

Abstract
The multifunctional cytochrome P450 17A1 (CYP17A1) plays a crucial role in the human steroid hormone synthesis (UniProtKB - P05093). It first carries out standard monooxygenase chemistry, converting PREG and PROG into 17-OH pregnenolone (17OH-PREG) and 17OH-PROG (17OH-PROG), utilizing a “Compound I” to initiate hydrogen abstraction and radical recombination in the classic “oxygen rebound” mechanism. Additionally, these hydroxylated products also serve as substrates in a second oxidative cycle which cleaves the 17–20 carbon–carbon bond to form dehydroepiandrosterone (DHEA) and androstenedione (AD), which are key precursors in the generation of the powerful androgens and estrogens. Interestingly, in humans, with 17OH-PREG this so-called lyase reaction is more efficient than for 17OH-PROG, based on Kcat/Km values. In the present work the asparagine residue at 202 position was replaced by serine, an alteration which can affect substrate orientation and control substrate preference for the lyase reaction. First, we report studies of solvent isotope effects for the N202S CYP17A1 mutant in the presence of 17OH-PREG and 17OH-PROG, which suggest that the ferric peroxo- species is the predominant catalytically active intermediate in the lyase step. This conclusion is further supported by employing a combination of cryoradiolysis and resonance Raman techniques to successfully trap and structurally characterize the key reaction intermediates, including the peroxo-, hydroperoxo- and the crucial peroxo-hemiketal intermediate. Collectively, these studies clarify the role of the N202 residue in manipulating substrate orientation, inducing active site structural changes that can potentially alter the H-bonding interactions with the key Fe-O-O fragment and the degree of protonation of the reactive ferric peroxo intermediate, thereby impacting lyase efficiency.
Keywords: Cytochrome P450, Human steroid metabolism, catalytic mechanism, CYP17A1
Graphical Abstract

Introduction
The multifunctional human cytochrome P450 CYP17A1 plays a central role in the steroidogenic pathway, catalyzing the 17α-hydroxylation of pregnenolone and progesterone and the subsequent 17,20 carbon-carbon bond scission of these hydroxylated products, referred to as lyase activity, to form dehydroepiandrosterone (DHEA) and androstenedione (AD) (Figure 1A).1–2 Hydroxylation and carbon-carbon bond cleavage represent multiple reactivities of CYP17A1, defining a key branch point at which steroid hormone precursors are directed towards corticoid production or committed to formation of androgens and estrogens.3 These two reactivities of CYP17A1 are present in the both the pregnenolone and progesterone pathways in human steroidogenesis and are essential for both glucocorticoid and androgen production. Consistent with its functional significance, polymorphisms affecting the activity of CYP17A1 are deleterious to normal human physiology, while CYP17A1 deficiency has been known to manifest as symptoms of hypertension4 and abnormal sexual development in afflicted individuals.5 Because of its key role in the generation of sex hormones, CYP17A1 has become a front-line target for inhibition in the treatment of androgen receptor positive prostate cancer.5–6 Publication of the first crystal structure of CYP17A1 with the inhibitor abiraterone acetate by Scott’s group was an important breakthrough,7 together with structures of CYP17A1with native substrates.8
Figure 1.
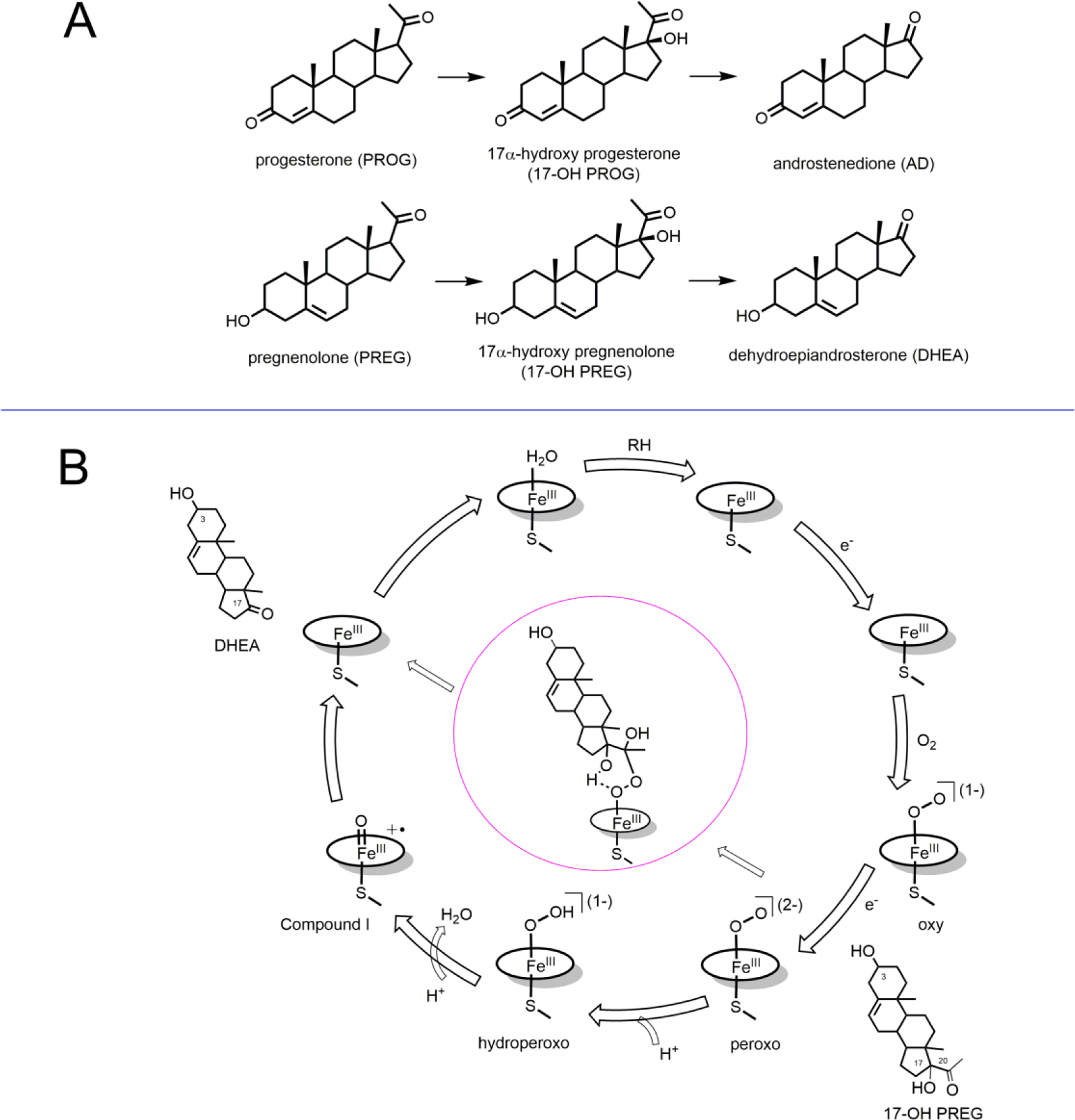
(A) Structure of substrates and products involved in human cytochrome P450 CYP17A1 lyase reactions. (B) Cytochrome P450 enzymatic cycle and the pathway of formation of peroxo hemiketal by attaching on C20 of the substrate (17OH-PREG).
