

Abstract
Acinetobacter baumannii is a Gram-negative bacteria associated with drug resistance and infection in healthcare settings. An understanding of both the biological roles and antigenicity of surface molecules of this organism may provide an important step in the prevention and treatment of infection through vaccination or the development of monoclonal antibodies. With this in mind, we have performed the multistep synthesis of a conjugation-ready pentasaccharide O-glycan from A. baumannii with a longest linear synthetic sequence of 19 steps. This target is particularly relevant due to its role in both fitness and virulence across an apparently broad range of clinically relevant strains. Synthetic challenges include formulating an effective protecting group scheme as well as the installation of a particularly difficult glycosidic linkage between the anomeric position of a 2,3-diacetamido-2,3-dideoxy-D-glucuronic acid and the 4-position of D-galactose.
Keywords: Carbohydrates, O-Glycans, Multistep synthesis, Drug-resistant bacteria
Graphical Abstract

Multistep chemical synthetic efforts have produced a pentasaccharide O-glycan from the drug-resistant Gram-negative pathogen Acinetobacter baumannii. Synthetic challenges include installation of the unusual uronic acid moiety as well as an effective protecting group scheme. This structure is critical for virulence, and synthetic material may provide insights on its biological roles and antigenicity.
Introduction
Acinetobacter baumannii is an opportunistic, Gram-negative pathogen infamous for the nosocomial infections that it causes especially with the infirm and immunocompromised as well as those who have suffered traumatic injury.[1–5] Multidrug resistance as well as extensive and even pandrug resistance (in which the bacterial pathogen in question is resistant to all approved antibiotics) are becoming increasingly prevalent with A. baumannii.[6–8] What is worse, despite extensive efforts, there are no approved vaccinations for the prevention of infection.[9.10] Meanwhile, the urgency for providing new solutions to prevention and treatment of A. baumannii infection has been emphasized by agencies such as the U.S. Centers for Disease Control and the World Health Organization. An underemphasized area in the quest to address the A. baumannii problem, in our opinion, is that of the glycans presented on the surface of this organism, be they capsular polysaccharides (CPS),[11] lipooligosaccharides (LOS),[11–13] or O-glycans.[14]
Vaccines based on the covalent conjugation of capsular polysaccharide portions, be they synthetic or harvested from laboratory production of bacteria, to carrier proteins (i.e. glycoconjugate vaccines) have been a triumph in the prevention of infection with bacterial pathogens such as Haemophilus influenzae, Streptococcus pneumoniae, and Neisseria meningitidis.[15–19] We have become interested in the chemical synthesis and immunological studies of A. baumannii glycans, especially those that might be expressed on a high proportion of clinically relevant strains. With regard to chemical synthesis, a few efforts in the area of A. baumannii glycan synthesis have been reported. In 2015, the Hashimoto group reported the synthesis of a tetrasaccharide repeating unit from the “O-antigen” isolated from A. baumannii serogroup O18.[20] However, the lack of the appropriate WaaL ligase gene in A. baumannii calls into question whether this was O-antigen or perhaps the repeating unit of a capsular polysaccharide.[21] In 2021, Zhang and Seeberger reported the synthesis of trisaccharide repeating units from the CPS of A. baumannii AB5075.[22] In early 2022, we reported the synthesis of the tetrasaccharide CPS repeating unit from A. baumannii D78.[23] Even more recently, Yang, Yin, and co-workers reported the synthesis and immunological evaluation of an LOS core structure from A. baumannii ATCC 17904.[24]
Of particular interest to us is another glycan not from the CPS or LOS of A. baumannii but a protein-linked pentasaccharide O-glycan (1, Scheme 1). Discovered by Feldman, Vinogradov, and co-workers in 2012, this structure (isolated and characterized as a conjugate to L-serine, L-glutamic acid, and L-alanine, i.e. “SEA“) was originally associated with A. baumannii ATCC 17978.[14] However, further study indicated that the “glycosylation machinery” associated with the O-glycan was “present in all clinical isolates tested as well as in all of the genomes sequenced.”[14] Disruption of the biosynthesis of this glycan resulted in defective biofilm formation as well as reduced fitness in a mouse sepsis model. Clearly, this O-glycan is of great importance to the virulence of clinically relevant strains of A. baumannii and could be an interesting subject for further immunological study. What is more, in 2017, Wu and co-workers determined that this same pentasaccharide is a repeating subunit from the CPS of A. baumannii strain SK44.[25] Antibodies recognizing the CPS were reported to provide protection against infection, and the same antibodies were found to bind to 62% of clinical isolates. These results, being originally puzzling to us before we were aware of the work of Feldman and Vinogradov,[14] are readily explained by the abundance of the pentasaccharide as a stand-alone O-glycan on most or perhaps all clinically relevant strains.
Scheme 1.
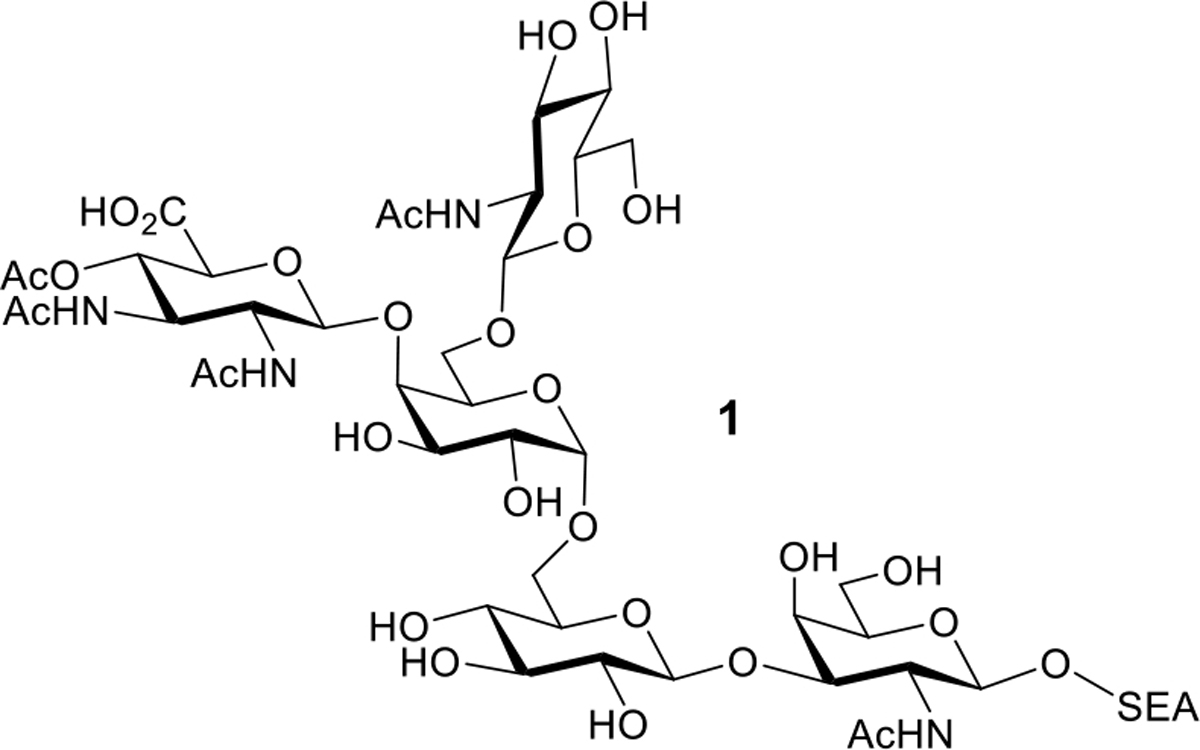
A. baumannii Pentasaccharide O-Glycan
With this knowledge in hand, we aimed to synthesize conjugation-ready analog 2 chemically for further immunological study (Scheme 2). The retrosynthetic analysis of 2 is depicted in Scheme 2. We reasoned that fully protected precursor 3 could be converted to 2 in two steps starting with functional group interchange of azide to acetamido on the left-most uronic acid moiety to generate the unusual 2,3-Diacetamido-4-Acetyl-2,3-Dideoxy-D-glucuronic Acid (DADA). We will further comment on the importance of this strategy below.
Scheme 2.
