

Abstract
Prostate cancer (PCa) is annually the most frequently diagnosed cancer in the male population. To date, the diagnostic path for PCa detection includes the dosage of serum prostate-specific antigen (PSA) and the digital rectal exam (DRE). However, PSA-based screening has insufficient specificity and sensitivity; besides, it cannot discriminate between the aggressive and indolent types of PCa. For this reason, the improvement of new clinical approaches and the discovery of new biomarkers are necessary. In this work, expressed prostatic secretion (EPS)-urine samples from PCa patients and benign prostatic hyperplasia (BPH) patients were analyzed with the aim of detecting differentially expressed proteins between the two analyzed groups. To map the urinary proteome, EPS-urine samples were analyzed by data-independent acquisition (DIA), a high-sensitivity method particularly suitable for detecting proteins at low abundance. Overall, in our analysis, 2615 proteins were identified in 133 EPS-urine specimens obtaining the highest proteomic coverage for this type of sample; of these 2615 proteins, 1670 were consistently identified across the entire data set. The matrix containing the quantified proteins in each patient was integrated with clinical parameters such as the PSA level and gland size, and the complete matrix was analyzed by machine learning algorithms (by exploiting 90% of samples for training/testing using a 10-fold cross-validation approach, and 10% of samples for validation). The best predictive model was based on the following components: semaphorin-7A (sema7A), secreted protein acidic and rich in cysteine (SPARC), FT ratio, and prostate gland size. The classifier could predict disease conditions (BPH, PCa) correctly in 83% of samples in the validation set. Data are available via ProteomeXchange with the identifier PXD035942.
Introduction
Prostate cancer (PCa) is the most common and the second lethal cancer in men with 191.930 new cases and 33.330 deaths in the United States in 2020.1 PCa diagnosis is based on the digital rectal exam (DRE) and on serum dosage of prostate-specific antigen (PSA),2 although the PSA test shows low specificity, sensitivity, and the inability to stratify patients.3 In fact, PSA is detectable at low levels in the blood circulation as the result of the diffusion through the prostate basal cells; conversely, with the PCa onset, PSA levels increase because the tumor changing the architecture of the gland leads to tissue flaking and PSA release.4 Unfortunately, the increase of PSA levels cannot be considered as a parameter that uniquely reflects the presence of PCa because PSA levels could also increase in other conditions such as prostatitis, gland inflammation, and benign prostatic hyperplasia (BPH).5 What further complicates PCa diagnosis is the fact that some patients with advanced PCa show levels of PSA comparable to those detected in patients with benign alterations. This leads to the necessity of developing new approaches to improve PCa diagnosis.
The aim of this work is to make an initial assessment of the utility of a proteomic profile of EPS-urine samples in helping the classification of PCa and BPH, two conditions that share the characteristic of increased PSA levels.
During the development of the experimental design, attention was mainly focused on two aspects: (i) to use an easily collectable sample as the starting material and (ii) to analyze the proteomic profile of each sample through a method able to detect proteins at low abundance. To fulfill the first point, proteomic analysis was performed on EPS-urine, a prostate proximal biofluid. EPS-urine6,7 is a sample collected after DRE, clinical practice that promotes the release of prostate-specific proteins in the biofluid. For this reason, EPS-urine analysis may hold promise for detecting proteins that give an early signal of an alteration of the health status of the gland. Considering that urinary proteins are dispersed in a large volume, we chose an approach that allowed sample concentration before the enzymatic digestion step: namely, the filter-aided sample preparation (FASP)8−10 protocol.
To obtain a deep proteome coverage, the urinary proteomic profile was investigated through data-independent acquisition (DIA), a sensitive quantitative method where all ions undergo MS/MS events. By sequencing all peptides present in the sample, even those at low abundance,11 the probability of detecting proteins that mediate molecular processes involved in the tumor increases. For this reason, the snapshot of the sample12 provided by DIA analysis represents a box to research potential molecular switches that trigger the tumor onset.
We developed and implemented a machine learning (ML) pipeline to select features and identify interesting patterns. Such a pipeline is based on a voting mechanism that ensembles the ML models, enhancing the prediction performance on average. In the literature, there are available pipelines similarly processing prostate cancer and (more general) biological data sets, with similar results.13−15
Materials and Methods
All chemicals used in the experiments described were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
Sample Collection
EPS-urine samples were obtained from the Urology Units of the Magna Graecia University of Catanzaro and from the Romolo Hospital Urology Unit. The study was approved by the Institutional Ethical Committee of the Magna Graecia University of Catanzaro, RP 41/2018; all patients provided their written informed consent for the analysis of EPS-urine samples. Overall, 73 specimens from PCa patients and 60 from BPH patients were collected after DRE.
The characteristics of patients enrolled in this work are summarized in Table 1.
Table 1. Median Values and Interquartile Ranges (IQR) of Main Clinical Variables in Our Sample Set.
| variables | PCa (n = 73) | BPH (n = 60) | p-value |
|---|---|---|---|
| age (years), median (IQR) | 69.0 (63.0–74.0) | 68.0 (62.0–72.0) | 0.19 |
| PSA (ng/mL), median (IQR) | 7.43 (5.66–13.12) | 2.72 (1.21–4.80) | <0.01 |
| PSA ratio (%), median (IQR) | 16.0 (13.0–22.0) | 36.0 (25.0–45.25) | <0.01 |
| prostate volume (cc), median (IQR) | 40.0 (30.0–50.00) | 57.0 (40.0–79.0) | <0.01 |
| Gleason score, n (%) | |||
| Gleason 6 (3 + 3) | 22 (31.0%) | N/A | |
| Gleason 7 (3 + 4) | 22 (31.0%) | N/A | |
| Gleason 7 (4 + 3) | 16 (22.5%) | N/A | |
| Gleason 8 | 9 (12.7%) | N/A | |
| Gleason 9 | 2 (2.8%) | N/A |
FASP Protocol
After collection, EPS-urine samples were centrifugated within 2 h of collection at 2100 rcf for 10 min to remove cellular debris; the supernatant was stored at −80 °C until use.