Mechanistic understanding of the lyase reaction catalyzed by CYP17A1 requires identifying the main pathway for product formation between two alternative possibilities. As shown in Figure 1B, one is the major catalytically active intermediate in P450 catalysis, the ferryl heme complex pi-cation radical, so-called Compound I.9–10 Being a strong oxidant, this intermediate is always considered as the most probable candidate responsible for P450 catalysis. Another pathway which involves C-C bond scission by direct attack of (unprotonated) peroxo-complex on a susceptible carbon site to form a ferric hemiketal intermediate (center, Figure 1B), as first suggested by Akhtar and coworkers,3, 11–13 currently is getting support from recent spectroscopic and functional studies. Resonance Raman, combined with cryoradiolysis, provided the first characterization of a trapped peroxo-hemiketal intermediate formed by peroxo-ferric complex and 17OH-PREG14 and 17OH-PROG15 in the wild-type (WT) CYP17A1 (Figure 1B). These results, together with the inverse hydrogen/deuterium (H/D) solvent isotope effect observed for these reactions in steady-state kinetics,15–16 strongly suggested the ferric-peroxo intermediate as the predominant catalytically active intermediate and the absence of or diminished role for the Compound I intermediate. Detailed QM/MM analysis of lyase catalytic mechanism also suggested a preferred pathway via the peroxo-ferric intermediate followed by formation of a hemiketal transitional complex and also pointed out the absence of a transition state in the abstraction of the allylic 17-hyrdroxyl hydrogen.17
The major challenge for using CYP17A1 inhibitors in clinical treatment is in targeting only the carbon-carbon cleavage or “lyase” step, while keeping the first hydroxylation step mostly unperturbed. This goal could be accomplished by developing more selective inhibitors, and some success has been recently achieved.18 Still, more selective inhibitors are needed in order to minimize the side effects of such treatment, and evaluation of critical structural features of substrate binding is one of the most important steps towards this goal.
A useful comparison of the functional properties of CYP17A1 from several mammalian sources was published by Usanov’s group,19 documenting an important structural feature strongly correlated with preference for the pregnenolone (PREG) verses progesterone (PROG) pathways.20 Human CYP17A1, together with bovine, bison and cat enzymes, have asparagine at position-202 (N202) and show a preference for 17OH-PREG in the lyase reaction, while guinea pig, horse, rat and African frog have various amino acids at this position and prefer 17OH-PROG as a substrate.19 This selectivity can be attributed to the hydrogen-bonding properties of the residue at 202 position. X-ray structures of CYP17A1 with all of the native substrates show the C-3 substituents (-OH in PREG or =O in PROG) within H-bonding distance with the N202 side chain in the human CYP17A1.8 Based on these observations, we mutated this N202 residue to serine, and evaluated the spectroscopic and functional properties of the dioxygen adduct of this N202S CYP17A1.21 Replacement of asparagine for serine caused reversal in product turnover rates, as reproduced in Table 1.
Table 1:
Kinetic Data; Reprinted with Permission from ref (21). Copyright 2018, American Chemical Society
| wild type | N202S | |
|---|---|---|
| Ks 17OH-PREG (µmol−1) | 0.17 | 1.5 |
| Ks 17OH-PROG (µmol−1) | 0.55 | 0.053 |
| Kcat 17OH PREG (min−1) | 0.41 | 0.12 |
| Kcat 17OH-PROG (min−1) | 0.20 | 0.49 |
| Kcat/Ks 17OH-PREG (min−1 µmol−1) | 2.41 | 0.08 |
| Kcat/Ks 17OH-PROG (min−1 µmol−1) | 0.36 | 9.25 |
In addition, resonance Raman spectra of the oxy-complex of the N202S mutant measured in the presence of these substrates indicated important perturbations in hydrogen-bonding pattern for both substrates. In order to gain further insight into the structural details critical for the substrate specificity and efficient lyase catalysis in CYP17A1, we performed further spectroscopic and functional studies of the catalytic intermediates in the N202S mutant which was observed to switch preference for Δ-4 and Δ-5 substrates.21 We compare resonance Raman spectra of the cryoreduced dioxygen complexes (peroxo states) and the steady-state kinetic properties of the mutant with both hydroxylation substrates and with the lyase substrates, 17OH-PROG and 17OH-PREG. We also measured the kinetic solvent isotope effect (KSIE) in this mutant in order to help define a role for the peroxo-ferric intermediate in lyase catalysis by this cytochrome P450.
Materials and Methods
Expression, purification and Nanodisc incorporation of N202S CYP17A1.
The expression of human CYP17A1, purification and assembly into Nanodiscs is described in several published works.16, 21–22
Activity KSIE measurements.
17OH-PREG lyase activity was quantitated in a reconstituted system containing 200 picomoles CYP17A1 in POPC Nanodiscs, and a four-fold molar excess of cytochrome b5 and CPR in 1 mL of 0.1 M potassium phosphate buffer, pH/pD 7.4 containing 50 mM NaCl and 50 µM 17OH-PREG (Millipore-Sigma). All the components were mixed and incubated for 20–30 minutes at 37°C; the reaction was initiated by addition of 200 nmol NADPH. The reactions were quenched after 10 minutes by addition of 50 µL of 9M H2SO4 and were stored at −80 °C until extraction. Solutions were thawed and 800 µL was delivered to a test tube and steroid product extracted via double extraction with 2 mL dichloromethane. Extracted steroids were dried under a stream of N2 and reconstituted in 1.5 mL 80% MeOH/H2O. The resultant mixture was purified via a second extraction using 750 µL of pentane and the MeOH/steroid layer was removed and evaporated to dryness under vacuum. The samples were then resuspended in 100 µl of N-Methyl-N-(trimethylsilyl) trifluoroacetamide, incubated at 60°C for 1 hour and analyzed on Agilent 7890A GC/5975C MS (GC/MS) instrument using deuterated DHEA (dehydroepiandrosterone-[d6], Isosciences, Ambler, PA) as an internal standard. 17OH-PROG lyase activity was measured using similar reaction mixtures under the same conditions, with 17OH-PROG (Millipore-Sigma) as a substrate. After quenching, products were extracted in dichloromethane, dried under a gentle stream of N2 and reconstituted in methanol prior to analysis by reversed phase HPLC using a C18 column (ACE). Separation was achieved using a linear gradient of 90:5:5 to 10:45:45 H2O/methanol/acetonitrile, and products were quantitated at 240 nm.
Preparation of samples for rR spectroscopy.
Samples contained 275 μM N202S CYP17A1 Nanodiscs in 100 mM potassium phosphate, pH=7.4; 250 mM sodium chloride; 30% (v/v) distilled glycerol; and 475 μM substrate, (PROG, PREG, 17OH-PROG or 17OH-PREG). The ferric samples were transferred into NMR tubes, deoxygenated under argon and reduced with a 1.5-fold molar excess of sodium dithionite in presence of 6.87 μM methyl viologen. Each sample was cooled for 1 min at −15°C in a dry ice-ethanol bath. Oxy-ferrous forms were generated by bubbling with 16O2 or 18O2 for 5s, followed by rapid freezing in liquid N2. Resonance Raman measurements, albeit of somewhat low signal-to-noise (S/N), were performed in order to verify formation of the dioxygen adducts. Frozen samples containing oxy-ferrous N202S CYP17A1 were subsequently radiolytically reduced at the Notre Dame Radiation Lab to the peroxo-state by a ~ 3 MRad dose of γ-rays in a Gammacell 200 60Co source for 5 hours while immersed in liquid nitrogen. Interrogation of initial intermediates generated at 77K was performed as described below at 77K. In order to generate later intermediates, annealing was carried out by immersing the sample tube containing irradiated CYP17A1 N202S into a pentane/liquid nitrogen bath at 165K for 1 minute, then quickly returning (a few seconds) to the liquid nitrogen bath.
Resonance Raman Measurements.
Samples of irradiated oxy ND: N202S CYP17A1 were acquired with either the 441.6 nm line from a He-Cd laser (IK Series He−Cd laser, Kimmon Koha Co., Ltd.) or the 406.7 nm line from a Kr+ laser (Coherent Innova Sabre Ion Laser). All spectra were measured using a Spex 1269 spectrometer equipped with a Spec-10 LN liquid nitrogen-cooled detector (Princeton Instruments, NJ). The slit width was set at 150 μm using a 1200/groove/mm grating; with this combination, the resultant spectral dispersion is 0.46 cm−1/pixel. The laser power incident on samples was ~1 mW and the NMR tubes were spun to avoid laser-induced heating and photodissociation. All measurements were done at 77K by immersing the tube in a double-walled quartz which is filled with liquid nitrogen. The total collection time for each spectrum is 7 hrs. Spectra were calibrated with data acquired for fenchone (Sigma-Aldrich, Wisconsin) and processed with Grams/32 AI software (Galactic Industries, Salem, NH). Note that, owing to the complexity of spectral data acquired for these types of measurements, where strongly enhanced heme modes also occur in the regions where internal modes of the Fe-O-O fragments are observed, the most effective way to visualize the targeted ν(Fe-O) and ν(O-O) modes is to generate [16O2-18O2] difference traces by subtracting the two absolute spectral traces, where all non-shifted features (i.e., those not associated with the Fe-O-O fragment) effectively cancel.