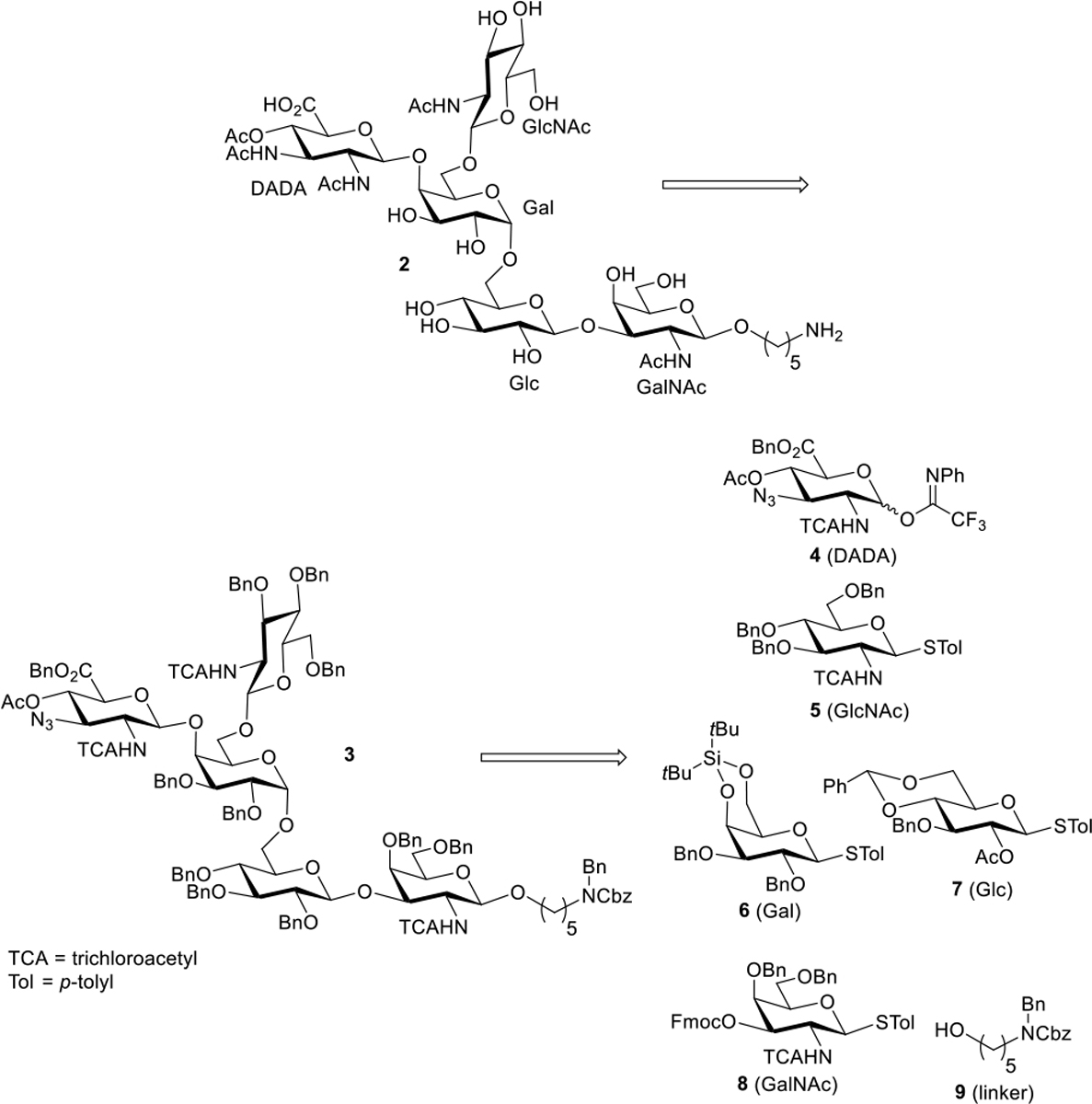
Targeted Product and Retrosynthesis
Hydrogenolysis of the resulting intermediate would reduce all trichloroacetamido groups to acetamido while removing all benzyl groups and the Cbz protecting group. The final target 2 is appended with a 5-aminopentyl linker at the reducing end to enable further conjugation to an array or a carrier protein. Disconnection of 3 results in 5 monomers designated as 4-8 (DADA, N-acetyl-D-glucosamine [GlcNAc], D-galactose [Gal], D-glucose [Glc], and N-acetyl-D-galactosamine [GalNAc], respectively) and linker precursor alcohol 9. Individual synthesis of these monomers would commence from commercially available building blocks such as D-galactosamine, D-glucose, D-galactose, and D-glucosamine.
Results and Discussion
Monosaccharide/Donor Synthesis
We begin the discussion with our synthesis of “DADA” donor 4 (Scheme 3) which began similarly to a synthetic sequence in Yin and Seeberger’s synthesis of a P. shigelloides CPS trisaccharide subunit.[26] Starting from known 2-deoxy-2-trichloroacetamido-D-glucose-1,2,4,6-tetraacetate 11,[27] glycosylation of allylic alcohol proceeded in 85% yield to generate a glycosylation product 12 which was subjected to methanolysis and then benzylidenation to 13 (91%, 2 steps). Noteworthy is that the anomeric selectivity incurred in the glycosylation was opposite to that reported by Yin and Seeberger.[26] However, this was not detrimental to the final outcome. Conversion of 13 through a Lattrell-Dax inversion resulted in D-allosamine 14 (83%, 2 steps) followed by an additional 3-position triflation and inversion with azide to generate 15. Hydrolytic removal of benzylidene was then proceeded by 6-position oxidation to carboxylic acid (BAIB, TEMPO) and benzyl ester formation (BnBr, NaHCO3) to afford 16 (59%, three steps).
Scheme 3.
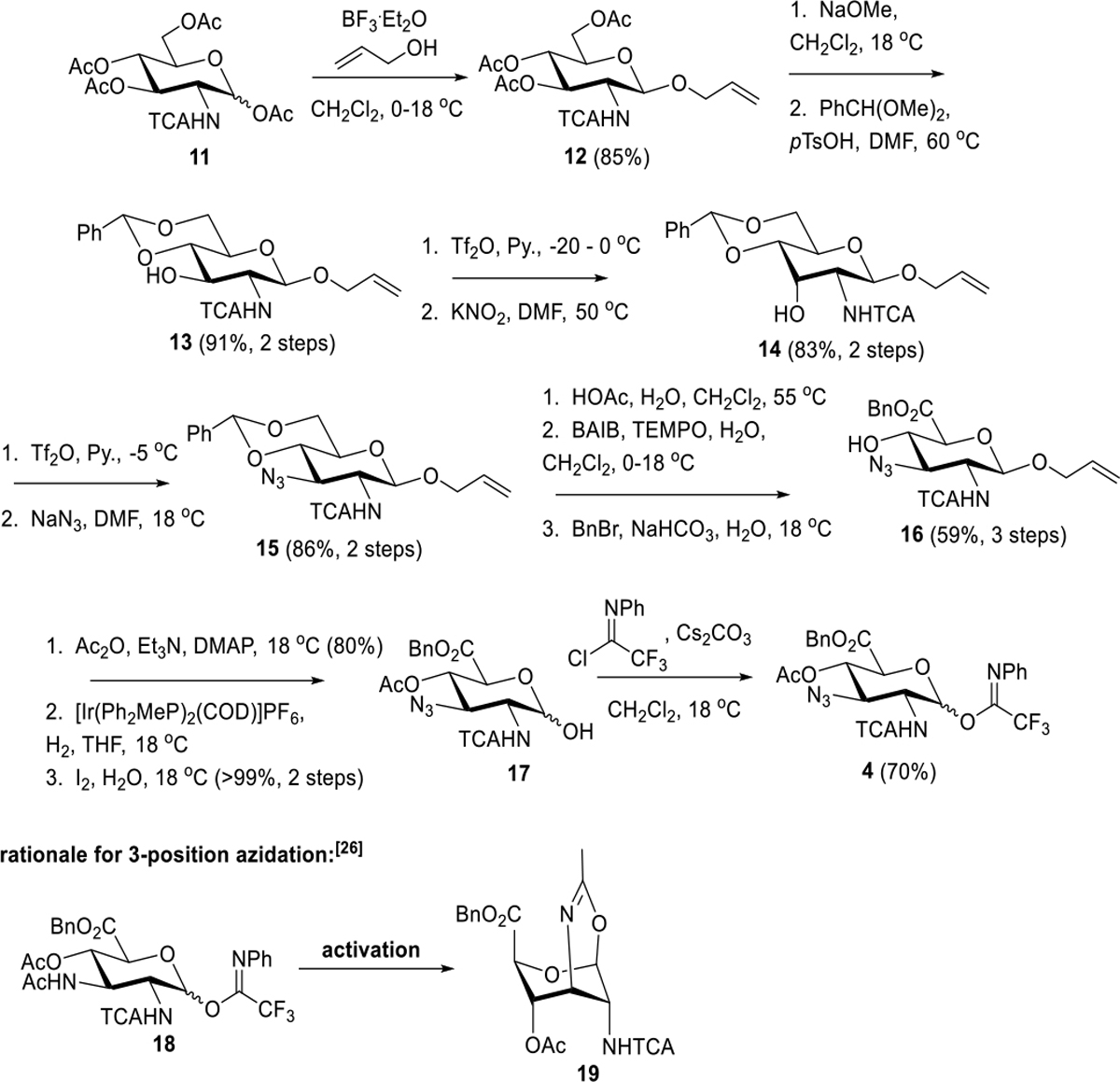
“DADA” Donor 4 Synthesis
Careful examination of the 1H NMR of 16 as well as the subsequent acetate (see compound 16a in the SI section) indicated that the glucuronate stereochemistry had been preserved despite the potential epimerizability of C5 in these intermediates. Noteworthy, too, is that C4 acetylation would preclude the implementation of acetates as permanent protecting groups during pentasaccharide assembly and global deprotection. Acetylation and then iridium-catalyzed alkene migration and oxidative hydrolysis of the resulting vinyl glycoside starting from 16 resulted in net deallylation and formation of reducing sugar 17. This intermediate was converted to N-phenyltrifluoroacetimidate donor 4 that was carried on to the assembly phase of the pentasaccharide synthesis. Our rationale for installing azido instead of amido at the 3-position of this donor was to avoid oxazine formation observed by Yin and Seeberger (see e.g. the conceivable but unwanted conversion of 18 to 19, Scheme 3)[26] through preemptive intramolecular attack of 3-position amide carbonyl on the anomeric position.
Our synthesis of GalNAc precursor 8 (Scheme 4) commenced from known thioglycoside 20[28] which underwent methanolysis followed by benzylidenation. In the latter case, promotion with CSA and α,α-dimethoxytoluene in DMF at 60 °C provided only partial conversion after 24 h. However, concentration of the reaction mixture and resubmission to conditions involving pTsOH·H2O in CH3CN resulted in complete conversion after just 30 minutes and afforded 21 in near-quantitative yield. At this stage, temporary TBDMS protection of 3-position hydroxyl enabled us to convert C4/C6 benzylidene to C4 benzyl with free C6 OH followed by benzylation of 6-position to generate 22 (83%, 3 steps). Subsequent TBDMS removal and reprotection of 3-position with Fmoc (which would have been susceptible to the previous Williamson etherification of 6-position under basic conditions) afforded donor 8 over a 2-step sequence (84%).
Scheme 4.
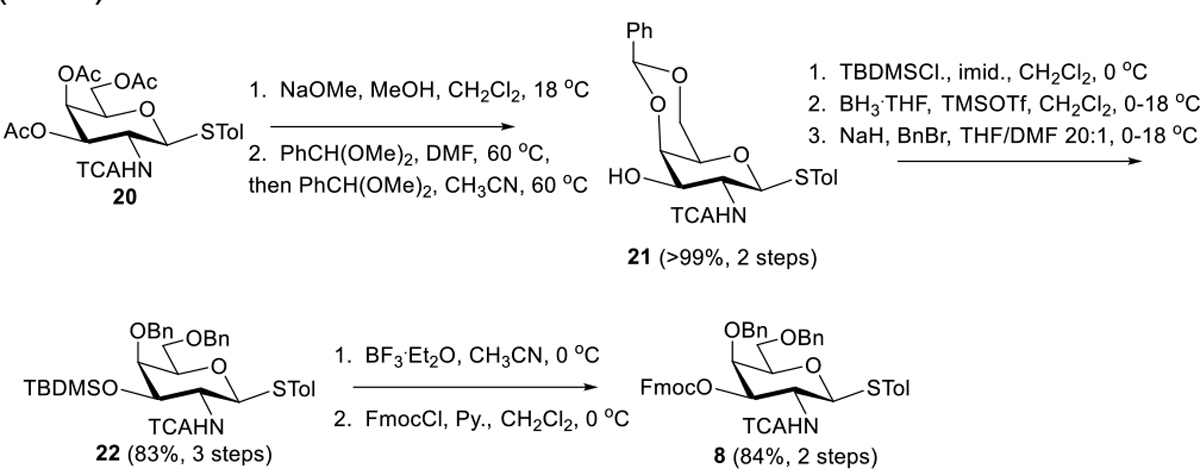
Synthesis of GalNAc Donor
Meanwhile, we conducted straightforward syntheses of donors 7 (Glc precursor, 6 steps from D-glucose pentaacetate), 6 (Gal precursor, three steps from p-thiocresyl galactose-2,3,4,6-tetraacetate), and 5 (GlcNAc precursor, 5 steps from D-glucosamine hydrochloride) as summarized in Scheme 5 (see SI for synthetic details). With all 5 of the monosaccharide donors in hand, we set our sights on the assembly phase of the pentasaccharide synthesis.