For FASP digestion, 500 μL of EPS-urine was diluted with 100 μL of the diluent (6% sodium dodecyl sulfate, SDS, 300 mM buffer Tris-HCl at pH 8.0 and 300 mM dithiothreitol, DTT) to achieve a final concentration of 1% SDS, 50 mM Tris-HCl, and 50 mM DTT. After the addition of denaturants, the samples were incubated at 95 °C for 10 min with gentle shaking. Subsequently, diluted EPS-urine samples (600 μL) were loaded onto a Microcon-10 Centrifugal Filter Unit (Millipore) and were processed as suggested by the manufacturer changing the wash volume from 100 to 200 μL, to more effectively remove detergent residues; the details of FASP digestion were reported in a previous study.16
After protein digestion, 15 μL of each EPS-urine digest was purified by strong cation exchange17 (SCX) StageTips to remove residues of the detergent. In detail, since salts prevent the binding of peptides to the SCX stationary phase, to reduce the salt concentration below 5 mM, the peptide solution was diluted 4-fold in 0.5% formic acid (FA) and 80% of acetonitrile (ACN) (wash solution 2). After purifying the samples as described previously,16 peptides were eluted in a volume of 7 μL and immediately diluted to 27 μL of 0.1% FA. By making this dilution, ∼1 μL of purified digest corresponded to 1 μL of the starting EPS-urine sample. This portion of the purified sample was only used for acquiring preliminary injections, exploited for the estimation of protein amount and for compiling the sample card (see below).
Protein Amount
To estimate the protein amount, 1 μL of the peptide mixture purified by StageTips was analyzed by LC-MS/MS, and the total area of all identified peptides was calculated; the value of the total area was interpolated with a calibration line built as follows: (i) starting from HeLa digest stock with a concentration of 100 ng/μL, five different solutions were prepared (1, 2.5, 7.5, 25, and 75 ng/μL), (ii) each solution was injected in duplicates using the same injection volume (2 μL), and (iii) the proteomic analysis was performed with the same LC-MS/MS acquisition method used for the preliminary injection of sample.
Raw files of HeLa injections and raw files of preliminary sample injections were analyzed in Proteome Discoverer 1.4 as described in the DDA Data Analysis section to identify and quantify peptides present in each sample; the total area was calculated by summing up the peak areas of all detected peptides. Total area values from each sample were interpolated with the external standard (HeLa digest) calibration curve to estimate protein concentration in the FASP digests. Based on this estimation, 2 μg of total proteins from each sample were purified for subsequent analyses (see below).
Sample Card
To obtain the first overview of the sample (sample card), the following parameters were evaluated: (i) the number of the identified proteins and peptides, (ii) the presence of prostate-specific proteins, and (iii) the total protein content.
An important reference for the sample card elaboration was the list of 49 proteins classified as “prostate enriched” in EPS-urine, which was provided by Principe et al.18 This list was further reduced by us using the BioGPS (www.biogps.org)19 database to select only prostate-specific proteins; after this data filtering, the number of proteins was reduced from 49 to 33. We assumed that the total protein intensity of these proteins (equal to 33) could provide an estimation of relative EPS content in the samples. For this reason, we introduced the EPS factor, a parameter calculated by dividing the total intensity of EPS proteins (33 proteins) by the total intensity of the identified proteins in the sample; the value of intensity was obtained by processing the raw files of preliminary injections with MaxQuant software (see below).
Besides the parameters just described, the following elements were included in the sample card: (i) the number of identified proteins and peptides, (ii) the protein amount, and (iii) the area under chromatogram (AUC) of preliminary injections (Xcalibur software, Thermo Scientific).
Encompassing so many elements, the sample card represented for us a snapshot of the sample and a valid means to make an initial quality assessment of each individual sample.
C18 StageTip Purification
After completing the preliminary injections, 2 μg of proteins were withdrawn from each sample and purified both by SCX StageTips, as described above, and by C18 StageTips17 to discard the salts deriving from SCX purification. In detail, 7 μL of the SCX eluate was acidified with 150 μL of 0.1% trifluoroacetic acid (TFA) and loaded on C18 StageTips. An elution volume of 10 μL was kept at 30 °C for 3 min in a speed-vac to reduce the volume to 2–3 μL. Afterward, 47 μL of 0.1% FA was added.
DIA Library
To achieve high proteome coverage, the EPS-urine samples were analyzed by DIA analysis.11−20 To generate the DIA spectral library, peptides from 22 EPS-urine samples (around 11 μg) were pooled and loaded on a StageTip made of two stacked disks of the C18 stationary phase; fractionation of peptides in basic reversed-phase mode was performed using solutions constituting 10 mM triethylammonium bicarbonate (TEAB), 0.2% ammonium hydroxide, and increasing concentrations of acetonitrile (ACN; 4, 8, 12, 16, 20, 24, 28, 32, 40, 80%).
The 10 fractions were analyzed by DDA mode, and the obtained identifications were used to build the DIA spectral library, a fundamental element for DIA analysis on the single samples.
LC-MS/MS Analysis
Peptides were separated by an Easy nLC-1000, chromatographic instrument coupled to a Q-Exactive “Classic” mass spectrometer (both from Thermo Scientific, Bremen, Germany).