Results
Kinetic Solvent Isotope Effect in Lyase Catalysis by N202S CYP17A1.
In order to probe the mechanistic origin of C-C bond scission catalyzed by the N202S mutant CYP17A1 with 17OH-PREG and 17OH-PROG as substrates, we compared the rates of catalysis in H2O and D2O buffers and calculated KSIE as a ratio (kH/kD) of these rates. Previously it was shown that KSIE serves as an important mechanistic probe of proton-dependent enzymatic catalysis in P450 enzymes15, 23–28 as well as in other enzymes and synthetic catalysts.29–31 Comparison of catalytic rates measured in H2O and D2O buffers provides a direct method for distinction between catalytic pathways that involve solvent dependent protonation and those that do not require protons from solvent. Formation of the high-valent ferryl intermediate (Compound 1), which is the main catalytic intermediate in most of P450 reactions, involves two protonation events and thus results in a “normal”, but masked, KSIE in the range of 1.3 – 3, typically observed in these reactions.24–26 However, if product formation follows an alternative pathway via peroxo-anion and peroxo-hemiketal intermediate, where protonation is not required, the reaction is faster in D2O than in H2O and an inverse KSIE is observed.15–16, 32 Here the proton dependent uncoupling pathway, involving distal oxygen protonation and release of hydrogen peroxide or cleavage and subsequent reduction of Compound 1 to water, is slowed in the presence of D2O resulting in an increased flux through the product forming channel. Comparison of the rates of product formation catalyzed by the N202S mutant of CYP17A1 revealed an inverse KSIE measured with both substrates; i.e. 0.39±0.03 with 17OH-PREG and 0.71±0.01 with 17OH-PROG. These numbers are close to the earlier results obtained for the WT CYP17A1 with the same substrates 0.3916 and 0.81.15 Interestingly, with 17OH-PROG the inverse KSIE is slightly more pronounced in the N202S mutant, suggesting more favorable positioning of this substrate for peroxo-driven catalysis due to the serine mutation at 202 position. This observation is consistent with our earlier spectroscopic and functional studies of N202S mutant.21
Structural Definition of Intermediates by resonance Raman spectroscopy.
The rR difference traces acquired for the cryoreduced oxy complex of the PROG bound sample of N202S CYP17A1 are shown in Figure 2. Trace A, acquired for samples prepared at 77K, exhibits two oxygen isotope sensitive modes in the 700–800 cm−1 region. One occurs at 802 cm−1 and shifts to 764 cm−1 upon 18O2 substitution, yielding an isotope shift of 38 cm−1, a value entirely consistent with a ν(O-O) stretching vibration; based on earlier studies of these P450 enzymatic intermediates, which included studies performed with deuterated water,14–15, 33 this mode shows no sensitivity to replacement of H- with D and is most reasonably assigned to a peroxo-intermediate. The corresponding ν(Fe-O) mode is expected in the region where the 553 cm−1 feature is observed and is seen to shift to 526 cm−1 for the 18O2 sample, as expected. Another ν(16O-16O) mode is seen at 775 cm−1, shifting by 38 cm−1 for the 18O2 samples, an isotope shift consistent with theory. It is noted that the corresponding ν(Fe-16O) mode occurs at 571 cm−1 and exhibits a 27 cm−1 shift to lower frequency with 18O substitution, a shift consistent with that expected for a Fe-O harmonic oscillator. Again, based on results of many studies of these types of samples, including studies in H2O and D2O,14–15 this set of ν(Fe-O) and ν(O-O) modes is quite consistent with the presence of a hydroperoxo- species. In summary, these spectral results document the formation of both the peroxo- and hydroperoxo- intermediates upon cryoradiolytic reduction of the dioxygen adduct of PROG bound N202S CYP17A1. The observation of a peroxo-intermediate for this PROG bound sample of the mutant protein, contrasts with the spectral results seen for the PROG bound sample of the wild-type (WT) protein, where no evidence was found for the presence of the initially formed peroxo-intermediate, with protonation to form the hydroperoxo- species occurring even at 77K; i.e., for the PROG bound intermediate, protonation of the ferric peroxo- intermediate is apparently less efficient for the N202S mutant than for the WT protein.
Fig 2.

The rR 16O2-18O2 difference trace irradiated dioxygen adducts of PROG bound CYP 17A1(N202S). Spectra were measured with 442.1nm excitation at 77K. Note that the peak near 674 cm−1 arises because of slightly different concentrations of the 16O2 and 18O2 intermediates, resulting in a residual heme internal mode in the positive side of the difference trace.
Turning attention to the spectra obtained for the N202S CYP17A1 sample bound with the lyase substrate, 17OH-PROG (trace A of Figure 3), two intermediates, the peroxo- and hydroperoxo- forms, were trapped at 77K, with each exhibiting a set of oxygen isotope sensitive bands in the expected region. The peroxo- species exhibits a ν(16O-16O) mode at 789 cm−1 with its ν(Fe-16O) mode being seen at 562 cm−1; the 18O isotope shifts to 751 cm−1 and 536 cm−1, respectively, are in reasonable agreement with expectations. The hydroperoxo- species shows a positive band at 770 cm−1, which downshifts by 38 cm−1 upon 18O substitution. In the lower frequency region, the associated ν(Fe-16O) occurs at 576 cm−1, with a ν(Fe-18O) mode shifting, as expected, by ~25 cm−1 to 551 cm−1.
Fig 3.

The rR 16O2-18O2 difference traces of irradiated dioxygen adducts of 17OH-PROG bound CYP17A1(N202S). (A) sample irradiated at 77K measured with 441.6 nm excitation line (B) sample after annealing to 165 K measured with 441.6 nm excitation line and (C) sample after annealing to 165 K measured with 406.7 nm excitation line. Note that the peak near 674 cm−1 arises because of slightly different concentrations of the 16O2 and 18O2 intermediates, resulting in a residual heme internal mode in the positive side of the difference trace.
Earlier studies of the WT CYP17A1 enzyme bound with the lyase substrates, employing 406 nm excitation, documented a set of vibrational modes that emerged upon annealing to higher temperatures, which are associated with a new “peroxo-like” intermediate, previously identified as the central peroxo-hemiketal species in Figure 1.18–19 Similar studies conducted here for the N202S mutant are shown in Figure 3. Trace B of Figure 3 exhibits the (16O2-18O2) difference spectrum for the 17OH-PROG bound sample annealed to 165K, using the 442 nm excitation line of the krypton laser. Here it is seen that the peroxo- intermediate has virtually disappeared, leaving only weak difference bands associated with small amounts of the hydroperoxo-intermediate, the ν(O-O) and ν(Fe-O) modes exhibiting the same frequencies as seen for the 77K sample. In the case of the WT enzyme, it had been shown that the disappearance of the peroxo-species upon annealing to 165K and higher was accompanied by the appearance of a new intermediate whose ν(Fe-O) and ν(O-O) modes, appearing at 573 cm−1 and 785 cm−1, were effectively enhanced with laser excitation at 406 nm, rather than 442 nm. Consequently, similar experiments, using the 406 nm excitation line of the krypton ion laser were conducted for the N202S mutant, the resulting (16O2-18O2) difference spectrum for the 17OH-PROG bound sample that had been annealed to 165K being shown in trace C of Figure 3 where it is seen that the ν(16O-16O) and ν(Fe-16O) modes of a new intermediate are observed at 781 cm−1 and 575 cm−1. These display the expected 16O/18O isotopic shifts and the difference trace also exhibits features at 1131/1066 cm−1 and 530/501 cm−1 that are associated with residual ferrous dioxygen (oxy) CYP17A1 that was not reduced during the exposure to the γ-ray source. As was explained previously,14–15 this new species, whose internal modes of the Fe-O-O- fragment are effectively enhanced with 406 nm excitation, is most reasonably attributed to the peroxo-hemiketal formed by attack of the initial peroxo-intermediate at the C20 carbonyl group of the lyase substrate.