Scheme 5.
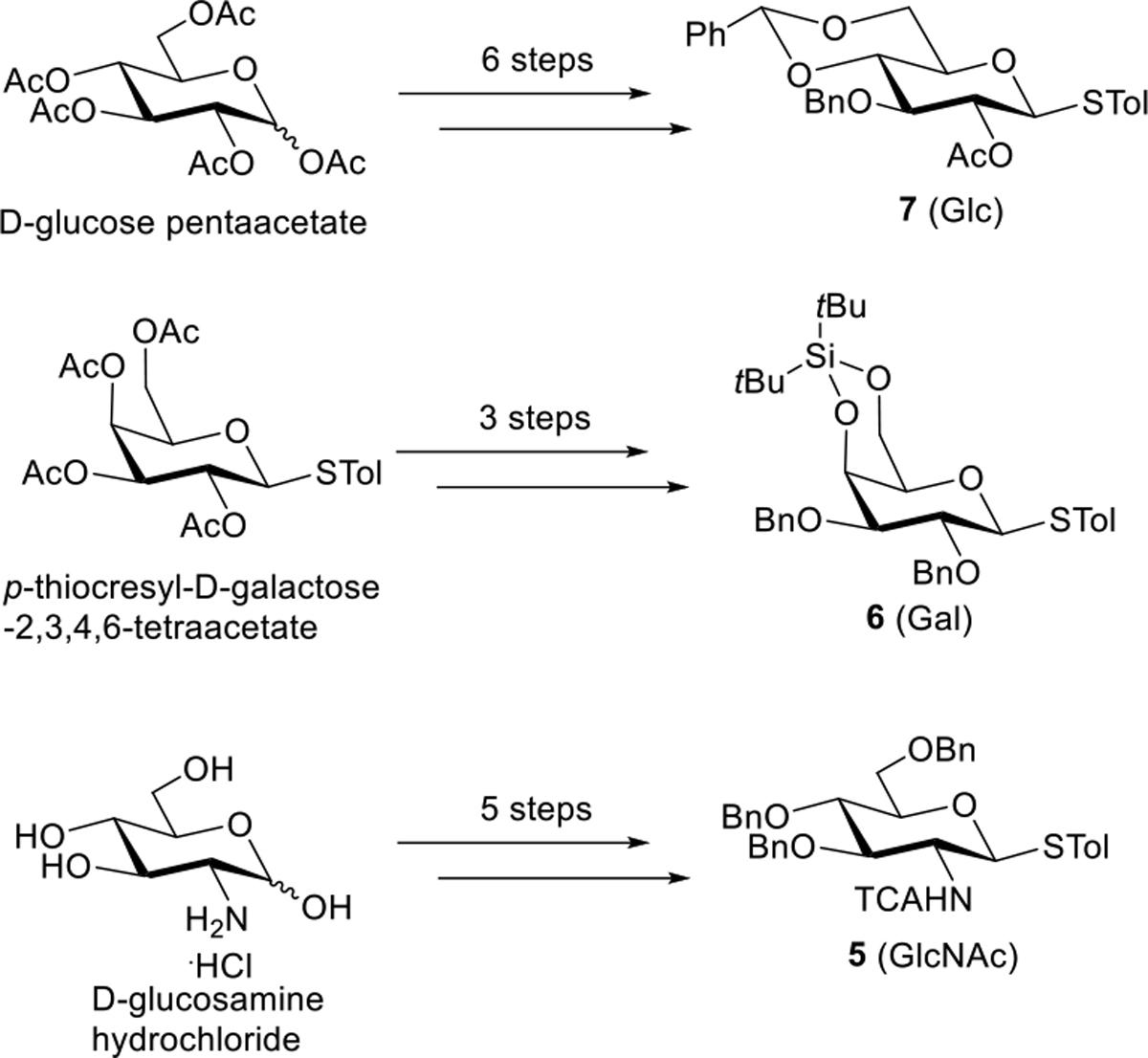
Synthesis of Glc, Gal, and GlcNAc Donors
Assembly
The first sequence in the assembly phase involved the synthesis of linker-appended disaccharide acceptor 26 (Scheme 6). Thiocresyl donor 8 was reacted with acceptor 9 using a set of glycosylation conditions that would serve us well throughout the assembly phase (NIS, AgOTf, CH2Cl2, low temperature)[29] to afford a glycoside which could not be purified away from succinimide. However, Fmoc was removed (92%, two steps) to generate 23 (as the only observed and 1,2-trans stereoisomer, 13C anomeric carbon δ 100.0 ppm) which was readily purified. An unmistakable HMBC correlation could be seen between the anomeric proton at δ 4.58 ppm and linker CH2-O carbon at δ 69.5, and correlations of this nature were observed in several subsequent intermediates, affirming the formation of this linkage. Reaction of acceptor 23 with donor 7 (NIS, AgOTf, CH2Cl2) afforded disaccharide 24 in high (95%) yield. Interchange of the Glc C2 protecting group was necessary at this stage as late-stage removal of C2 Ac would be incompatible with the C4 Ac on DADA.
Scheme 6.
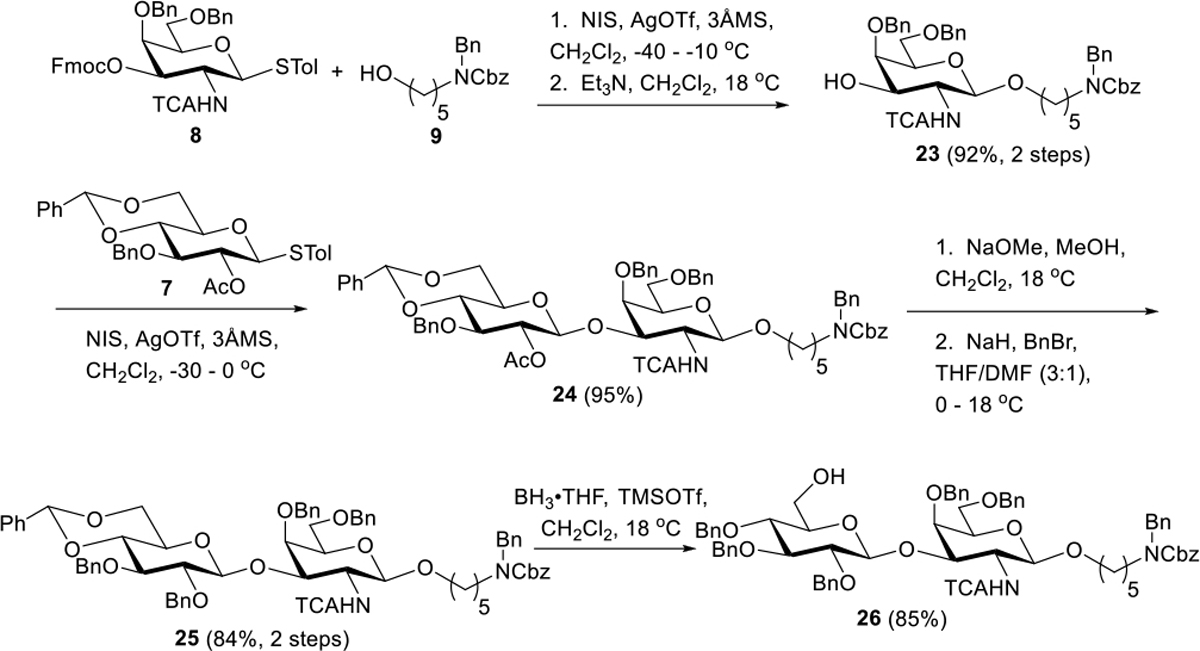
Synthesis of Disaccharide Acceptor 26
Thus, methanolysis and benzylation under Williamson conditions provided disaccharide 25 (84%, two steps). At this stage, NMR analysis confirmed 1,2-trans stereochemistry of the newly formed glycosidic linkage with the Glc anomeric carbon at δ 104.3. Noteworthy, too, was the downfield chemical shift of the GalNAc C3 from δ 71.0 in 23 to δ 76.1 in 25. Further, an HMBC correlation between Glc anomeric proton at δ 4.56 and GalNAc C3 at δ 76.1 could be observed, establishing the 1→3 linkage between Glc and GalNAc. Finally, benzylidene migration to the C4 of Glc was effected with BH3·THF and TMSOTf to produce acceptor 26. Regioselectivity could be confirmed with an HMBC correlation between the Glc C6 hydroxyl proton (δ 1.67 ppm) and the C6 carbon (δ 61.8 ppm) as well as COSY between the hydroxyl proton and C6 protons at δ 3.71 and δ 3.83 ppm.
Assembly from disaccharide 26 to fully protected pentasaccharide 3 was carried out in four steps according to Scheme 7. With disaccharide acceptor 26 in hand, we performed glycosylation with donor 6 (NIS, AgOTf, CH2Cl2) to generate a trisaccharide (compound 26a, see SI) in a modest 68% yield. A downfield shift of the Glc C6 13C signal from δ 61.8 ppm in 26 to δ 66.4 ppm in the trisaccharide provided evidence of connectivity. Meanwhile, the anomeric proton of the newly installed Gal was observed at δ 5.01 ppm with the anomeric carbon at δ 98.4 ppm. Highly noteworthy was that HMBC correlations between the Gal anomeric proton and the C6 carbon of Glc could be seen while further HMBC correlations between the Gal anomeric carbon and Glc C6 proton multiplets between δ 3.73 and 3.83 ppm could be observed. With this connectivity determined, removal of di-tert-butylsilylene (DTBS) from Gal produced diol trisaccharide acceptor 27 in 86% yield.