For preliminary analysis, 1 μL of the peptide mixture was separated using a linear gradient of 75 min at a flow rate of 230 nL/min on a 15 cm, 75 μm i.d., in-house-made column packed with 3 μm C18 silica particles (Dr. Maisch). The binary gradient was performed using mobile phase A (0.1% FA, 2% ACN) and mobile phase B (0.1% FA and 80% ACN). Peptide elution was obtained at a flow rate of 230 nL/min and ramped from 6% B to 42% B in 60 min and from 42% B to 100% B in an additional 8 min; the column was cleaned by running 100% B for 5 min. For preliminary analysis, the Q-Exactive mass spectrometer operated in DDA mode using a top-12 method. The MS full scan range was 350–1800 m/z, with a resolution of 70 000, an ACG target of 1e6, and a maximum injection time of 50 ms. The mass window for precursor ion isolation was 1.6 m/z, with a resolution of 35 000, an AGC target of 1e5, a maximum injection time of 120 ms, an HCD fragmentation at normalized collision energy of 25, and dynamic exclusion of 15 s.
For the construction of the spectral library, the 10 fractions obtained by high-pH reversed-phase C18 fractionation were separated using a linear gradient of 140 min at a flow rate of 230 nL/min on a 15 cm, 75 μm i.d., in-house-made column packed with 3 μm C18 silica particles. Peptide elution was obtained using a gradient from 3% B to 25% B in 90 min, from 25% B to 40% B in 30 min, from 40% B to 100% B in 8 min, and then at 100% B for 10 min. The mass spectrometer was acquired in DDA mode using the same parameters described above.
Each EPS-urine sample was analyzed in DIA mode with the same chromatographic method used for fraction analysis with a unique shrewdness: every 10 analyses, at the end of the gradient, 100% B was maintained for 70 min instead of 10; this procedure allowed for more effective regeneration of the column and, consequently, longer chromatographic performance. The DIA method enclosed 26 windows with a full scan at resolution of 17 500 (AGC target of 1e6 and maximum injection time of 50 ms) and DIA scans with 35 000 (AGC target of 5e5, maximum injection time of 120 ms, and normalized collision energy of 25). In detail, the total number of windows was 26, including 20 windows with an isolation width of 20 m/z, 5 windows with an isolation width of 50 m/z, and 1 window with an isolation width of 200 m/z. The resulting m/z range was from 350 to 1200 Th.
DDA Data Analysis
The raw files of preliminary injections were analyzed by Proteome Discoverer 1.4 (Thermo Fisher Scientific, Bremen, Germany),21 using Sequest as the search engine, and the Human Uniprot Complete proteome database, downloaded on March 2016 and containing 42.013 sequences. This analysis was performed using an MS tolerance of 15 ppm, an MS/MS tolerance of 0.02 Da, trypsin as an enzyme, and a maximum of two missed cleavage sites. Oxidation of methionines (+15.995 Da) was set as dynamic modification, whereas carbamidomethylation of cysteines (+57.021) was the only static modification. The false discovery rate (FDR) for peptide identification was assessed by a percolator; the cutoff value was set at 0.01. Quantification at the peptide level was achieved within Proteome Discoverer using the “event detector” (mass precision 2 ppm) and “precursor ion area detector” nodes to calculate the peptide peak area.
To elaborate the sample card, the same raw files of preliminary injections were processed in MaxQuant software (version 1.6.1.0)22 using the following settings: protein database Human Complete proteome (see above), an MS tolerance of 6 ppm, an MS/MS tolerance of 20 ppm, trypsin/P as an enzyme, and two missed cleavages. Carbamidomethylation of cysteines was set as static modification, and oxidation of methionine and protein N-terminal acetylation were allowed as variable modifications. FDR was set to 0.01, and only peptides with ≥7 amino acid residues were selected for identification. At least one unique peptide was necessary to identify a protein.
DIA Data Processing
For spectral library generation, the raw files of C18 high-pH reversed-phase fractionation were analyzed in Spectronaut by setting the Q-value cutoff to 0.01 with a minimum of 3 and a maximum of 6 fragment ions.
The raw files of the DIA acquisition were imported in Spectronaut 13.0 with no file conversion, and the obtained identifications were filtered by a Q-value of 0.01. Protein quantification was performed using “major group quantity” with a minimum of 1 and a maximum of 10 peptides and setting the Q-value percentile at 0.45 for data filtering. The intensity for each protein was calculated by summing fragment ion peak areas. In the end, DIA analysis performed in Spectronaut gave us a matrix with the quantified proteins in the different samples.
To cross-correlate proteomic data with clinical information, the matrix with the quantified proteins in each patient was merged with the following clinical parameters: patient age, total PSA, ratio PSA free/total PSA (FT ratio), and prostatic gland size.
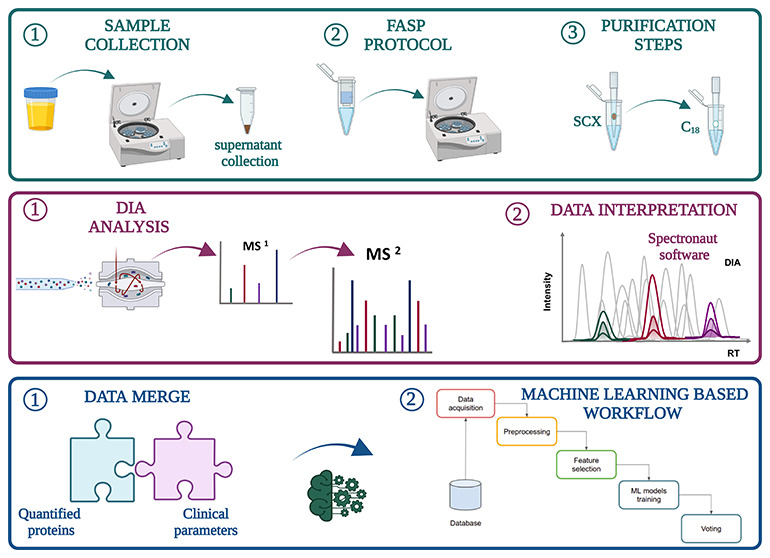
A framework for extracting and analyzing features from clinical and mass spectrometry data was developed. The framework can be summarized in modules, as reported in Figure 1.