Figure 4 displays the rR-enhanced difference trace obtained for the PREG bound N202S CYP17A1 samples. It shows two clear positive bands at 800 cm−1 and 777 cm−1 in the higher frequency region, both shifting as expected (39 and 38 cm−1) upon 18O2 substitution. These are most reasonably assigned to the ν(O-O) modes of the peroxo- and hydroperoxo- intermediates, respectively. The corresponding ν(Fe-O) modes are seen at 557 cm−1 (peroxo-) and 573 cm−1 (hydroperoxo-) with the expected isotopic shifts for the 18O2 containing samples, the overall behavior of the spectral patters being almost identical with that seen for the WT enzyme in the presence of PREG.
Fig 4.
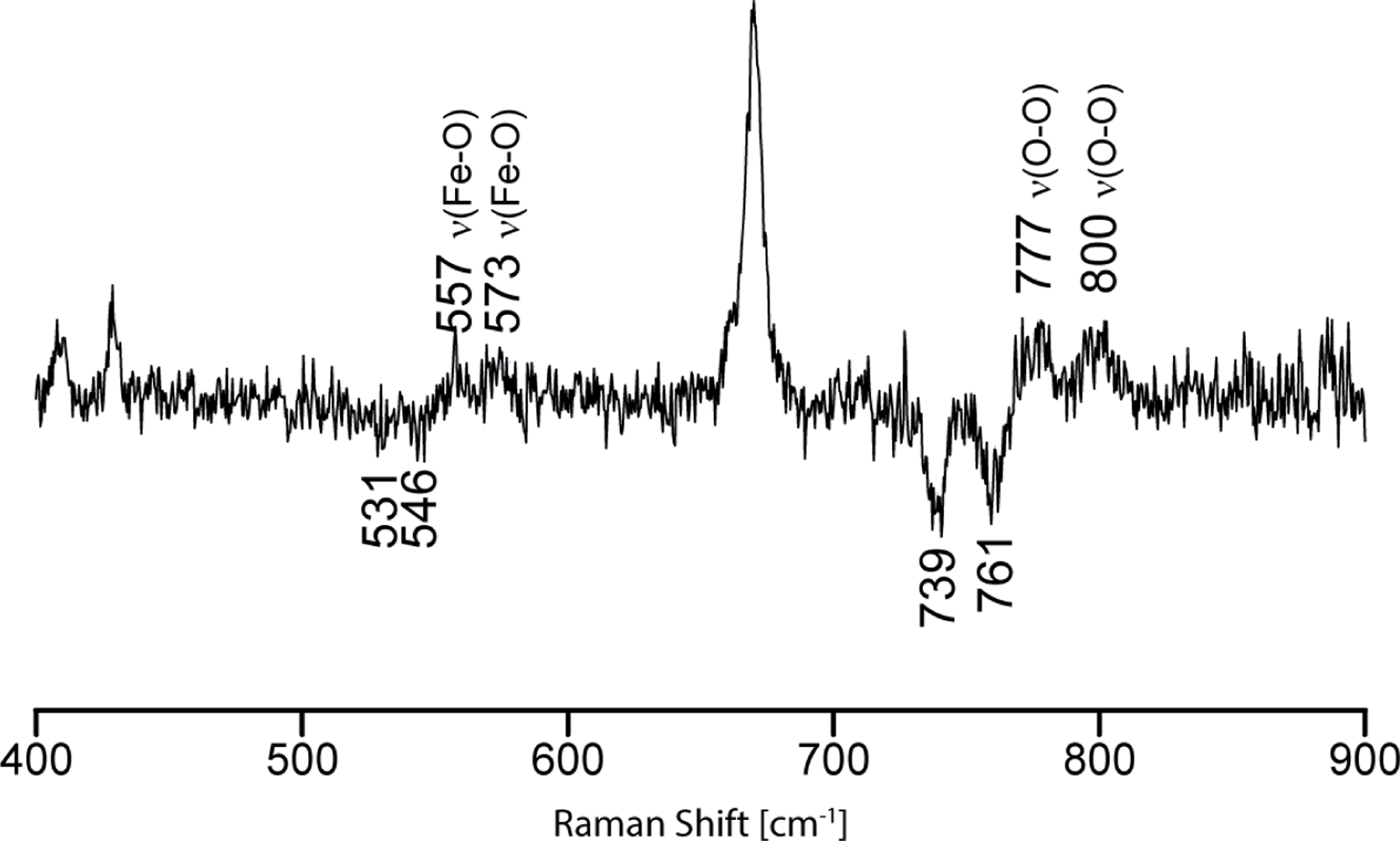
The rR 16O2-18O2 difference trace for irradiated dioxygen adducts of PREG bound CYP17A1(N202S). Spectra were measured with 442.1nm excitation line at 77K. Note that the peak near 674 cm−1 arises because of slightly different concentrations of the 16O2 and 18O2 intermediates, resulting in a residual heme internal mode in the positive side of the difference trace.
The rR spectra obtained for the cryoreduced sample of N202S CYP17A1 bound with the lyase substrate, 17OH-PREG, are shown in Figure 5. In the difference trace a ferric peroxo-intermediate was confirmed by observation of the ν(16O −16O) mode at 796 cm−1 (Δ16/18= 38 cm−1), while its ν(Fe-16O) mode is seen at 545 cm−1 and shifts down by 25 wavenumbers upon 18O2 substitution (Δ16/18= 25 cm−1). Interestingly, evidence for a hydroperoxo species is also obtained, with a ν(16O −16O) mode at 779 cm−1 (Δ16/18= 41 cm−1) and a ν(Fe −16O) mode at 562 cm−1 (Δ16/18= 27 cm−1). Thus, unlike the WT protein, which showed no evidence for formation of a hydroperoxo-intermediate, the N202S mutant forms substantial amounts of this species. One initially puzzling aspect of these data for the 17OH-PREG bound mutant, is that the ν(Fe-O) mode for the hydroperoxo-intermediate occurs about 10 cm−1 lower than those seen for the other substrates (571–576 cm−1). However, it must be recalled that the ν(Fe-O) mode for the peroxo- intermediate of both the WT and mutant proteins, appears 8–12 cm−1 lower than that for the PREG bound sample as a consequence of the 17OH fragment providing an H-bonding interaction to the proximal oxygen of the Fe-Op-Ot fragment. Clearly, the data acquired suggest that this proximal H-bond persists upon protonation of the terminal oxygen to form the hydroperoxo-intermediate.
Fig 5.

The rR 16O2-18O2 difference traces of irradiated dioxygen adducts of 17OH-PREG bound CYP17A1(N202S). (A) sample irradiated at 77K measured with 441.6 nm excitation line (B) sample after annealing to 165 K measured with 441.6 nm excitation line and (C) sample after annealing to 165 K measured with 406.7 nm excitation line. Note that the peak near 674 cm−1 arises because of slightly different concentrations of the 16O2 and 18O2 intermediates, resulting in a residual heme internal mode in the positive side of the difference trace.