Scheme 7.
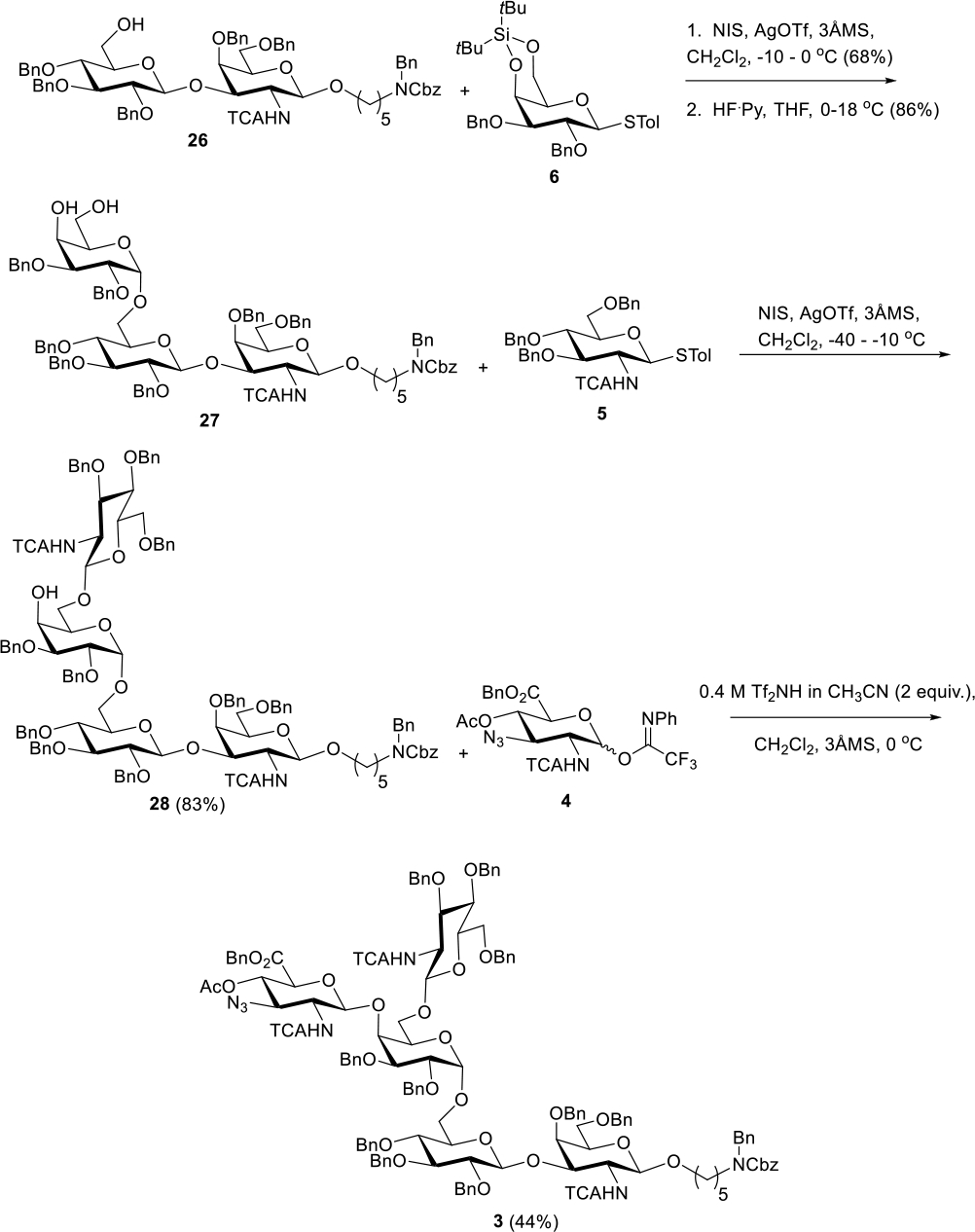
Asssembly of Fully Protected Pentasaccharide
Selective glycosylation of diol 27 was predicated on the increased nucleophilicity of Gal C6 OH relative to C4. While we have encountered problems with this type of assumption in the recent past,[23] this strategy served us well in this particular synthesis. Thus, reaction of 27 with donor 5 (NIS, AgOTf, CH2Cl2) afforded tetrasaccharide 28 in 83% yield. HMBC correlations between the GlcNAc anomeric proton at δ 4.76 ppm and the Gal C6 carbon at δ 69.5 ppm as well as HMBC correlations between the GlcNAc anomeric carbon at δ 100.5 ppm and Gal C6 protons at δ 4.00 and 3.78 ppm confirmed the new 1→6 linkage. Further, TOCSY confirmed that the assigned Gal C4 OH proton at δ 2.62 ppm was part of the same spin system as the Gal C6 and anomeric protons.
Due to the electronic and steric deactivation associated with the C4 OH of Gal in 28 along with the potentially low intrinsic reactivity of electronically deactivated DADA donor 4, we approached the final glycosylation with trepidation. Multiple attempts at glycosylation using promotion with TMSOTf simply resulted in silylation of the Gal C4 OH of 28. Triflic acid resulted in the formation of unidentified by-products and <20% of the desired pentasaccharide with catalytic quantities and complete decomposition of 4 with stoichiometric quantities. Given its comparable acidity to triflic acid and demonstrated differences in reactivity and behavior of trifluoromethanesulfonimide anion relative to trifluoromethanesulfonate,[30–32] we considered the use of trifluoromethanesulfonimide (Tf2NH) as an alternative strong acid for the activation of 4. We found that addition of a 0.4 M CH3CN solution of 2 equivalents of Tf2NH to a CH2Cl2 solution of 28 and 4 at 6 °C resulted in rapid consumption of 4 and glycosylation of 28 to produce 3 whereas use of catalytic quantities of Tf2NH did not promote glycosylation. The yields we observed were typically modest with this difficult glycosylation (30s-40s % yield), with a characteristic yield of 44% indicated in Scheme 7 and the accompanying procedure in the Supplementary Information. However, highly noteworthy is that unreacted 28 can be recovered from chromatographic purification of reaction mixtures and recycled for further use.
Global Deprotection
The final deprotection sequence proved to be straightforward with the exception of the final purification (Scheme 8). Treatment of 3 with a mixture of thioacetic acid and pyridine effected the conversion of the 3-position azide on “DADA” to acetamido. The resulting product (3a, see SI) was afforded in 87 % yield. The final hydrogenolysis employed Pd(OH)2/C with a solvent mixture of CH2Cl2, H2O, and EtOH under 1 ATM of H2 and successfully removed all Bn groups and Cbz and replaced chlorine atoms with H as predicted. Purification first with sephadex LH-20 stationary phase removed all but one unidentified impurity.
Scheme 8.
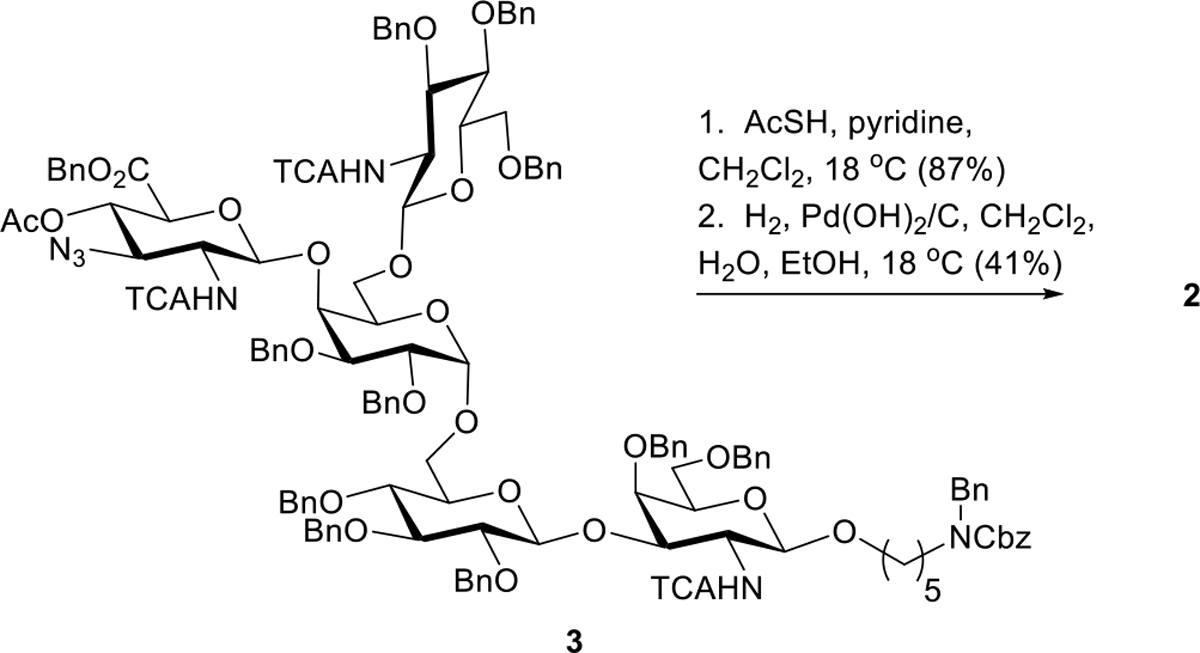
Global Deprotection
A subsequent purification using polyacrylamide beads (Bio-Gel P-2) as the stationary phase successfully provided pure desired product 2 in a modest 41% yield. This current approach has given us access to milligram quantities of 2 which will be sufficient for future immunological and other studies. The synthesis of 2 proceeded with a longest linear sequence of 19 steps starting from known donor 20 (Scheme 4).
Both 1D and 2D NMR analysis of 2 was critical for establishing the glycosidic linkage between the anomeric position of DADA and the C4 position of Gal which was not easily discerned from spectroscopic analysis of 3. In particular, consideration of HSQC, TOCSY, HSQC-TOCSY, and HMBC of 2 led to the identification of a key HMBC correlation between the DADA anomeric proton at δ 4.89 and the Gal C4 at δ 77.8. Furthermore, comparison especially of the 1H NMR and HSQC spectra of 2 to those of Feldman and Vinogradov’s original isolate (conjugate of the pentasaccharide to a tripeptide consisting of L-serine, L-glutamate, and L-alanine, i.e. SEA)14 showed striking similarities (see SI, pg. S195, reference 14). The totality of the NMR analysis performed on 2 and intermediates leading up to its synthesis as well as comparison to Feldman and Vinogradov’s data strongly bolster the original structural assignment of the pentasaccharide. Furthermore, Seeberger and co-workers published the synthesis of 2 a mere 5 days after we initially submitted this work for review.[33] Comparison of spectroscopic data indicated that the compounds synthesized by us and by Seeberger and co-workers are identical.