Figure 1.

ML-based workflow.
Mass spectrometry data were gathered and collected by the data acquisition module. Also, patients’ clinical information was collected from electronic health records (EHRs) enclosing patient age, total PSA, FT ratio, and prostate gland size. Such data were merged based on patient id and stored in a database (see DB module in Figure 1). Patients’ merged data were then preprocessed, and only information useful for defining and creating the ML methods was selected. Since the clinical process generated potentially incomplete data sets, the preprocessing module also identified missing data and generated synthetic values according to data distribution. Data were then filtered and scaled to prepare the data set for analysis via ML models.23 The approach was based on the ensembling of ML models whose results were integrated by voting ML approaches. The ML process was implemented in Python language. Python scripts were based on the scikit-learn library and run on Google collab for training and validation phases.
To build a predictive model, our data were divided into two groups: a data set constituting 121 samples (90% of data) and a validation set constituting 12 samples (10% of data). The data set of 121 samples was in turn split into a training set and a testing set, according to a 10-fold cross-validation strategy. In detail, the training set was used to train the ML models in classifying the samples, while the testing set was used to evaluate the prediction power of the predictive model. Finally, the accuracy of the model was evaluated using the validation set (12 samples).
Results and Discussion
In the first stage of this work, 73 PCa and 60 BPH EPS-urine samples were analyzed in DDA mode. On average, 991 proteins with 3940 peptides and 991 proteins with 3965 peptides were identified in BPH and PCa groups, respectively. To summarize the main qualitative characteristics of our sample set, these data together with the EPS factor, i.e., the percentage of protein intensity ascribed to prostate-specific proteins (33 proteins; Table S1), were enclosed in the sample card (Table S2); in particular, the EPS factor was an index of the secretory capacity of the gland.
After this first exploration of the data set in data-dependent mode, the proteomic profile of each sample was thoroughly investigated by DIA analysis. DIA data were processed by Spectronaut, identifying and quantifying 2615 proteins. The number of quantified proteins was high compared to previous studies;24 this high proteome coverage resulted in a higher probability of detecting tissue-specific proteins, possibly related to PCa. To confirm this hypothesis, our protein list was compared to a panel (composed of 135 proteins) generated after matching the proteins contained in our spectral library to (i) the list of “Elevated genes of PCa” from Protein Atlas (www.ProteinAtlas.org), (ii) the list of “Prostate Cancer”-related proteins from BioGPS (www.biogps.org), and (iii) the “Prostate enriched proteins” from Protein Atlas. This comparison showed an overlap of 73% (99 of 135), demonstrating that a relevant part of the PCa-related proteome originally present in our library was detected in DIA single LC-MS/MS injections.
The 2615 quantified proteins were filtered by Spectronaut setting the Q-value percentile to 0.45; this parameter required that the matrix only included proteins quantified in at least 60 samples (45% of 133). By setting this filter, a matrix composed of 1670 proteins was obtained (Table S3); these 1670 proteins were quantified in all samples; thus, no data imputation was required for MS data. The list of quantified urinary proteins obtained by Spectronaut (1670) was merged with the following clinical information: patient age, total PSA, PSA free, FT ratio, and prostate gland size (Table S4); this strategy has allowed us to build a more complete picture, encompassing clinical and proteomics data, for each patient.
The full matrix was loaded from the database containing both clinical and proteomics data and stored in a Python Dataframe variable. Both clinical and mass spectrometry data were then preprocessed. We note that in the case of missing values for clinical-related information (e.g., prostate gland size, age, total PSA), missing and invalid value samples were updated by data distribution average values. Imputation regarded a very small number of records (8 out of a total of 600 records). No imputation was needed for MS data because the criteria adopted for protein filtering (i.e., valid values in at least 45% of samples) returned a full matrix with no missing values. Categorical values were converted into numeric classes, finally normalizing all numeric values in the 0–1 range. After the preprocessing phase, a feature selection algorithm has been applied, to reduce the number of inputs for the development of the prediction model. The pipeline included a feature selection module, which implemented the following models: (i) Pearson correlation coefficient: (ii) Chi-square test; (iii) RFE (recursive feature elimination); (iv) random forest; and (v) logistic regression. Pearson correlation was used to evaluate the correlation between couples of features; the objective was to exclude redundant variables and only retrieve independent ones. Thus, this phase was used to identify the most statistically significant features (i.e., columns of the data set) according to each model’s predictive performance and rank them according to a relevance score (i.e., how many models agreed on its relevance). We used the above reported feature selection models and focused on the features on which all of the methods agree. This reduced the total number of variables for multivariate analysis to four best candidates: semaphorin-7A (sema7A), secreted protein acidic and rich in cysteine (SPARC), FT ratio, and prostate gland size (see Figure 2D).
Figure 2.