As was the case for the sample bound with the 17OH-PROG, rR spectra were acquired, after annealing to 165K, of the cryoreduced sample of the oxy form of the N202S mutant of CYP17A1 bound with 17OH-PREG. The difference trace (Figure 5.B) acquired for the annealed sample, employing 442 nm excitation, which enhances only the modes of the initially formed peroxo- and hydroperoxo- intermediates, shows that the peroxo-intermediate has disappeared, leaving only weak difference features observed at 778/738 cm−1, which are associated with the ν(O-O) mode of the hydroperoxo-species; the ν(Fe-O) modes of this sample were too weak to be observed. As is the case of the WT enzyme,14–15 when employing the 406 nm excitation line for this annealed sample (Figure 5, trace C), a new intermediate is seen to have formed, exhibiting its ν(O-O) and ν(Fe-O) difference features at 786/744 cm−1 and 576/549 cm−1. These spectral features are consistent with attack of the initially formed peroxo-intermediate at the C20 carbonyl of the 17OH-PREG, giving rise to the peroxo-hemiketal species depicted in Figure 1, which eventually converts to the DHEA product.14–15
Discussion
CYP17A1 effectively catalyzes two distinct types of reactions, the hydroxylation of pregnenolone (PREG) and progesterone (PROG) via a Compound 1 intermediate and a C-C bond cleavage (lyase) conversion of these 17-hydroxylated substrates, 17OH-PREG and 17OH-PROG. Previous studies of the WT protein provided significant evidence that these lyase reactions occur via attack of a reactive peroxo- intermediate on the susceptible C20 carbonyl of the lyase substrates.14–15 Of special interest is the fact that the efficiency for lyase activity for OH PREG is much greater than that for 17OH-PROG,19 prompting us, and others, to seek a structural basis for this differential reactivity.3, 11, 14–15, 22, 34–35
In a peroxo-anion mediated lyase reaction the nucleophilic reactivity of the Fe-Op-Ot is lowered if the terminal oxygen (Ot) is tied up in a hydrogen bond.17 As shown in this and earlier works,15–18, 21, 36–39 this critical interaction is quite effectively revealed by rR spectroscopy, with H-bonding to the Op causing an ~ 10 cm−1 decrease in the ν(Fe-O) frequency, relative to that for a non-H-bonded peroxo-fragment. Conversely, an increase of about ~10 cm−1 is observed for H-bonding to the terminal oxygen Ot.14–15, 22, 39 In addition to the importance of the inherent reactivity of the ferric peroxo-intermediate, another important factor affecting the lyase reaction is the efficiency of protonation of the active site Fe-O-O peroxo fragment, producing the hydroperoxo-intermediate (Fe-O-O-H), which would effectively divert the peroxo-intermediate from the lyase cycle. It is important to note here that protonation of the active Fe-O-O fragment occurs via the proton shuttle network present in all cytochromes P450,40 typically involving an acid/alcohol pair. It is noted that in the case of CYP17A1, this acid/alcohol assembly is associated with the E305 and T306 residues.41–42 Again, rR spectroscopic studies of these cryoradiolytically generated intermediates are well suited to document this process, providing direct interrogation of the extent of protonation of the ferric-peroxo species via the measured relative intensities of the peroxo- and hydroperoxo- rR signals. Obviously, yet another factor controlling the efficiency of the lyase reaction is the precise distance between the nucleophilic terminal oxygen (Ot) and the susceptible electrophilic C20 carbonyl carbon. Unfortunately, this is one factor that is not revealed by the RR technique. In this context, it is useful to compare the rR data acquired for the N202S mutant and WT protein.
All of the rR spectral data acquired here for the cryoreduced samples of the oxy complexes of the N202S mutant of CYP17A1, along with those previously obtained for the WT enzyme, are summarized in Table 2. Also, the spectral difference traces in the ν(O-O) region are displayed in Figure 6; for clarity, color-coded simulated spectra are employed, which faithfully reproduce the experimental data obtained for these systems.14–15 Inspection of Table 2 shows that the peroxo intermediate of the 17OH-PREG bound WT protein exhibits a ν(Fe-O) mode whose frequency is shifted down by 8 cm−1 compared to that seen for the non-H-bonding PREG sample, confirming the presence of an H-bond to the proximal oxygen of the Fe-Op-Ot fragment, whereas that seen for the 17OH-PROG bound sample shifts to higher frequency by approximately 8 cm−1 compared to that expected for the PROG bound sample, behavior reflecting an H-bonding interaction to the terminal oxygen (Ot). These rR spectroscopic results revealing differential H-bonding schemes for the two lyase substrates are also supported by crystallographic studies by Scott and coworkers,8 which indicate that 17OH-PROG is positioned higher in the pocket, consistent with H-bonding of the 17OH fragment to the terminal oxygen of the Fe-Op-Ot fragment, while 17OH-PREG can be situated closer to the Fe-O-O fragment, positioning the 17OH fragment for an H-bond to the proximal oxygen of the Fe-O-O fragment. Further support for this view is the fact that such relative positioning of the two hydroxylase substrates, PREG and PROG, is also likely (vide infra), and would be consistent with the observation that hydroxylation of PROG yields about 20–25% of the alternate product, 16OH-PROG, whereas PREG hydroxylation yields no traces of 16OH-PREG.43–44
Table 2.
Summary of the ν(Fe–O) and ν(O–O) Modes in peroxo and hydroperoxo forms for Wild-Type (WT)and N202S CYP17A1.
| WT CYP17A1 | |||||
|---|---|---|---|---|---|
| PROG | PREG | 17OH-PROG | 17OH-PREG | ||
| Peroxoanion | ν(O-O) (cm−1) | 802 | 790 | 796 | |
| ν(Fe-O) | [554]a | 554 | 562 | 546 | |
| hydroperoxo | ν(O-O) | 772 | 775 | 771 | none |
| ν(Fe-O) | 575 | 572 | 576 | ||
| New intermediate | ν(O-O) (cm−1) | NA | NA | 785 | 791 |
| ν(Fe-O) | NA | NA | 573 | 579 | |
|
N202S CYP17A1 | |||||
| PROG | PREG | 17OH-PROG | 17OH-PREG | ||
| Peroxoanion | ν(O-O) (cm−1) | 802 | 800 | 789 | 796 |
| ν(Fe-O) | 553 | 557 | 562 | 545 | |
| hydroperoxo | ν(O-O) | 775 | 777 | 770 | 779 |
| ν(Fe-O) | 571 | 573 | 576 | 562 | |
| New intermediate | ν(O-O) (cm−1) | NA | NA | 781 | 786 |
| ν(Fe-O) | NA | NA | 575 | 576 | |
The peroxo intermediate was not trapped; however, work with the dioxygen complexes of PROG bound and PREG bound CYP17A1, showed virtually identical ν(Fe-O) stretching frequencies. Thus, the frequency observed for the peroxo- intermediate of PREG bound enzyme is listed as “expected” here.
Figure 6.

Summary of different intermediates trapped during irradiation and annealing process in CYP17A1 wild-type and N202S mutant, in the presence of 17OH-PROG and 17OH-PREG. Note: spectra were simulated by employing 50%/50% Lorentzian and Gaussian function.
Turning attention to the rR spectral data provided in Table 2 for the N202S mutant, it is seen that the observed behavior of the ν(Fe-O) and ν(O-O) modes of the peroxo- intermediate of the mutant are virtually identical to those observed for the WT protein; i.e., the ν(Fe-O) shifts down (by ~10 cm−1) for the 17OH-PREG bound sample, with that for the 17OH-PROG bound sample shifting up by ~10 cm−1. It is noted that this behavior contrasts with earlier work reporting the rR spectral results for the dioxygen adducts of the N202S mutant,21 which provided evidence for altered H-bonding interactions with the Fe-O-O fragment of this ferrous-dioxygen (oxy) complex, exhibiting an upshift of ν(Fe-O) for the 17OH-PREG substrate and a significant downshift for the 17OH-PROG sample, the essential result being that the ν(Fe-O) modes for the two lyase substrates have the same frequency. The present results show that, for these peroxo-intermediates, the H-bonding patterns are not significantly altered for the mutant; i.e., a significant downshift for 17OH-PREG and a significant upshift for 17OH-PROG bound mutant compared to the samples with the hydroxylase substrates.14–15 The most reasonable interpretation of this differing behavior is that the mutation induced a shift of substrate positioning, which results in a general weakening of H-bonding to the superoxide fragment of the Fe-O-O linkage in the oxy intermediate; however, stronger H-bonding interactions are expected for the more nucleophilic peroxo-intermediate, with rR spectroscopy showing them to be quite similar to those within the WT protein.