Conclusions
Herein, we have detailed the (longest linear sequence of 19 steps from known donor 20) synthesis and characterization of a conjugation-ready pentasaccharide O-glycan from A. baumannii ATCC 17978. Challenges included the synthesis of a “DADA” donor not predisposed to unwanted oxazine formation as well as the assembly of a branched trisaccharide substructure consisting of GlcNAc, DADA, and Gal. In particular, we had to resort to an unusual set of conditions to promote the formation of the congested glycosidic linkage between the anomeric position of DADA and the 4-position of Gal. Further, the presence of acetate at the 4-position of DADA necessitated careful development of a viable protecting group scheme. Both the 1D and 2D NMR analysis of synthetic intermediates and final product 2 confirm our structure and bolster the original structural assignment made by Feldman and Vinogradov. While the full range of functions for this O-glycan is not fully understood, evidence strongly suggests its importance for both the fitness and virulence in multiple (perhaps most) clinically relevant strains of A. baumannii. Synthetic access to the O-glycan pentasaccharide may open new chapters in understanding the molecular biology of A. baumannii, studying immune responses to infection with it, and the development of vaccines to prevent infection.
Experimental Section
General Information.
Solvents and reagents used were obtained from commercial sources. Dichloromethane used in glycosylation was freshly obtained from a solvent purification system (PureSolv 400–5). Powdered 3 Å Molecular sieves from BTC used in all glycosylations were activated by flame drying under vacuum, allowed to cool while capped under vacuum, and then backfilled with nitrogen gas just before they were transferred into the reaction vessel with a flame dried spatula (mass of 3 Å MS used was determined by difference). Room temperature was 18 °C most of the time. A temperature of 0 °C was achieved by using ice or ice water. Temperatures below 0 °C were achieved by using dry ice and isopropyl alcohol. Specific temperature ranges were achieved by varying the amount of dry ice used. Oven-dried pear-bottom-shaped flasks (P.B.F) with a tiny oven-dried stir bar were especially important in running glycosylation reactions under concentrated conditions. Column chromatography was performed using silica gel (60 Å) from SiliCyle. Thin layer chromatography (TLC) was performed using silica gel (60 Å) with F254 indicator on aluminum sheets purchased from Merck. Prep TLC was performed using “Analtech silica gel GF UV254 20*20 1000 micron” from Miles Scientific. A 3/1 dichloromethane (DCM)/Ethyl acetate (EtOAc) solvent mixture stock solution is referred to as ‘DE’ throughout this document (especially important in TLC and column chromatography). A hand-held UV lamp was used to visualize compounds on TLC and Prep TLC. Staining of compounds on TLC was performed by immersing developed TLC in p-anisaldehyde stain then drying excess stain on both sides of the TLC plate using a paper towel then heating the aluminum side of the TLC plate using a heat gun until colored spots appear on the side with silica gel. 1H NMR, 13C NMR (DEPT and APT), COSY, HMBC, HSQC, TOCSY, and HOHAHA experiments were performed using a Bruker AV-400, Bruker AV-500 NMR, or a Bruker AVANCE Neo 700 MHz spectrometer. Deuterated solvents used for NMR were obtained from Cambridge Isotope Labs (CDCl3), Acros organics (d-MeOD), and Sigma Aldrich (D2O). HRMS was performed using an Agilent 6210 electrospray time-of-flight mass spectrometer. Optical rotation data were obtained using a JASCO P-2000 instrument.
Allyl glycoside DADA precursor 12.
To a stirred solution of 1127 (2-deoxy-2-trichloroacetamido-D-glucose-1,3,4,6-tetraacetate, 2.94 g, 5.97 mmol) in dry dichloromethane (12 mL) in a pear shaped bottom flask (PBF), freshly activated (by flame drying under vacuum) 3 Å molecular sieves (1.80 g) was added. The flask was capped with a septum then carefully purged with nitrogen gas three times. Allyl alcohol (2.5 ml, 36 mmol) was then added under nitrogen atmosphere and then the flask was transferred into an ice bath. BF3 Et2O (8.0 ml, 65 mmol) was then added dropwise under nitrogen atmosphere over the course of approximately two minutes. After five minutes, the reaction flask was removed from the ice bath and allowed to gradually warm to room temperature. After 48 hrs, TLC indicated total consumption of 11. The organic solution was poured into a beaker with ice water (50 ml), then the mixture was filtered through a fritted funnel. The organic and aqueous layers were separated and then the organic layer was washed with 10 ml saturated NaHCO3, then dried using Na2SO4, then concentrated to give crude material that was purified by flash chromatography (40–65% DE/hexanes (DE= 3/1 DCM/EtOAc)) to give 2.494 g of 12 (white foam, 85% yield). Characterization of this compound matched that reported in the literature.27
Benzylidene-protected allyl glycoside DADA precursor 13.
To a stirred solution of 12 (5.76 g, 11.7 mmol) in 3.5/1 MeOH/DCM (45ml), 5M NaOMe (4.0 ml, 20 mmol) was added dropwise. The reaction was allowed to stir under nitrogen atmosphere at room temperature. After 50 minutes, the reaction was quenched by portion-wise addition of 16 g amberlite IR120 beads. The mixture was filtered, and the filtrate concentrated using a rotary evaporator to give a concentrate. The concentrate was then co-evaporated with 2.0 ml toluene three times and then placed under high vacuum for approximately 10 minutes to give crude material 12a. To 12a in a round bottom flask (R.B.F) with a stir bar, 20.0 ml of DMF and 4.0 ml (27 mmol) benzaldehyde dimethyl acetal were added, respectively. Once all 12a had dissolved, 0.4989 g (2.897 mmol) of TsOH H2O was added. A reflux condenser was attached, and the reaction was then allowed to proceed at 60 °C using an oil bath under nitrogen atmosphere. After 48 hours, the reaction was concentrated using a rotary evaporator. The concentrate was dissolved in 150 ml EtOAc. The new organic solution was washed twice with 250 ml water. The organic phase was separated and dried using Na2SO4, then concentrated to give a crude material that was purified by flash chromatography (30–45% EtOAc/hexanes) to give 4.8407g of 13 (white solid, 91% two-step yield). Characterization of this compound matched that reported in the literature.27
Epimerized allyl glycoside DADA precursor 14.
To a stirred solution of 13 (5.9124 g, 13.060 mmol) in dichloromethane (35ml) at −20 °C, pyridine (10.0 ml, 124 mmol) was added. The reaction flask was then flushed with nitrogen gas for 5 seconds. A solution of triflic anhydride (4.7 ml, 28 mmol, diluted in 10 ml DCM) was then added dropwise. After 10 minutes, the reaction was allowed to stir at 0 °C. After an additional 80 minutes, TLC indicated total consumption of 13. The reaction mixture was diluted with 600 ml DCM (product not very soluble in DCM) then the organic solution was washed with 125 ml 1M HCl, then washed with 100 ml water and then finally 100 ml saturated NaHCO3. The organic phase was dried using Na2SO4 then concentrated to give crude material 13a (7.46 g) that was used in the next step without further purification. To a stirred solution of crude material 13a from the previous procedure in DMF (50.0 ml), KNO2 (6.21 g, 73.0 mmol) was added. A reflux condenser was attached, and the reaction was then allowed to proceed at 50 °C using an oil bath under nitrogen atmosphere. After 3 hours, the reaction mixture was diluted using 600 ml EtOAc to give a solution that was washed with 800 ml water. The organic phase was dried using Na2SO4 then concentrated to give a crude material that was purified by flash chromatography (20–25% EtOAc/hexanes) to give 4.9356 g of 14 (white foam, 83% two step yield).
Azidoglycoside DADA precursor 15.
To a stirred solution of 14 (2.1875 g, 4.8320 mmol) in dichloromethane (12.2 ml) at −5 °C, pyridine (4.0 ml, 50 mmol) was added. The pear-shaped flask was then purged and backfilled with nitrogen gas (thrice). A solution of triflic anhydride (2.2 ml, 13 mmol, diluted in 6 ml DCM) was then added dropwise. After two hours, TLC showed that most of the starting material was consumed. More Tf2O solution (1.2 ml, 7.1 mmol, diluted in 3.0 ml DCM) was added dropwise. After an additional 5 h, TLC showed that all starting material was consumed. The reaction mixture was diluted with 20 ml DCM, washed with 1M HCl solution (100 ml), and the organic phase was separated then washed with saturated NaHCO3 solution (30 ml). The organic phase was once again separated and dried using Na2SO4 then concentrated to give crude material 14a that was used in the next step without further purification. To a stirred solution of crude material 14a above in DMF (25 mL), NaN3 (3.97 g, 61.1 mmol) was added portion-wise at 18 °C. After 16 hours, TLC showed total consumption of 14a. The reaction mixture was diluted with EtOAc (200ml) to give a solution that was washed with 300ml water. The organic phase was then dried using Na2SO4 then concentrated to give a crude material that was purified by flash chromatography (25–35% DE/hexanes (DE = 3/1 DCM/EtOAc)) to give 1.9794 g of 15 (white foam, 86% two step yield).
Uronic ester DADA precursor 16.