Key steps of our workflow: (A) EPS-urine sample collection and FASP protocol, (B) elaboration of the sample card by DDA analysis, (C) high-pH reversed-phase C18 fractionation for spectral library generation and DIA analysis by Spectronaut, and (D) bioinformatics analysis by ML models. Created with BioRender.com.
Random forest, support vector machine, decision tree, K-nearest neighbors classifier, and logistic regression ML models were trained by measures referred to the selected features, as described in the Materials and Methods section. Briefly, 90% of the data set was used for training and testing using a 10-fold cross-validation approach, whereas 10% of the data set (12 samples) was dedicated to the validation phase using hard/soft voting strategies. The models were evaluated on the testing set based on the following measures: AUC, F1, specificity, and sensitivity; the relative values are reported in Table 2. We integrated all five models through two voting-based strategies: (i) hard voting and (ii) soft voting. Hard voting counted models that agreed on the predicted classes, whereas soft voting weighted models based on predictive accuracy. For instance, considering the hard voting approach, if four ML models out of five predicted the PCA class for a certain input, then the PCA class was adopted as a result. In the soft voting approach, each ML model prediction (i.e., PCA or BPH class) was weighted by the F1 performance measure (as per column “F1” of Table 2). The classifier could predict disease conditions (BPH, PCa) correctly in 83% of samples in the validation set (10 out of 12 samples).
Table 2. AUC, F1, Accuracy, Specificity, and Sensitivity Values Obtained for Each ML Model.
| AUC | F1 | accuracy | specificity | sensitivity | |
|---|---|---|---|---|---|
| random forest | 0.710 | 0.733 | 0.711 | 0.716 | 0.704 |
| logistic regression | 0.779 | 0.830 | 0.793 | 0.910 | 0.648 |
| KNN | 0.729 | 0.768 | 0.736 | 0.791 | 0.667 |
| SVM | 0.707 | 0.802 | 0.736 | 0.970 | 0.444 |
| decision tree | 0.777 | 0.814 | 0.785 | 0.851 | 0.704 |
The discrimination power of our model was compared to other biomarker discovery efforts on PCa published in the last decade. Glycoproteomic analysis performed on serum by Cima et al.25 allowed elaborating a signature based on four glycoproteins able to classify PCa and BPH patients with an AUC of 0.726: a value comparable to the one obtained by PSA alone within the same sample set (0.730). The authors combined the four-protein signature and PSA into a single predictive model, which showed an AUC of 0.840. This study represents a milestone in PCa biomarker discovery by MS, though glycopeptide profiling performed on serum presents advantages and disadvantages, as described in our recent review.26
Proximal biofluids, such as EPS-urine, being physically closer to the tumor, may contain proteins secreted or shed from cancer cells. A notable effort in EPS-urine analysis has been reported by Kim et al.27 Starting from a previously compiled database of EPS-enriched proteins, they narrowed down the attention to 34 candidates of potential diagnostic and prognostic value, which were assayed by selected reaction monitoring (SRM). This analysis generated a predictive model based on six peptides able to separate controls from PCa patients with an AUC of 0.77. In their case, the proteomic model performed remarkably better than PSA alone (AUC = 0.67). Furthermore, to better investigate the molecular dynamics of PCa, urine was also studied from the point of view of its metabolomic profile28 to detect metabolites capable of promoting tumor growth.29
The model described here had the strength of a “high-coverage” proteomic analysis combined with several clinical parameters. Though the starting number of quantified proteins was very high (1670), the four-feature signature included just two proteins. Their involvement in cancer development was already established.30,31 As can be appreciated in Figure 3, their average abundance in EPS-urine was very low. Their detection was facilitated by the extended dynamic range provided by the DIA scanning mode.
Figure 3.

Ranking plot of identified proteins by DIA analysis. Prostate-specific proteins (33) are indicated in violet, while the two components of our model (SPARC and Sema7A) are in purple.
Univariate analysis and dot plots for the two proteins and the two clinical variables are reported in Figure 4; levels of both proteins were found, on average, decreased in the PCa group.
Figure 4.
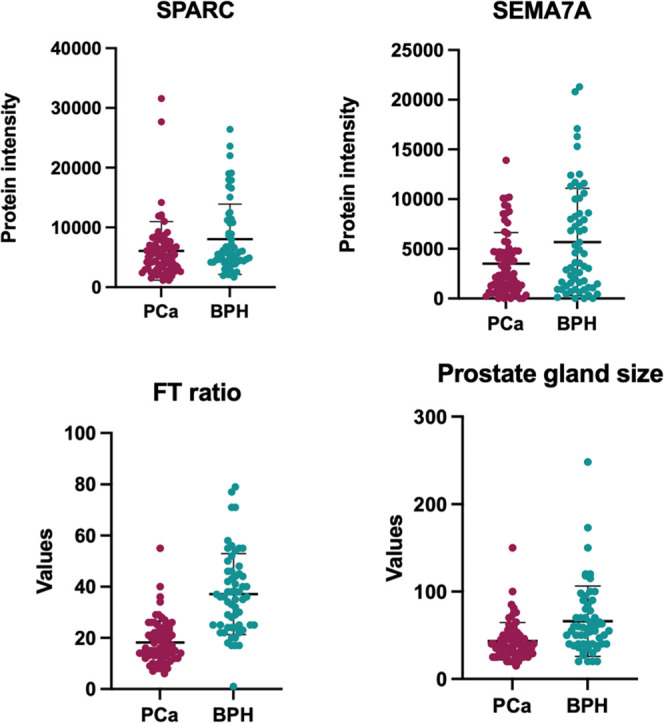
This panel shows the dot plots relative to univariate analysis for the two proteins (SPARC and Sema7a) and the two clinical variables (FT ratio and prostate gland size); all these variables showed, on average, a significant decrease (p-value < 0.05) in PCa samples with respect to BPH.