Clearly, multiple factors are important, including the dynamic positioning of the susceptible C20 carbonyl atom for each substrate and the relative efficiency of protonation of the reactive Fe-O-O peroxo- fragment. Fortunately, though the technique is “silent” regarding the precise positioning of the carbonyl fragment, rR detection can provide useful information regarding the degree of protonation of the ferric-peroxo intermediate. In the spectral traces depicted in panels A and B of Figure 6, it is seen that the initial result of cryoradiolysis (77K) of the oxy adduct of wild type CYP17A1 bound with 17OH-PROG is the generation of the peroxo-intermediate, accompanied by some conversion, even at 77K, to the hydroperoxo-intermediate. In contrast, at this temperature, the sample formed with 17OH-PREG showed no evidence for generation of the hydroperoxo-intermediate. Furthermore, upon annealing of both samples to 165K, facilitating proton transfer,45–48 the spectral data for the 17OH-PROG sample provide evidence for conversion of the peroxo- intermediate to more of the hydroperoxo-intermediate, whereas the sample bound with 17OH-PREG indicates further loss of the peroxo-intermediate, but still no evidence for generation of the hydroperoxo-species. Inasmuch as the initially generated peroxo-fragment (Fe-O-O) can react with the susceptible C20 carbonyl or be diverted from the lyase reaction by protonation to form the hydroperoxo- species, this lack of hydroperoxo- formation for the 17OH-PREG bound sample can be reasonably considered to reflect the relatively greater reactivity of the Fe-O-O fragment; i.e., the driving force for reaction with the C20 carbonyl overriding the inherent tendency for protonation of the terminal oxygen of the Fe-O-O fragment. On the other hand, the failure to form the hydroperoxo- intermediate in the presence of 17OH-PREG could also be ascribed to a possible decrease in protonation efficiency for this substrate relative to that for the 17OH-PROG bound species.
Turning attention to the corresponding rR spectral data acquired with the N202S mutant, shown in panels C and D of Figure 6, the most noticeable change compared to the WT protein (panels A and B) is that the sample bound with 17OH-PREG now provides evidence that the peroxo-intermediate readily converts to the hydroperoxo-species even at 77K and continues to generate more of this species at 165K, leaving no trace of the peroxo-intermediate. The frequency data compiled in Table 2 strongly suggest that the mutation caused no substantial changes in the H-bonding interactions with the Fe-O-O fragment compared to the WT enzyme, and therefore rather insignificant differences in the relative nucleophilicity of the terminal oxygen of the Fe-O-O fragment. Therefore, it is most reasonable to attribute the decrease in lyase activity for the 17OH-PREG sample of the mutant to the increased efficiency of protonation, the consequence of which is to divert the reactive peroxo-fragment from lyase reactivity by generating the hydroperoxo- intermediate. In order to gain further insight into the factors controlling the degree of protonation of the ferric-peroxo-intermediate, it is helpful to refer to recent work from Scott and coworkers.35
As illustrated in Figure 7, in the case of the WT sample bound with 17OH-PREG, the 3-OH substituent of the substrate is situated to donate a hydrogen bond to the carbonyl fragment of N202, while the amide fragment of this residue is positioned to interact with the E305 residue. Quite relevant to this issue is the well-established behavior of CYP101 and its D251N mutant, noting that comparison of crystal structures of CYP101 and CYP17A1 indicate similar positioning of the D251/T252 pair with that of the E305/T306 pair of CYP17A1.12, 49–51 The WT CYP101 exhibits very efficient proton transfer to the active site ferric peroxo-intermediate, rapidly forming the hydroperoxo-intermediate even at 77K.52 Replacement of the aspartate carboxylate fragment with the amide fragment of the asparagine residue in the D251N mutant leads to a rather drastic reduction in protonation of the reactive ferric peroxo- intermediate, which is then effectively trapped and characterized.33 Clearly, it is plausible that the association of the amide fragment of N202 of WT CYP17A1 with the 305 glutamate carboxylate plays a similar role in restricting protonation of the ferric peroxo-intermediate in WT CYP17A1 as the D251N replacement in CYP101.53–54
Figure 7.

Proposed schematic of interaction between the C3 alcohol/keto group and the side chain amide of Asn202 and mutated Ser202, and their nearby Glu305 residue.
While efficient protonation of the ferric peroxo-intermediate is essential for promoting well-coupled hydroxylase activity, it is clearly detrimental to lyase reactivity. Referring to Figure 7, when 17OH-PROG is the substrate in the WT CYP17A1 active site, the 3-carbonyl fragment of the substrate accepts a hydrogen bond from the amide fragment of the N202 residue, thereby removing the asparagine amide interaction with the E305 residue, leading to more facile protonation of the Fe-O-O fragment, resulting in substantially decreased lyase efficiency for 17OH-PROG relative to 17OH-PREG. Supporting this argument, it is noted that this decreased efficiency for hydroperoxo-formation for the 17OH-PREG sample of the WT enzyme, relative to the 17OH- PROG bound sample, would be expected to be mirrored for the corresponding PREG and PROG bound samples. This expectation is borne out in the rR spectra of the cryoreduced samples of the WT enzyme, published elsewhere.14–15 There it was shown that while substantial amounts of the peroxo- intermediate are observed for the PREG- bound sample,14 virtually no amount of peroxo is observed for the PROG bound sample, even at 77K, with only the hydroperoxo intermediate being observed.15 Furthermore, these latter results are entirely consistent with the reported decrease in hydroxylase activity for PREG, compared to PROG,19 owing to more facile formation of the hydroperoxo intermediate, facilitating Compound I formation, with the latter substrate. In conclusion, in the case of the N202S mutant, the amide fragment is removed, eliminating interference with the likely E305/T306 proton shuttle for the 17OH-PREG bound sample, thereby diminishing lyase efficiency, an observation consistent with the rR documented increase in the generation of the hydroperoxo- intermediate for the 17OH-PREG bound sample.
This analysis accounting for the decreased lyase efficiency for the 17OH-PREG bound N202S mutant, mainly owing to increased protonation of the reactive ferric peroxo-intermediate, does not address the increased lyase efficiency observed for the 17OH-PROG bound mutant. First, it is noted that the rR data show that the ν(Fe-O) stretching mode patterns are comparable for the mutant protein, implying that the inherent nucleophilicity of the Fe-O-O peroxo fragment is not substantially altered. However, comparison of traces A and C in the top panel of Figure 6 reveals that the protonation of the ferric peroxo-fragment is actually greater for the N202S mutant than for the WT, an observation that would argue for decreased, rather than increased lyase efficiency for 17OH-PROG. One possible explanation for the observed increase in lyase efficiency for the 17OH-PROG bound mutant, unfortunately not addressable by the rR spectroscopic studies conducted here, is repositioning of the substrate in a manner which brings the susceptible C20 carbonyl closer to the reactive terminal oxygen of the peroxo fragment.
In conclusion, comparative analysis of the present results acquired for the N202S variant of CYP17A1 with those previously obtained for the WT enzyme provides a more comprehensive picture of the relative lyase activity of the enzyme for the two lyase substrates. The rR studies performed here, and elsewhere for the WT enzyme,14–15 reveal that the mutation does not substantially alter the inherent nucleophilicity of the terminal oxygen of the Fe-O-O fragment of the mutant protein for either lyase substrate, showing relatively unchanged H-bonding patterns for the ν(Fe-O) modes. On the other hand, the mutation does result in a more efficient protonation of the ferric peroxo-intermediate for 17OH-PREG bound mutant compared to the WT protein, owing to the removal of the amide fragment of the N202 residue, which apparently induces a conformational change that alters the proton shuttle. The basis for the observed increase in lyase efficiency for the 17OH-PROG bound mutant is less clear, but possibly attributable to slight rearrangement of the substrate in the active site, which bring the susceptible C20 carbonyl closer to the terminal oxygen of the Fe-O-O peroxo fragment.