To a stirred solution of 15 (0.5292 g, 1.108 mmol) in DCM (2.0 ml), AcOH (11.0 ml) and water (2.0 ml) were added. A reflux condenser was attached, and the reaction was then allowed to proceed at 55 °C using an oil bath under nitrogen atmosphere. After 4 h, TLC showed total consumption of 15. The reaction mixture was concentrated to give crude material that was purified by flash chromatography (45–60% EtOAc/hexanes) to give 0.3901 g of 15a (white solid). To a stirred solution of 15a (0.3801 g, 0.9756 mmol) in DCM (20.0 ml) at 0 °C, water (2.0 ml), (diacetoxyiodo)benzene (PIDA, 0.7850 g, 2.437 mmol), and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 78.5 mg, 0.502 mmol) were added, respectively. After 40 minutes, the ice bath was removed and the reaction was allowed to warm to room temperature. After an additional 3 h, TLC showed total consumption of starting material 15a. The reaction mixture was transferred into a separatory funnel where aqueous and organic layers were separated. The organic phase was concentrated to give crude material 15b that was used in the next step without further purification. To a stirred solution of crude material 15b in DMF (25 ml) at 18 °C, NaHCO3 (0.8 g, 10 mmol), and BnBr (0.90 ml, 7.6 mmol) were added. After 15 h, TLC showed total consumption of starting material 15b. The reaction mixture was concentrated using rotary evaporator then diluted with EtOAc (20.0 ml). The resultant solution was washed with water 50 ml once. The organic phase was then dried using Na2SO4 then concentrated to give a crude material that was purified by flash chromatography (40–60% DE/hexanes (DE= 3/1 DCM/EtOAc)) to give 0.3243 g of 16 (white foam, 59% three step yield).
Uronic ester DADA precursor 17.
To a stirred solution of 16 (0.7800 g, 1.580 mmol) in DCM (10.0 ml) at 18 °C, triethylamine (4.0 ml (29 mmol), 4-dimethylamino pyridine (DMAP) (44.15 mg, 0.3614 mmol), and acetic anhydride (2.4 mL, 25 mmol) were added, respectively. After 80 minutes, TLC showed total consumption of starting material 16. The reaction mixture was diluted with 20 ml DCM, then the organic solution was washed with 70 ml 1M HCl. The organic phase was dried using Na2SO4 then concentrated to give crude material that was purified by flash chromatography (15–25% EtOAc/hexanes) to give 0.6791 g of 16a (yellow oil, 80% yield). 16a (0.6791 g, 1.268 mmol) was stirred in THF (5.0 mL) at 18 °C in a pear-bottom-shaped flask (PBF) under nitrogen atmosphere. In a separate 4ml Wheaton vial, hydrogen gas was bubbled through a red solution of (1,5-Cyclooctadiene) bis(methyldiphenylphosphine)iridium(I) hexafluorophosphate (55.4 mg, 0.0655 mmol) in THF (2.0 ml) until the solution became colorless with all iridium catalyst dissolved. This solution was then transferred dropwise using a syringe to the PBF-containing solution of 16a in THF. After 80 minutes, TLC showed total consumption of starting material 16a and formation of a new (less polar) spot. Additional THF (1 ml), and water (4.0 ml) were added to the reaction mixture followed by portion-wise addition of iodine (673.7 mg, 2.654 mmol). After 30 minutes, TLC showed that all the less polar spot was consumed and a more polar spot (lactol product) was formed. The reaction mixture was then diluted with 30.0 mL EtOAc then washed once with 30.0 mL saturated solution of Na2S2O3. The organic phase was dried using N2SO4 then concentrated to give crude material that was purified by flash chromatography (20–40% EtOAc/hexanes) to give 0.6215 g of 17 (yellow solid, 99% yield).
DADA donor 4.
To a stirred solution of 17 (0.4418 g, 0.8913 mmol) in dichloromethane (5.0 ml), Cs2CO3 (1.6202 g, 4.9727 mmol)) was added followed by dropwise addition of 2,2,2-trifluoro-N-phenylacetimidoyl chloride42 (0.3020 g, 1.455 mmol). The reaction was allowed to run at room temperature (18 °C) under nitrogen atmosphere. 30 minutes later, TLC indicated that the reaction was complete. Stirring was discontinued and the reaction diluted with 20 ml DCM and then washed with 10 ml water once. The organic phase was dried using Na2SO4 then concentrated to give a crude material that was purified by flash chromatography (20–30% Et2O/hexanes) to give 0.4163 g of 4 (white foam, 70%).
GalNAc thioglycoside 21.
To 2034 (para-cresyl-1-thio-β-D-2-deoxy-2-trichloroacetamidogalactose-3,4,6-triacetate, 6.3425 g, 11.390 mmol) dissolved in (4/1) MeOH/DCM (50 ml), a 5M NaOMe solution in MeOH (1.0 ml) was added dropwise while stirring. After 30 minutes, TLC indicated complete formation of a more polar compound. The reaction mixture was neutralized by addition of 8.2 g of Dowex resin (50WX8 200–400 MESH). The mixture was filtered, then the filtrate was concentrated to give crude triol, which was then co-evaporated with 3ml toluene two times then placed under high vacuum for 30 minutes. Crude triol was then dissolved in DMF (30 ml). Benzaldehyde dimethyl acetal (2.6 mL, 17 mmol) and camphor sulfuric acid (1.18 g, 5.08 mmol) were then added into the flask, respectively. A reflux condenser was attached, and the reaction was then allowed to proceed at 60 °C using an oil bath under nitrogen atmosphere. After 10 hrs, TLC showed very minimal consumption of triol 20a. The reaction was concentrated as is and co-evaporated with 3 ml toluene twice. To the concentrate in R.B.F, acetonitrile (30 ml), benzaldehyde dimethyl acetal (3.0 ml, 20 mmol) and TsOH·H2O (0.49 g, 2.8 mmol) were added, respectively. A reflux condenser was once again attached, and the reaction was then allowed to proceed at 60 °C using an oil bath under nitrogen atmosphere. After just 50 minutes, TLC showed that all triol 20a was consumed and product 21 formed. The reaction was allowed to cool to room temperature, then Et3N (2.0 ml) was added dropwise to quench the reaction. The mixture was concentrated using a rotary evaporator. To the crude concentrate, 150 ml EtOAc was added to give a solution that was washed with water (120 ml). The organic phase was dried using Na2SO4 then concentrated to give crude material that was purified by flash chromatography (30–60% EtOAc/hexanes) to give 5.85 g of 21 (99% two-step yield). Characterization of this compound matched that reported in the literature.34
GalNAc thioglycoside 22.
To a stirred solution of 21 (1.9936 g, 3.8425 mmol) in DCM (15.0 mL) at 0 °C, tert-butyldimethylsilyl chloride (1.65 g, 10.9 mmol), and imidazole (0.91 g, 13 mmol) were added, respectively. The reaction was then allowed to warm to room temperature. After 14 hours, TLC showed total consumption of starting material 21. The reaction mixture was placed in an ice bath and quenched by dropwise addition of MeOH (2.0 mL). The mixture was then diluted with DCM (20 mL) then washed with a saturated solution of NH4Cl (50 ml) followed by a wash with a saturated NaCl solution (20 mL). The organic phase was separated and dried using Na2SO4 then concentrated to give crude material 21a34 that was used in the next step without further purification. To a stirred solution of crude 21a from the previous procedure in DCM (17.0 mL) at 0 °C under nitrogen atmosphere, BH3·THF (17.0 mL, 17.0 mmol) was added dropwise until effervescence stopped. Trimethylsilyl trifluoromethanesulfonate (TMSOTf,100.0 μL, 0.55 mmol) was then added dropwise using a micro-syringe. After 5 minutes, the reaction was allowed to warm to room temperature (18 °C). After 90 minutes, TLC showed complete consumption of 21a and formation of a more polar spot (21b). The reaction mixture was quenched with saturated NaHCO3 (15 mL) dropwise at 0 °C followed by addition of 5 mL MeOH. 50 mL DCM was added and the resultant mixture with white suspension was washed with water (150 mL). The organic phase was separated and dried using Na2SO4 and then concentrated to give crude material 21b that was used in the next step without further purification. To a stirred solution of crude material 21b in 20:1 THF/DMF (21 ml) at 0 °C, benzyl bromide (1.37 mL,11.5 mmol) was added. This was followed by portion-wise addition of 60% NaH suspended in mineral oil (1.55 g, 38.8 mmol). After 5 minutes, the reaction was allowed to warm to room temperature (18 °C). After 4 hours, TLC showed total consumption of starting material 21b and formation of a less polar spot (22). The reaction flask was transferred into an ice bath then quenched by dropwise addition of 3.0 mL MeOH followed by dropwise addition of water (7.0 mL) until gas evolution stopped. The mixture was then diluted with EtOAc (40 ml) then washed with water (50 ml). The organic phase was separated and then dried using Na2SO4 and then concentrated to give crude material 22 that was purified by flash chromatography (8–25% EtOAc/hexanes) to give 2.3222 g of 22 (white solid, 83% three step yield).
GalNAc thioglycoside donor 8.
To a stirred solution of 22 (2.2874 g, 3.1540 mmol) in acetonitrile (25.0 mL) at 0 °C under nitrogen atmosphere, BF3·Et2O (0.50 mL, 4.1 mmol) was added dropwise. After 1 hour, TLC showed total consumption of starting material 22 and formation of a more polar spot (22a). The reaction mixture was quenched by addition of Saturated NaHCO3 (5 mL) then filtered through Celite. The filtrate was concentrated then diluted with DCM (20 mL) and water (5 mL) to enable separation of organic and aqueous phases. The organic phase was dried using Na2SO4 and then concentrated to give crude 22a that was used in the next step without further purification. To crude 22a from the previous procedure in a R.B.F with a stir bar, DCM (10 mL) and fluorenylmethyloxycarbonyl chloride (1.08 g, 4.17 mmol) were added, respectively. The reaction mixture was stirred at 0 °C, then pyridine (0.95 mL, 12 mmol) was added dropwise. After 4 hours, TLC showed total consumption of 22a and formation of less polar spot (8). The reaction was quenched by addition of 1M HCl (5 mL) dropwise. The reaction mixture was then diluted with 20 mL DCM and washed with 20 mL saturated NaHCO3. The organic phase was separated and dried using Na2SO4 and then concentrated to give crude material that was purified by flash chromatography (20–30%, DE/hexanes (DE= 3/1 DCM/EtOAc)) to give 2.1703 g of 8 (white solid, 83% two-step yield).
Glc thioglycoside donor 7.