Both proteins enclosed in the predictive model showed decreased levels in PCa patients compared to BPH. Since the digital rectal exam is only performed on patients with elevated PSA levels, no healthy subjects were recruited in this study. Thus, no information could be acquired on the levels of SPARC and SEMA7A in EPS-urine from healthy subjects. Consequently, no comparison can be made between the levels of these proteins in healthy subjects and PCa or BPH groups. As a result, this model can only be applied following a positive PSA test readout to discriminate between benign and cancerous disease. First-level evaluation aimed at discriminating between healthy subjects and patients with prostatic disease will be carried out by means of well-established PSA serum testing.
The two proteins belonging to the model harbor different, often tissue-dependent molecular functions in cancer development. SEMA7A belongs to the semaphorin family, which comprises proteins involved both in physiological events (growth and migration of nervous cells, and assembly of cytoskeleton) and in neoplastic mechanisms (cell invasion and migration).30 A possible explanation of SEMA7A higher levels in EPS-urine from BPH patients is that this protein could play a protective role against cancer development. It is known from the literature that this protein is an immune semaphorin, modulating several immunoinflammatory processes. In particular, it has been identified as a regulator of the effector phase of the T-cell-mediated inflammatory response. Since the T-cell response plays a critical role in anticancer surveillance, this might suggest an involvement of this protein in preventing cancer development. Nevertheless, in other tumors, this protein was involved in promoting migration, invasion, and angiogenesis;32 in fact, some studies showed that elevated levels of SEMA7A are associated with the progression of breast cancer.32,33 Concerning PCa, one work has found increased levels (over 18-fold) of SEMA7A following the overexpression of ERG in mouse prostate organoids.34 Based on this evidence, SEMA7A seems to have a role in establishing the microenvironment of premalignant, ERG-positive prostate lesions. Nevertheless, no experimental evidence relative to its expression or its potential role in PCa development has been reported.
SPARC is a glycoprotein belonging to cellular matrix proteins, and it is involved in tissue remodeling.35 SPARC’s role in PCa is quite debatable, since its levels of expression may point in opposite directions depending on if the tumor or the stroma is considered. For example, the upregulation of SPARC in the tumor is correlated to the epithelial-to-mesenchymal transition (EMT) and to events specific to malignant phenotype such as metastasis;36 contrariwise, SPARC of stromal derivation appears to hinder the growth of PCa cells.37,38 Since the role of this protein is strictly related to its localization (tumoral/stromal) and since the origin of SPARC found in EPS-urine is unknown, no conclusive interpretation of data can be made at this stage.
Conclusions
All in all, our experimental design could be considered a starting point to investigate the potential of DIA-based proteomic analysis of EPS-urine in the context of PCa. DIA analysis provided an extended dynamic range, which allowed the detection of low-abundance proteins. The combination of clinical and proteomic variables yielded a classifier comprising four variables: SPARC, SEMA7A (both from proteomics data), FT ratio, and prostate gland size (clinical parameters). This classifier had AUC values higher than those of PSA alone in three out of five of the ML approaches used, but with higher specificity. Both PSA alone and our classifier could predict disease conditions (BPH, PCa) correctly in 83% of samples in the validation set.
Despite the promising results, a larger validation cohort is needed to assess the ability of our predictive model to discriminate between PCa from BPH patients. Though the FASP/DIA protocol achieves a wide proteome coverage, it is relatively laborious and time-consuming and thus might not be the best approach for validation on a larger sample cohort. Alternative approaches based on ELISA assays on the two protein candidates and a total protein content measurement could be more appealing for validation at a larger scale.
Acknowledgments
This work was supported by MIUR (Ministero Università Ricerca, PRIN 2017 to M.G.) and by POR Calabria FESR 2014–2020, action 1.2.2, “INNOPROST”. The TOC figure was created with BioRender.com.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c05487.
Table S1: intensity of prostate-specific proteins calculated by MaxQuant software; Table S2: sample card; Table S3: list of quantified proteins in Spectronaut by setting Q-value percentile to 0.45; and Table S4: clinical information of enrolled patients (XLSX)
Accession Codes
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the data set identifier PXD035942. Reviewer account details: (i) reviewer_pxd035942@ebi.ac.uk (Username), (ii) G2RDjjgn (Password).