Acknowledgments
We acknowledge grant support from the National Institutes of Health, MIRA R35GM118145 (SGS), R01 GM110428 (JRK, SGS) and GM125303 (JRK). The authors thank Dr. Jay A. LaVerne, Notre Dame Radiation Laboratory (Notre Dame University, IN), a facility of the US Department of Energy, Office of Basic Energy Science for his assistance with the cryo-irradiation procedures.
Footnotes
Accession Code
Human CYP17A1: P05093
References
- 1.Nakajin S; Hall PF, Microsomal Cytochrome-P-450 from Neonatal Pig Testis - Purification and Properties of a C21-Steroid Side-Chain Cleavage System (17-Alpha-Hydroxylase-C17,20 Lyase). J. Biol. Chem 1981, 256 (8), 3871–3876. [PubMed] [Google Scholar]
- 2.Nakajin S; Shively JE; Yuan PM; Hall PF, Microsomal Cytochrome-P-450 from Neonatal Pig Testis - 2 Enzymatic-Activities (17-Alpha-Hydroxylase and C-17,C-20-Lyase) Associated with One Protein. Biochemistry 1981, 20 (14), 4037–4042. [DOI] [PubMed] [Google Scholar]
- 3.Akhtar M; Wright JN; Lee-Robichaud P, A review of mechanistic studies on aromatase (CYP19) and 17 alpha-hydroxylase-17,20-lyase (CYP17). J. Steroid Biochem. Mol. Biol 2011, 125 (1–2), 2–12. [DOI] [PubMed] [Google Scholar]
- 4.Ackermann D; Pruijm M; Ponte B; Guessous I; Ehret G; Escher G; Dick B; Al-Alwan H; Vuistiner P; Paccaud F; Burnier M; Pechere-Bertschi A; Martin PY; Vogt B; Mohaupt M; Bochud M, CYP17A1 Enzyme Activity Is Linked to Ambulatory Blood Pressure in a Family-Based Population Study. Am. J. Hypertens 2016, 29 (4), 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sivonova MK; Jurecekova J; Tatarkova Z; Kaplan P; Lichardusova L; Hatok J, The role of CYP17A1 in prostate cancer development: structure, function, mechanism of action, genetic variations and its inhibition. Gen. Physiol. Biophys 2017, 36 (5), 487–499. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y; Sawyers CL; Scher HI, Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 2008, 8 (4), 440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeVore NM; Scott EE, Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012, 482 (7383), 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrunak EM; DeVore NM; Porubsky PR; Scott EE, Structures of Human Steroidogenic Cytochrome P450 17A1 with Substrates. J Biol Chem 2014, 289 (47), 32952–32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshimoto FK; Gonzalez E; Auchus RJ; Guengerich FP, Mechanism of 17 alpha,20-lyase and new hydroxylation reactions of human cytochrome P450 17A1. O-18 labeling and oxygen surrogate evidence for a role of a perferryl oxygen. J. Biol. Chem 2016, 291 (33), 17143–17164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guengerich FP; Yoshimoto FK, Formation and Cleavage of C-C Bonds by Enzymatic Oxidation Reduction Reactions. Chem. Rev 2018, 118 (14), 6573–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akhtar M; Corina D; Miller S; Shyadehi AZ; Wright JN, Mechanism of the Acyl-Carbon Cleavage and Related Reactions Catalyzed by Multifunctional P-450s - Studies on Cytochrome-P-450(17-Alpha). Biochemistry 1994, 33 (14), 4410–4418. [DOI] [PubMed] [Google Scholar]
- 12.Lee-Robichaud P; Akhtar ME; Akhtar M, An analysis of the role of active site protic residues of cytochrome P-450s: mechanistic and mutational studies on 17 alpha-hydroxylase-1 7,20-lyase (P-450(17 alpha) also CYP17). Biochem. J 1998, 330, 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LeeRobichaud P; Kaderbhai MA; Kaderbhai N; Wright JN; Akhtar M, Interaction of human CYP17 (P-450(17 alpha), 17 alpha-hydroxylase-17,20-lyase) with cytochrome b(5): Importance of the orientation of the hydrophobic domain of cytochrome b(5). Biochem. J 1997, 321, 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mak PJ; Gregory MC; Denisov IG; Sligar SG; Kincaid JR, Unveiling the crucial intermediates in androgen production. Proc. Natl. Acad. Sci. U.S.A 2015, 112 (52), 15856–15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mak PJ; Duggal R; Denisov IG; Gregory MC; Sligar SG; Kincaid JR, Human Cytochrome CYP17A1: The Structural Basis for Compromised Lyase Activity with 17-Hydroxyprogesterone. J. Am. Chem. Soc 2018, 140 (23), 7324–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gregory MC; Denisov IG; Grinkova YV; Khatri Y; Sligar SG, Kinetic Solvent Isotope Effect in Human P450 CYP17A1-Mediated Androgen Formation: Evidence for a Reactive Peroxoanion Intermediate. J. Am. Chem. Soc 2013, 135 (44), 16245–16247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonomo S; Jorgensen FS; Olsen L, Mechanism of Cytochrome P450 17A1-Catalyzed Hydroxylase and Lyase Reactions. J Chem Inf Model 2017, 57 (5), 1123–1133. [DOI] [PubMed] [Google Scholar]
- 18.Bonomo S; Hansen CH; Petrunak EM; Scott EE; Styrishave B; Jorgensen FS; Olsen L, Promising Tools in Prostate Cancer Research: Selective Non-Steroidal Cytochrome P450 17A1 Inhibitors. Sci. Rep 2016, 6, 29468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilep AA; Sushko TA; Usanov SA, At the crossroads of steroid hormone biosynthesis: The role, substrate specificity and evolutionary development of CYP17. Biochim Biophys Acta Proteins Proteom 2011, 1814 (1), 200–209. [DOI] [PubMed] [Google Scholar]
- 20.Wada A; Mathew PA; Barnes HJ; Sanders D; Estabrook RW; Waterman MR, Expression of Functional Bovine Cholesterol Side-Chain Cleavage Cytochrome-P450 (P450scc) in Escherichia-Coli. Arch. Biochem. Biophys 1991, 290 (2), 376–380. [DOI] [PubMed] [Google Scholar]
- 21.Gregory MC; Mak PJ; Khatri Y; Kincaid JR; Sligar SG, Human P450 CYP17A1: Control of Substrate Preference by Asparagine 202. Biochemistry 2018, 57 (5), 764–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregory M; Mak PJ; Sligar SG; Kincaid JR, Differential Hydrogen Bonding in Human CYP17 Dictates Hydroxylation versus Lyase Chemistry. Angew. Chem. Int. Ed 2013, 52 (20), 5342–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swinney DC; Mak AY, Androgen Formation by Cytochrome-P450 Cyp17 - Solvent Isotope Effect and Pl Studies Suggest a Role for Protons in the Regulation of Oxene Versus Peroxide Chemistry. Biochemistry 1994, 33 (8), 2185–2190. [DOI] [PubMed] [Google Scholar]
- 24.Vidakovic M; Sligar SG; Li HY; Poulos TL, Understanding the role of the essential Asp251 in cytochrome P450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Biochemistry 1998, 37 (26), 9211–9219. [DOI] [PubMed] [Google Scholar]
- 25.Xiang H; Tschirret-Guth RA; de Montellano PRO, An A245T mutation conveys on cytochrome P450(eryF) the ability to oxidize alternative substrates. J. Biol. Chem 2000, 275 (46), 35999–36006. [DOI] [PubMed] [Google Scholar]