To a RBF charged with a stir bar, β-D-Glucose pentaacetate (7a, 11.71 g, 30.00 mmol) and p-thiocresol (5.74 g, 46.2 mmol) were dissolved in dichloromethane (125 mL). The R.B.F was capped followed by purging and backfilling with nitrogen gas three times. The reaction mixture was then stirred at 0 °C before BF3·Et2O (12.0 mL, 97.2 mmol) was added dropwise. Five minutes after the addition was complete, the ice bath was removed and the reaction was allowed to warm to room temperature (18 °C). After 10 hours, TLC indicated complete consumption of starting material 7a and formation of product 7b. 50 mL of saturated NaHCO3 was added, and the mixture was allowed to stir for 20 minutes. The organic phase was then separated and dried using Na2SO4 then concentrated using a rotary evaporator to give crude material that was purified by flash chromatography (30–45% EtOAc/hexanes) to give 13.3 g of 7b (white solid, 98% yield). Characterization of this compound matched that reported in the literature.35 To 7b (7.6934 g, 16.928 mmol) dissolved in 5:1 MeOH/DCM (60 ml), a 5M NaOMe solution in MeOH (1.0 mL) was added dropwise while stirring. After 30 minutes, TLC indicated complete formation of a more polar compound. The reaction mixture was neutralized by addition of 7.18 g of Dowex resin (50WX8 200–400 MESH). The mixture was filtered, then the filtrate was concentrated to give crude tetraol 7c, which was then co-evaporated with 5ml toluene two times then placed under high vacuum for 30 minutes. Crude tetraol was then dissolved in DMF (35 mL). Benzaldehyde dimethyl acetal (9.0 mL, 60.0 mmol) and p-TsOH·H2O (1.72 g, 9.95 mmol), were then added into the flask, respectively. A reflux condenser was attached, and the reaction was then allowed to proceed at 65 °C using an oil bath under nitrogen atmosphere. After 12 hours, the reaction was allowed to cool to room temperature, then Et3N (1.0 ml) was added dropwise to quench the reaction. The mixture was concentrated using a rotary evaporator. To the crude concentrate, 100 mL EtOAc was added to give a solution that was washed with water (200 mL). The organic phase was dried using Na2SO4 then concentrated to give crude material that was purified by flash chromatography (30–100% EtOAc/hexanes) to give 2.8213 g of 7d (45% two-step yield). Characterization of this compound matched that reported in the literature.35 To 7d (2.80 g, 7.48 mmol) in R.B.F with a stir bar, toluene (60 mL) was added. The reaction mixture was stirred at 0 °C before dibutyltin oxide (2.9192 g, 11.728 mmol) was added. A reflux condenser was attached, and the reaction was heated to reflux using an oil bath under nitrogen atmosphere. After 13 hours, the reaction mixture was concentrated, and DMF (40 ml) was added. The resulting reaction mixture was then stirred at 0 °C (using cold bath) before benzyl bromide (2.66 mL, 22.4 mmol) and cesium fluoride (3.4080 g, 22.436 mmol) were added, respectively. The cold bath was removed and the reaction allowed to gradually warm to room temperature. After 6 hours, stirring ceased and EtOAc (300 mL) was added to the reaction mixture. This was followed by washing using 300 mL water once. The organic phase was separated and dried using Na2SO4 then concentrated to give crude material that was purified by flash chromatography (20–50% EtOAc/hexanes) to give 3.4784 g of 7e (white solid, >99% yield).36 To a stirred solution of 7e (1.0384 g, 2.2352 mmol) in DCM (10.0 mL) at 18 °C, 4-dimethylamino pyridine (DMAP,10.0 mg, 0.082 mmol), triethylamine (0.8 mL, 6 mmol), and acetic anhydride (0.80 mL, 8.4 mmol) were added, respectively. After 2 hours, TLC showed total consumption of starting material 7e. The reaction mixture was diluted with 20 ml DCM, then the organic solution was washed with 20 mL 1M HCl then separated and washed with 50 mL saturated NaHCO3. The organic phase was dried using Na2SO4 then concentrated to give crude material that was purified by flash chromatography (15–20% DE/hexanes (DE=3/1 DCM/EtOAc)) to give 0.6188 g of 7 (white solid, 55% yield). Characterization of this compound matched that reported in the literature.37
Gal thioglycoside donor 6.
To 6a38 (para-cresyl-1-thio-β-D-galactose-2,3,4,6-tetraacetate, 4.11 g, 9.04 mmol) dissolved in (2/1) MeOH/DCM (15 ml), a 5M NaOMe solution in MeOH (1.0 mL) was added dropwise while stirring at 18 °C. After 30 minutes, TLC indicated complete formation of a more polar compound. The reaction mixture was neutralized by addition of 5.01 g of Dowex resin (50WX8 200–400 MESH). The mixture was filtered, then the filtrate was concentrated to give crude tetraol which was then co-evaporated with 2 mL toluene two times then placed under high vacuum for 10 minutes to give crude tetraol 6b that was used in the next step without further purification. To 6b from the previous procedure in an R.B.F with a stir bar, pyridine (25 mL) was added. The flask was capped with a rubber septum and then purged and backfilled with nitrogen gas three times. The flask was then transferred to a −30 °C cold bath. Di-tert-butylsilyl bis(trifluoromethanesulfonate) (3.8 mL, 12 mmol) was then added dropwise. The reaction was then allowed to warm gradually to 0 °C. After 2 hours, TLC showed consumption of 6b. The reaction was then quenched by addition of 5.0 mL MeOH dropwise. The reaction mixture was then concentrated to give a material that was redissolved in EtOAc (100 mL). This solution was washed with 1M HCl (100 mL) followed by a wash with 100 mL saturated NaHCO3 solution. The organic phase was separated and dried using Na2SO4 then concentrated to give crude material 6c that was used in the next step without further purification. To a stirred solution of 6c from the previous procedure in DMF (40 mL) at 0 °C, benzyl bromide (5.0 mL, 42 mmol) was added followed by portion-wise addition of 60% NaH suspended in mineral oil (4.5 g, 110 mmol) over a period of two minutes. The reaction was allowed to stir under nitrogen atmosphere while warming to room temperature. After 18 h, the reaction was transferred to an ice bath then quenched by dropwise addition of water (10 mL) over a period of 10 minutes until gas evolution stopped. The mixture was diluted with 100mL EtOAc, then washed once with 250 mL water. The organic phase was dried using Na2SO4 then concentrated to give a crude material that was purified by flash chromatography (0–10% EtOAc/hexanes) to give 2.3599 g of 6 (white solid, 43% three-step yield). Spectroscopic data matched that reported in the literature.39
GlcNAc thioglycoside donor 5.
To 5a40 (para-cresyl-1-thio-β-D-2-deoxy-2-trichloroacetamidoglucose-3,4,6-triacetate, 7.86 g, 14.1 mmol) dissolved in (1.5/1) MeOH/DCM (37 mL), a 5M NaOMe solution in MeOH (1.8 mL) was added dropwise while stirring. After 30 minutes, TLC indicated complete formation of a more polar compound. The reaction mixture was neutralized by addition of 7.82 g of Dowex resin (50WX8 200–400 MESH). The mixture was filtered, then the filtrate was concentrated to give crude tetraol, which was then co-evaporated with 4mL toluene two times then placed under high vacuum for 20 minutes to give crude 5b that was used in the next step without further purification. To a stirred solution of 5b from the previous procedure in THF (80 mL) at 0 °C, benzyl bromide (10.0 mL, 84 mmol) was added followed by portion-wise addition of 60% NaH suspended in mineral oil (7.00 g, 175 mmol ) over a period of one minute. The reaction was allowed to stir under nitrogen atmosphere while warming to room temperature. After 21 h, the reaction was transferred to an ice bath then quenched by dropwise addition of water (15 mL) over a period of 8 minutes until gas evolution stopped. The mixture was diluted with 50 mL EtOAc, then concentrated to give a crude material that was re-dissolved in 200 mL EtOAc, the new organic solution was washed with 150 mL water. The organic phase was separated and dried using Na2SO4 then concentrated to give crude material that was purified by flash chromatography (20–40% EtOAc/hexanes) to give 3.7013 g of 5 (white solid, 37% two-step yield).
Glycoside 23.
To a stirred solution of thioglycosyl donor 8 (1.0198 g, 1.2239 mmol) and N-benzyl-N-benzyloxycarbonyl-5-aminopentan-1-ol acceptor 941 (0.5481 g, 1.674 mmol) in dichloromethane (15.0 mL) in a pear-shaped bottom flask (PBF) was added activated (by flame drying under vacuum) powdered 3 Å molecular sieves (1.3002 g, from BTC) at room temperature. The PBF was immediately capped using a septum, and the mixture allowed to stir at room temperature (18 °C) for 17 minutes. The PBF was then transferred into a −50 °C cold bath, and N-Iodosuccinimide (0.6051 g, 2.690 mmol) was added in one portion. After 5 minutes, silver triflate (0.1557 g, 0.6060 mmol) was added in one portion (note: while adding both NIS and AgOTf solids, the septum was removed and the respective solids added very fast while making sure they drop directly on the DCM solution and septum was put back on fast). The reaction was allowed to slowly warm to 0 °C (note: color change to brownish-red, indicating glycosylation, started to occur when temperature was above −20 °C as confirmed by TLC). After 2 hours, TLC indicated complete consumption of donor 8. The reaction was filtered through celite while rinsing with 60 mL dichloromethane. The filtrate was washed vigorously with 5 mL saturated Na2S2O3 solution. The organic phase was separated and dried using Na2SO4 and then concentrated to give crude 23a that was purified by flash chromatography (20–30% EtOAc/petroleum ether) to give 1.2239 g of 23a (white foam, ~96% yield, has inseparable succinimide impurity) that was used in the next step without further purification. To a stirred solution of 23a (1.1825 g, 1.1409 mmol) in CH2Cl2 (12.0 mL) at room temperature (18 °C) under nitrogen atmosphere, Et3N (6.0 mL, 43 mmol) was added dropwise. After 3 hours, TLC showed total consumption of starting material 23a and formation of a more polar spot (23). The reaction mixture was diluted with 50 mL CH2Cl2 then washed with 50.0 mL 1M HCl. The organic phase was separated and dried using Na2SO4 and then concentrated to give crude 23 that was purified by flash chromatography (45–60% DE/petroleum ether, DE = 3/1 DCM/EtOAc) to give 0.9190 g of 23 (colorless oil, 92% yield, two steps).
Disaccharide 24.
