
FIGURE 3.
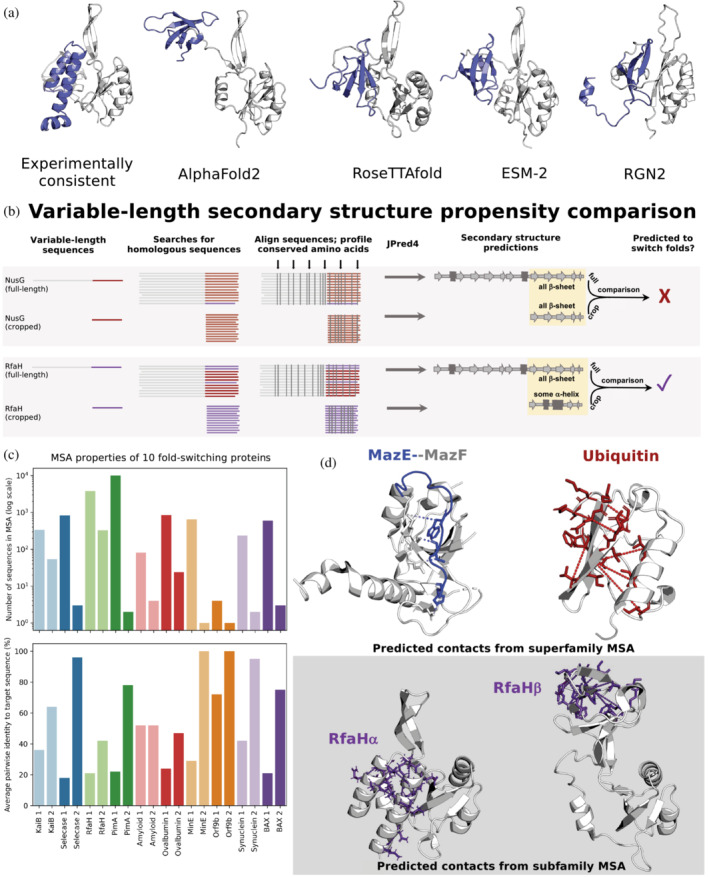
Predictive features of fold‐switching proteins. (a) All state‐of‐the‐art deep‐learning‐based methods fail to predict the experimentally characterized α‐helical ground state of Variant 5, a member of the NusG family with ≤29% sequence identity to its homologs with experimentally determined structures (Porter et al., 2022). The 3D helical model of Variant 5 (left), generated using Rosetta‐CM (Song et al., 2013) with RfaH (PDB 5OND, Chain A) as a template, is consistent with its chemical‐shift‐derived secondary structures (Porter et al., 2022). In contrast, all state‐of‐the‐art methods, including trRosetta (Du et al., 2021), EVCouplings (Hopf et al., 2019), and PHYRE2 (Kelley et al., 2015; shown in Porter et al., 2022), predict the activated β‐roll fold. In all cases, Variant 5's fold‐switching CTD is slate while its single‐fold NTD is gray. (b) Contrastingly, Variant 5's ground state α‐helical fold was successfully inferred from variable‐length secondary structure propensity comparison. While both full‐length and cropped NusG sequences have similar amino acid conservation patterns (gray vertical lines, top gray panel), conservation patterns differ for full‐length and cropped RfaH (gray vertical lines, bottom gray panel). Similar/different full‐length and cropped conservation patterns lead to similar/different secondary structure predictions, suggesting that NusG does not switch folds (top) while RfaH does (bottom). These different patterns likely result from different multiple sequence alignment (MSA) homogeneities. The sequence distributions depicted are for illustrative purposes only since true sequence distributions are unknown. (c) For 10 fold‐switching proteins successfully predicted by JPred4 (Mishra et al., 2021), full‐length alignments yielding secondary structure prediction 1 are deeper (plot above) and more diverse (plot below), indicating the presence of both fold‐switching and single‐fold sequences. In contrast, cropped sequence MSAs, yielding secondary structure prediction 2, are shallower (plot above) and more similar to the target sequence (plot below), reflecting fold‐switching subfamily properties. (d). Some single‐fold, fold‐switching, and intrinsically disordered proteins coevolve differently. The bacterial IDP MazE (blue, PDB ID: 5CQX, Chain C) coevolves with its folded binding partner, MazF (gray, PDB ID: 5CQX, Chain A); predicted contacts taken from Pancsa et al. (2018). Stablizing intrachain contacts coevolve in single‐folding ubiquitin (red, PDB ID: 1UBQ, Chain A); predicted contacts generated using GREMLIN (Balakrishnan et al., 2011; Kamisetty et al., 2013). In contrast, GREMLIN can successfully predict inter‐residue contacts unique to both folds of RfaH when using an MSA composed of sequences that yield similar partially helical JPred4 predictions (Porter et al., 2022).