- 26.Makris TM; von Koenig K; Schlichting I; Sligar SG, Alteration of P450 distal pocket solvent leads to impaired proton delivery and changes in heme geometry. Biochemistry 2007, 46 (49), 14129–14140. [DOI] [PubMed] [Google Scholar]
- 27.Pearl NM; Wilcoxen J; Im S; Kunz R; Darty J; Britt RD; Ragsdale SW; Waskell L, Protonation of the Hydroperoxo Intermediate of Cytochrome P450 2B4 Is Slower in the Presence of Cytochrome P450 Reductase Than in the Presence of Cytochrome b5. Biochemistry 2016, 55 (47), 6558–6567. [DOI] [PubMed] [Google Scholar]
- 28.Jiang YY; He X; de Montellano PRO, Radical intermediates in the catalytic oxidation of hydrocarbons by bacterial and human cytochrome P450 enzymes. Biochemistry 2006, 45 (2), 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hangasky JA; Saban E; Knapp MJ, Inverse Solvent Isotope Effects Arising from Substrate Triggering in the Factor Inhibiting Hypoxia Inducible Factor. Biochemistry 2013, 52 (9), 1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davydov R; Matsui T; Fujii H; Ikeda-Saito M; Hoffman BM, Kinetic isotope effects on the rate-limiting step of heme oxygenase catalysis indicate concerted proton transfer/heme hydroxylation. J. Am. Chem. Soc 2003, 125 (52), 16208–16209. [DOI] [PubMed] [Google Scholar]
- 31.Dunford HB; Hewson WD; Steiner H, Horseradish-Peroxidase .29. Reactions in Water and Deuterium-Oxide - Cyanide Binding, Compound-I Formation, and Reactions of Compound-I and Compound-Ii with Ferrocyanide. Can. J. Chem 1978, 56 (22), 2844–2852. [Google Scholar]
- 32.Litzenburger M; Lo Izzo R; Bernhardt R; Khatri Y, Investigating the roles of T224 and T232 in the oxidation of cinnamaldehyde catalyzed by myxobacterial CYP260B1. FEBS Lett 2017, 591 (1), 39–46. [DOI] [PubMed] [Google Scholar]
- 33.Denisov IG; Mak PJ; Makris TM; Sligar SG; Kincaid JR, Resonance Raman Characterization of the Peroxo and Hydroperoxo Intermediates in Cytochrome P450. J. Phys. Chem. A 2008, 112 (50), 13172–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akhtar MW, Neville J., Acyl-Carbon Bond Cleaving Cytochrome P450 Enzymes: CYP17A1, CYP19A1 and CYP51A1. In Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450, Springer International: Switzerland, 2015; pp 107–130. [DOI] [PubMed] [Google Scholar]
- 35.Yadav R; Petrunak EM; Estrada DF; Scott EE, Structural insights into the function of steroidogenic cytochrome P450 17A1. Mol. Cell. Endocrinol 2017, 441 (C), 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y; Grinkova Y; Gregory MC; Denisov IG; Kincaid JR; Sligar SG, Mechanism of the Clinically Relevant E305G Mutation in Human P450 CYP17A1. Biochemistry 2021, 60 (43), 3262–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y; Denisov IG; Sligar SG; Kincaid JR, Substrate-Specific Allosteric Effects on the Enhancement of CYP17A1 Lyase Efficiency by Cytochrome b5. J. Am. Chem. Soc 2021, 143 (10), 3729–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y; Denisov IG; Grinkova YV; Sligar SG; Kincaid JR, P450 CYP17A1 Variant with a Disordered Proton Shuttle Assembly Retains Peroxo-Mediated Lyase Efficiency. Chem. Eur. J 2020, 26 (70), 16846–16852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spiro TG; Soldatova AV; Balakrishnan G, CO, NO and O-2 as vibrational probes of heme protein interactions. Coord. Chem. Rev 2013, 257 (2), 511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagano S; Poulos TL, Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam - Implications for the dioxygen activation mechanism. J. Biol. Chem 2005, 280 (36), 31659–31663. [DOI] [PubMed] [Google Scholar]
- 41.Martinis SA; Atkins WM; Stayton PS; Sligar SG, A Conserved Residue of Cytochrome-P-450 Is Involved in Heme-Oxygen Stability and Activation. J. Am. Chem. Soc 1989, 111 (26), 9252–9253. [Google Scholar]
- 42.Gerber NC; Sligar SG, Catalytic Mechanism of Cytochrome-P-450 - Evidence for a Distal Charge Relay. J. Am. Chem. Soc 1992, 114 (22), 8742–8743. [Google Scholar]
- 43.Swart P; Swart AC; Waterman MR; Estabrook RW; Mason JI, Progesterone 16-Alpha-Hydroxylase Activity Is Catalyzed by Human Cytochrome-P450 17-Alpha-Hydroxylase. J. Clin. Endocrinol. Metab 1993, 77 (1), 98–102. [DOI] [PubMed] [Google Scholar]
- 44.Auchus RJ, Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J. Steroid Biochem. Mol. Biol 2017, 165, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kappl R; Hohnberlage M; Huttermann J; Bartlett N; Symons MCR, Electron-Spin and Electron Nuclear Double-Resonance of the [Feo2]- Center from Irradiated Oxyhemoglobin and Oxymyoglobin. Biochim Biophys Acta 1985, 827 (3), 327–343. [Google Scholar]
- 46.Davydov R; Ledbetter-Rogers A; Martasek P; Larukhin M; Sono M; Dawson JH; Masters BSS; Hoffman BM, EPR and ENDOR characterization of intermediates in the cryoreduced oxy-nitric oxide synthase heme domain with bound L-arginine or N-G-hydroxyarginine. Biochemistry 2002, 41 (33), 10375–10381. [DOI] [PubMed] [Google Scholar]
- 47.Davydov R; Makris TM; Kofman V; Werst DE; Sligar SG; Hoffman BM, Hydroxylation of camphor by-reduced oxy-cytochrome P450cam: Mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J. Am. Chem. Soc 2001, 123 (7), 1403–1415. [DOI] [PubMed] [Google Scholar]
- 48.Davydov R; Macdonald IDG; Makris TM; Sligar SG; Hoffman BM, EPR and ENDOR of catalytic intermediates in cryoreduced native and mutant oxy-cytochromes P450cam: Mutation-induced changes in the proton delivery system. J. Am. Chem. Soc 1999, 121 (45), 10654–10655. [Google Scholar]
- 49.Graham SE; Peterson JA, Sequence alignments, variabilities, and vagaries. Methods Enzymol 2002, 357, 15–28. [DOI] [PubMed] [Google Scholar]
- 50.Sherbet DP; Tiosano D; Kwist KM; Hochberg Z; Auchus RJ, CYP17 mutation E305G causes isolated 17,20-lyase deficiency by selectively altering substrate binding. J. Biol. Chem 2003, 278 (49), 48563–48569. [DOI] [PubMed] [Google Scholar]
- 51.Khatri Y; Gregory MC; Grinkova YV; Denisov IG; Sligar SG, Active site proton delivery and the lyase activity of human CYP17A1. Biochem. Biophys. Res. Commun 2014, 443 (1), 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Denisov IG; Mak PJ; Makris TM; Sligar SG; Kincaid JR, Resonance Raman Characterization of the Peroxo and Hydroperoxo Intermediates in Cytochrome P450. J. Phys. Chem. A 2008, 112 (50), 13172–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mak PJ; Denisov IG; Victoria D; Makris TM; Deng T; Sligar SG; Kincaid JR, Resonance Raman Detection of the Hydroperoxo Intermediate in the Cytochrome P450 Enzymatic Cycle. J. Am. Chem. Soc 2007, 129 (20), 6382–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benson DE; Suslick KS; Sligar SG, Reduced oxy intermediate observed in D251N cytochrome P450(cam). Biochemistry 1997, 36 (17), 5104–5107. [DOI] [PubMed] [Google Scholar]