glennie MJ. Agonistic CD40 Antibodies and Cancer Therapy [Internet]. Clinical Cancer Research. 2013. page 1035–43. Available from: 10.1158/1078-0432.ccr-12-2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piechutta M, Berghoff AS. New emerging targets in cancer immunotherapy: the role of Cluster of Differentiation 40 (CD40/TNFR5). ESMO Open [Internet]. 2019;4:e000510. Available from: 10.1136/esmoopen-2019-000510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell [Internet]. 2017;171:1301–15.e14. Available from: 10.1016/j.cell.2017.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sodir NM, Kortlever RM, Barthet VJA, Campos T, Pellegrinet L, Kupczak S, et al. MYC Instructs and Maintains Pancreatic Adenocarcinoma Phenotype. Cancer Discov [Internet]. 2020;10:588–607. Available from: 10.1158/2159-8290.CD-19-0435 [DOI] [PubMed] [Google Scholar]
- 43.Muthalagu N, Monteverde T, Raffo-Iraolagoitia X, Wiesheu R, Whyte D, Hedley A, et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov [Internet]. 2020;10:872–87. Available from: 10.1158/2159-8290.CD-19-0620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, et al. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol [Internet]. 2011;187:4835–43. Available from: 10.4049/jimmunol.1102048 [DOI] [PubMed] [Google Scholar]
- 45.Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, et al. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A [Internet]. 2019;116:1361–9. Available from: 10.1073/pnas.1812266115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review). Oncol Lett [Internet]. 2011;2:583–9. Available from: 10.3892/ol.2011.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bagchi S, Yuan R, Engleman EG. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol [Internet]. 2021;16:223–49. Available from: 10.1146/annurev-pathol-042020-042741 [DOI] [PubMed] [Google Scholar]
- 48.Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol [Internet]. 2020;20:25–39. Available from: 10.1038/s41577-019-0218-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maeda T, Hiraki M, Jin C, Rajabi H, Tagde A, Alam M, et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res [Internet]. 2018;78:205–15. Available from: 10.1158/0008-5472.CAN-17-1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vartuli RL, Zhou H, Zhang L, Powers RK, Klarquist J, Rudra P, et al. Eya3 promotes breast tumor-associated immune suppression via threonine phosphatase-mediated PD-L1 upregulation. J Clin Invest [Internet]. 2018;128:2535–50. Available from: 10.1172/JCI96784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med [Internet]. 2020;71:47–58. Available from: 10.1146/annurev-med-062518-045435 [DOI] [PubMed] [Google Scholar]
- 52.Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol [Internet]. 2015;15:203–16. Available from: 10.1038/nri3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer [Internet]. 2008;8:976–90. Available from: 10.1038/nrc2231 [DOI] [PubMed] [Google Scholar]
- 54.Allen-Petersen BL, Sears RC. Mission Possible: Advances in MYC Therapeutic Targeting in Cancer. BioDrugs [Internet]. 2019;33:539–53. Available from: 10.1007/s40259-019-00370-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data for this project is publicly available in the GEO database at GSE215010 and GSE215006.