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Cancer Statistics, 2020. CA, Cancer J. Clin. 2020, 70, 7–30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Wang M. C.; Valenzuela L. A.; Murphy G. P.; Chu T. M. Purification of a Human Prostate Specific Antigen. J. Urol. 2017, 197, S148–S152. 10.1016/j.juro.2016.10.100. [DOI] [PubMed] [Google Scholar]
- Lumen N.; Fonteyne V.; De Meerleert G.; Ost P.; Villeirs G.; Mottrie A.; De Visschere P.; De Troyer B.; Oosterlinck W. Population Screening for Prostate Cancer: An Overview of Available Studies and Meta-Analysis. Int. J. Urol. 2012, 19, 100–108. 10.1111/j.1442-2042.2011.02912.x. [DOI] [PubMed] [Google Scholar]
- Kulasingam V.; Diamandis E. P. Strategies for Discovering Novel Cancer Biomarkers through Utilization of Emerging Technologies. Nat. Clin. Pract. Oncol. 2008, 5, 588–599. 10.1038/ncponc1187. [DOI] [PubMed] [Google Scholar]
- Tosoian J.; Loeb S. PSA and beyond: The Past, Present, and Future of Investigative Biomarkers for Prostate Cancer. Sci. World J. 2010, 10, 1919–1931. 10.1100/tsw.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake R. R.; Elschenbroich S.; Lopez-Perez O.; Kim Y.; Ignatchenko V.; Ignatchenko A.; Nyalwidhe J. O.; Basu G.; Wilkins C. E.; Gjurich B.; Lance R. S.; Semmes O. J.; Medin J. A.; Kislinger T. In-Depth Proteomic Analyses of Direct Expressed Prostatic Secretions. J. Proteome Res. 2010, 9, 2109–2116. 10.1021/pr1001498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake R. R.; White K. Y.; Fuller T. W.; Igwe E.; Clements M. A.; Nyalwidhe J. O.; Given R. W.; Lance R. S.; Semmes O. J. Clinical Collection and Protein Properties of Expressed Prostatic Secretions as a Source for Biomarkers of Prostatic Disease. J. Proteomics 2009, 72, 907–917. 10.1016/j.jprot.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal Sample Preparation Method for Proteome Analysis. Nat. Methods 2009, 6, 359–362. 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Zhao M.; Li M.; Yang Y.; Guo Z.; Sun Y.; Shao C.; Li M.; Sun W.; Gao Y. A Comprehensive Analysis and Annotation of Human Normal Urinary Proteome. Sci. Rep. 2017, 7, 3024 10.1038/s41598-017-03226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski J. R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal. Chem. 2016, 88, 5438–5443. 10.1021/acs.analchem.6b00859. [DOI] [PubMed] [Google Scholar]
- Gallien S.; Duriez E.; Demeure K.; Domon B. Selectivity of LC-MS/MS Analysis: Implication for Proteomics Experiments. J. Proteomics 2013, 81, 148–158. 10.1016/j.jprot.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Gillet L. C.; Navarro P.; Tate S.; Röst H.; Selevsek N.; Reiter L.; Bonner R.; Aebersold R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 2012, 11, O111.016717 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y.; Wang X.; Dai Q.; Wang Y.; Wang Q.; Peng S.; Wei X.; Qiu J.; Salahub D. R.; Xiong Y.; Wei D. Q. MDA-GCNFTG: Identifying MiRNA-Disease Associations Based on Graph Convolutional Networks via Graph Sampling through the Feature and Topology Graph. Brief. Bioinform. 2021, 22, bbab165 10.1093/bib/bbab165. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Wang X.; Zhou D.; Wei D. Q.; Peng S. SVPath: An Accurate Pipeline for Predicting the Pathogenicity of Human Exon Structural Variants. Brief. Bioinform. 2022, 23, bbac014 10.1093/bib/bbac014. [DOI] [PubMed] [Google Scholar]
- Sadhu A.; Bhattacharyya B. Common Subcluster Mining in Microarray Data for Molecular Biomarker Discovery. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 348–359. 10.1007/s12539-017-0262-3. [DOI] [PubMed] [Google Scholar]
- Prestagiacomo L. E.; Gabriele C.; Morelli P.; Rota M. A.; Alba S.; Cuda G.; Damiano R.; Gaspari M. Proteomic Profile of EPS-Urine through FASP Digestion and Data-Independent Analysis. J. Vis. Exp. 2021, 4, e62512 10.3791/62512. [DOI] [PubMed] [Google Scholar]
- Rappsilber J.; Mann M.; Ishihama Y. Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using StageTips. Nat. Protoc. 2007, 2, 1896–1906. 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Principe S.; Kim Y.; Fontana S.; Ignatchenko V.; Nyalwidhe J. O.; Lance R. S.; Troyer D. A.; Alessandro R.; Semmes O. J.; Kislinger T.; Drake R. R.; Medin J. A. Identification of Prostate-Enriched Proteins by in-Depth Proteomic Analyses of Expressed Prostatic Secretions in Urine. J. Proteome Res. 2012, 11, 2386–2396. 10.1021/pr2011236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Orozco C.; Boyer J.; Leglise M.; Goodale J.; Batalov S.; Hodge C. L.; Haase J.; Janes J.; Huss J. W.; Su A. I. BioGPS: An Extensible and Customizable Portal for Querying and Organizing Gene Annotation Resources. Genome Biol. 2009, 10, R130 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Hüttenhain R.; Collins B.; Aebersold R. Mass Spectrometric Protein Maps for Biomarker Discovery and Clinical Research. Expert Rev. Mol. Diagn. 2013, 13, 811–825. 10.1586/14737159.2013.845089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsburn B. C. Proteome Discoverer-a Community Enhanced Data Processing Suite for Protein Informatics. Proteomes 2021, 9, 15 10.3390/proteomes9010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Iguyon I.; Elisseeff A. An Introduction to Variable and Feature Selection. J. Mach. Learn. Res. 2003, 3, 1157–1182. 10.1162/153244303322753616. [DOI] [Google Scholar]
- Adachi J.; Kumar C.; Zhang Y.; Olsen J. V.; Mann M. The Human Urinary Proteome Contains More than 1500 Proteins, Including a Large Proportion of Membrane Proteins. Genome Biol. 2006, 7, R80 10.1186/gb-2006-7-9-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cima I.; Schiess R.; Wild P.; Kaelin M.; Schuffler P.; Lange V.; Picotti P.; Ossola R.; Templeton A.; Schubert O.; Fuchs T.; Leippold T.; Wyler S.; Zehetner J.; Jochum W.; Buhmann J.; Cerny T.; Moch H.; Gillessen S.; Aebersold R.; Krek W. Cancer Genetics-Guided Discovery of Serum Biomarker Signatures for Diagnosis and Prognosis of Prostate Cancer. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 3342–3347. 10.1073/pnas.1013699108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriele C.; Prestagiacomo L. E.; Cuda G.; Gaspari M. Mass Spectrometry-Based Glycoproteomics and Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 5222 10.3390/ijms22105222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Jeon J.; Mejia S.; Yao C. Q.; Ignatchenko V.; Nyalwidhe J. O.; Gramolini A. O.; Lance R. S.; Troyer D. A.; Drake R. R.; Boutros P. C.; Semmes O. J.; Kislinger T. Targeted Proteomics Identifies Liquid-Biopsy Signatures for Extracapsular Prostate Cancer. Nat. Commun. 2016, 7, 11906 10.1038/ncomms11906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euceda L. R.; Andersen M. K.; Tessem M. B.; Moestue S. A.; Grinde M. T.; Bathen T. F. NMR-Based Prostate Cancer Metabolomics. Methods Mol. Biol. 2018, 1786, 237–257. 10.1007/978-1-4939-7845-8_14. [DOI] [PubMed] [Google Scholar]
- Bruzzone C.; Loizaga-Iriarte A.; Sánchez-Mosquera P.; Gil-Redondo R.; Astobiza I.; Diercks T.; Cortazar A. R.; Ugalde-Olano A.; Schäfer H.; Blanco F. J.; Unda M.; Cannet C.; Spraul M.; Mato J. M.; Embade N.; Carracedo A.; Millet O. 1H NMR-Based Urine Metabolomics Reveals Signs of Enhanced Carbon and Nitrogen Recycling in Prostate Cancer. J. Proteome Res. 2020, 19, 2419–2428. 10.1021/acs.jproteome.0c00091. [DOI] [PubMed] [Google Scholar]
- Mastrantonio R.; You H.; Tamagnone L. Semaphorins as Emerging Clinical Biomarkers and Therapeutic Targets in Cancer. Theranostics 2021, 11, 3262–3277. 10.7150/THNO.54023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira-Barros E. G.; de Branco L. C.; Da Costa N. M.; Nicolau-Neto P.; Palmero C.; Pontes B.; Ferreira do Amaral R.; Alves-Leon S. V.; Marcondes de Souza J.; Romão L.; Fernandes P. V.; Martins I.; Takiya C. M.; Ribeiro Pinto L. F.; Palumbo A.; Nasciutti L. E. GLIPR1 and SPARC Expression Profile Reveals a Signature Associated with Prostate Cancer Brain Metastasis. Mol. Cell. Endocrinol. 2021, 528, 111230 10.1016/j.mce.2021.111230. [DOI] [PubMed] [Google Scholar]
- Song Y.; Wang L.; Zhang L.; Huang D. The Involvement of Semaphorin 7A in Tumorigenic and Immunoinflammatory Regulation. J. Cell. Physiol. 2021, 236, 6235–6248. 10.1002/jcp.30340. [DOI] [PubMed] [Google Scholar]
- Black S. A.; Nelson A. C.; Gurule N. J.; Futscher B. W.; Lyons T. R. Semaphorin 7a Exerts Pleiotropic Effects to Promote Breast Tumor Progression. Oncogene 2016, 35, 5170–5178. 10.1038/onc.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzoni M.; De Felice D.; Beccaceci G.; Di Donato G.; Foletto V.; Genovesi S.; Bertossi A.; Cambuli F.; Lorenzin F.; Savino A.; Avalle L.; Cimadamore A.; Montironi R.; Weber V.; Carbone F. G.; Barbareschi M.; Demichelis F.; Romanel A.; Poli V.; Del Sal G.; Julio M. K.; de Gaspari M.; Alaimo A.; Lunardi A. ETS-Related Gene (ERG) Undermines Genome Stability in Mouse Prostate Progenitors via Gsk3β Dependent Nkx3.1 Degradation. Cancer Lett. 2022, 534, 215612 10.1016/j.canlet.2022.215612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brekken R. A.; Sage E. H. SPARC, a Matricellular Protein: At the Crossroads of Cell-Matrix. Matrix Biol. 2000, 19, 569–580. 10.1016/S0945-053X(00)00105-0. [DOI] [PubMed] [Google Scholar]
- DeRosa C. A.; Furusato B.; Shaheduzzaman S.; Srikantan V.; Wang Z.; Chen Y.; Siefert M.; Ravindranath L.; Young D.; Nau M.; Dobi A.; Werner T.; McLeod D. G.; Vahey M. T.; Sesterhenn I. A.; Srivastava S.; Petrovics G. Elevated Osteonectin/SPARC Expression in Primary Prostate Cancer Predicts Metastatic Progression. Prostate Cancer Prostatic Dis. 2012, 15, 150–156. 10.1038/pcan.2011.61. [DOI] [PubMed] [Google Scholar]
- Kapinas K.; Lowther K. M.; Kessler C. B.; Tilbury K.; Lieberman J. R.; Tirnauer J. S.; Campagnola P.; Delany A. M. Bone Matrix Osteonectin Limits Prostate Cancer Cell Growth and Survival. Matrix Biol. 2012, 31, 299–307. 10.1016/j.matbio.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriquez C.; Cancila V.; Ferri R.; Sulsenti R.; Fischetti I.; Milani M.; Ostano P.; Gregnanin I.; Mello-Grand M.; Berrino E.; Bregni M.; Renne G.; Tripodo C.; Colombo M. P.; Jachetti E. Castration-Induced Downregulation of SPARC in Stromal Cells Drives Neuroendocrine Differentiation of Prostate Cancer. Cancer Res. 2021, 81, 4257–4274. 10.1158/0008-5472.CAN-21-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.