To a stirred solution of thioglycosyl donor 7 (488.4 mg, 0.9641 mmol) and galactosyl acceptor 23 (749.4 mg, 0.9204 mmol) in dichloromethane (4.0 mL) in a pear-shaped bottom flask (P.B.F) was added activated (by flame drying under vacuum) powdered 3 Å molecular sieves (0.8103 g, from BTC). The PBF was immediately capped using a septum and the mixture allowed to stir at room temperature (18 °C) for 10 minutes. The PBF was then transferred into a −25 °C cold bath and N-Iodosuccinimide (0.4345 g, 1.931 mmol) was added in one portion and the inside wall of the PBF was rinsed with 1.0 mL DCM to ensure all added material had been transferred to the reaction mixture. After 5 minutes, silver triflate (0.1075 g, 0.4184 mmol) was added in one portion and the inside wall of the PBF was rinsed with another 1 mL DCM (Note: While adding both NIS and AgOTf solids, the septum was removed and the respective solids added very fast while making sure they drop directly into the DCM solution and the septum was put back on fast). The reaction was allowed to slowly warm to 0 °C (note: color change to brownish-red, indicating glycosylation, started to occur when temperature was above −20 °C as confirmed by TLC). After 15 minutes, TLC indicated complete consumption of donor 7. The reaction was filtered through celite while rinsing with 30 mL dichloromethane. The filtrate was washed vigorously with 15 ml saturated Na2S2O3 solution. The organic phase was separated and dried using Na2SO4 and then concentrated to give crude 24 that was purified by flash chromatography (40–60% DE/petroleum ether, DE= 3/1 DCM/EtOAc) to give 1.0015 g of 24 (white foam, 91% yield).
Disaccharide 25.
To 24 (210.8 mg, 0.1762 mmol) dissolved in 1.5:1 MeOH/DCM (5.0 mL), a 5M NaOMe solution in MeOH (0.5 mL) was added dropwise while stirring at room temperature. After 10 hours, TLC indicated complete formation of a more polar compound. The reaction mixture was neutralized by addition of 4.35 g of Dowex resin (50WX8 200–400 MESH). The mixture was filtered through celite while rinsing with DCM (10 mL), then the filtrate was concentrated to give crude material which was then co-evaporated with 1mL toluene three times then placed under high vacuum for 20 minutes to give crude 24a that was used in the next step without further purification. To a stirred solution of 24a from the previous procedure in THF (3.0 mL) at 0 °C, benzyl bromide (0.4 mL, 3 mmol) was added followed by portion-wise addition of 60% NaH suspended in mineral oil (0.64 g, 16 mmol). The reaction was allowed to stir under nitrogen atmosphere while warming to room temperature. After 1 h, TLC showed that starting material 24a was still intact. 1 mL of DMF was added and reaction allowed to continue stirring. After an additional 1 hour, TLC now showed that all the starting material 24a was consumed to form a less polar spot (25). The reaction was diluted with 10 mL EtOAc then transferred into an ice bath. The reaction was then quenched by dropwise addition of water (5 mL) until gas evolution stopped. The mixture was diluted with 50 mL EtOAc, and washed with water (20 mL). The organic phase was separated, dried using Na2SO4 and then concentrated to give a crude material that was purified by flash chromatography (40–55% DE/petroleum ether, DE= 3/1 DCM/EtOAc) to give 184.4 mg of 25 (yellow oil, 84% two step yield).
Disaccharide 26.
To a stirred solution of 25 (184.4 mg, 0.1481 mmol) in dry dichloromethane (1 mL) under nitrogen atmosphere at 18 °C, was added 1M BH3·THF (0.7 mL, 0.7 mmol) dropwise followed by TMSOTf (3.0 μL, 0.017 mmol). After 3 hours, TLC showed that all of the starting material 25 was consumed to form a more polar spot (26). The reaction mixture was then transferred to an ice bath and quenched by dropwise addition of saturated NaHCO3 (0.5 mL) and then further quenched by dropwise addition of MeOH (0.5 mL) until gas evolution stopped. The mixture was diluted with dichloromethane (5.0 mL) then washed once with water (5 mL). The organic layer was separated, dried using Na2SO4, and concentrated to give crude material that was purified by flash chromatography (60–90% DE/petroleum ether, DE= 3/1 DCM/EtOAc) to give 156.5 mg of 26 (white foam, 85% yield).
Trisaccharide 27.
To a stirred solution of thioglycosyl donor 6 (407.2 mg, 0.6710 mmol) and galactosyl acceptor 26 (659.7 mg, 0.5292 mmol) in dichloromethane (3.0 mL) in a pear-shaped bottom flask (PBF) was added activated (by flame drying under vacuum) powdered 3 Å molecular sieves (0.4888 g, from BTC). The PBF was immediately capped using a septum and the mixture allowed to stir at room temperature (18 °C) for 18 minutes. The PBF was then transferred into a −10 °C cold bath and N-Iodosuccinimide (0.3044 g, 1.353 mmol) was added in one portion. After 4 minutes, silver triflate (38.9 mg, 0.151 mmol) was added in one portion and the inside wall of the PBF was rinsed quickly with 0.2 mL DCM (Note: While adding both NIS and AgOTf solids, the septum was removed and the respective solids added very fast while making sure that they dropped directly into the DCM solution and septum was put back on fast.). The reaction was allowed to slowly warm to 10 °C. After 85 minutes, TLC indicated complete consumption of donor 6. The reaction was filtered through celite while rinsing with 30 mL dichloromethane. The filtrate was washed vigorously with 8.0 mL saturated Na2S2O3 solution. The organic phase was separated and dried using Na2SO4 and then concentrated to give crude 26a that was purified by flash chromatography (40–65% DE/petroleum ether, DE= 3/1 DCM/EtOAc) to give 0.8970 g of 26a (thick yellowish oil, 98% yield). To a stirred solution of 26a (0.8869 g, 0.5128 mmol) in THF (4.0 mL) at 0 °C in a scintillation vial, 70% HF-pyridine (0.3 mL) was added. The reaction was allowed to stir and warm to room temperature (18 °C) under nitrogen atmosphere. 76 minutes later, TLC indicated complete consumption of starting material 26a. The reaction was quenched by dropwise addition of saturated NaHCO3 (5 mL). The mixture was then diluted with EtOAc (30 mL) then washed with 10mL saturated NaHCO3 (aq.) once. The organic phase was dried using Na2SO4, then concentrated to give a crude material that was purified by flash chromatography (40–65% DE/petroleum ether, DE= 3/1 DCM/EtOAc) to give 0.5705 g of 27 (colorless gum, 70% yield).
Tetrasaccharide 28.
To a stirred solution of thioglycosyl donor 5 (329.5 mg, 0.4700 mmol) and diol acceptor 27 (569.2 mg, 0.3582 mmol) in dichloromethane (8.0 mL) in a scintillation vial was added activated (by flame drying under vacuum) powdered 3 Å molecular sieves (0.9692 g, from BTC). The PBF was immediately capped using a septum and the mixture allowed to stir at room temperature (18 °C) for 14 minutes. The vial was then transferred into a −35 °C cold bath. After 6 minutes, N-Iodosuccinimide (0.1999 g, 0.8885 mmol) was added in one portion. 5 minutes later, silver triflate (48.4 mg, 0.188 mmol) was added in one portion and the inside wall of the PBF was rinsed quickly with 0.3 mL DCM (Note: While adding both NIS and AgOTf solids, the septum was removed and the respective solids added very fast while making sure that they drop directly into the DCM solution and septum was put back on fast). The reaction was allowed to slowly warm to −10 °C. After 110 minutes, TLC indicated complete consumption of donor 5. The reaction was filtered through celite while rinsing with 20 mL dichloromethane. The filtrate was washed vigorously with 10.0 mL saturated Na2S2O3 solution. The organic phase was separated and diluted with 50 mL DCM then washed vigorously with water (40 mL) twice (to get rid of succinimide). The organic phase was once again separated and dried using Na2SO4 and then concentrated to give crude 28 that was purified by flash chromatography (35–50% EtOAc/petroleum ether) to give 0.6450 g of 28 (yellowish white foam, 83% yield).
Pentasaccharide 3.
To a stirred solution of donor 4 (13.7 mg, 0.0205 mmol) and tetrasaccharide acceptor 28 (22.2 mg, 0.0103 mmol) in dichloromethane (0.25 mL) in a pear-shaped flask, powdered 3 Å molecular sieves (44.4 mg, activated by flame drying under vacuum) were added. The flask was flushed with nitrogen gas for 5 seconds and then put in a 6 °C cold bath. 55 μL of 0.414 M triflimide solution in CH3CN (0.023 mmol) was then added dropwise. After 34 minutes, TLC showed that all donor 4 was consumed. The reaction was quenched by addition of three drops of Et3N then filtered through celite while rinsing with 5 mL dichloromethane. The filtrate was concentrated using a rotary evaporator to give crude material that was purified using preparative TLC (38% EtOAc/hexanes) to give 11.9 mg of pentasaccharide 3 (white solid, 44% yield).
Final product 2.
To a stirred solution of pentasaccharide 3 (27.7 mg, 0.0105 mmol) in 0.1 mL CH2Cl2 in a pear-shaped flask, pyridine (0.6 mL) and thioacetic acid (0.35 mL) were added. The flask was flushed with nitrogen gas for 5 seconds and allowed to continue stirring. Reaction progress was monitored using FTIR by checking for disappearance of the azido signal (around 2106 cm −1). After 18 h, stirring was stopped and the reaction mixture was concentrated then co-evaporated with toluene to give crude material (AcSH was still present due to its high boiling point). The mixture was transferred into a scintillation vial while diluting with 2.0 mL dichloromethane. A low stream of pressurized nitrogen gas was carefully blown over the mixture for 2 hours to get rid of most of the AcSH. The remaining AcSH was removed by loading the crude material to a silica gel column, then running 50 mL of CH2Cl2 through it to wash out AcSH (yellow eluent). 1–2% MeOH/DCM was then passed through the column to elute 3a (26.2 mg) which was again pre-purified by preparative TLC using 31% acetone in petroleum ether to give 24.4 mg of clean 3a (white solid, 87% yield). To material 3a (16.1 mg, 0.00605 mmol) in a 4 mL Wheaton vial with a stir bar, dichloromethane (0.1 mL), EtOH (0.5 mL), and H2O (0.3 mL, HPLC grade) were added. The mixture was stirred briefly (2 minutes), then Pd(OH)2 (101.0 mg) was added in one portion. The vial was capped with a rubber septum, then hydrogen gas in a balloon was bubbled carefully through the mixture for approx. 45 seconds (this was done using two long, thin reusable syringe needles (Popper 7174 deflected point septum penetration hypodermic needles 22 * 2” (50.8 MM)) where the one attached to balloon was in contact with the reaction mixture on the sharp end while the other needle was impaled through the septum and acting as a hydrogen gas outlet). The reaction was then allowed to continue stirring at room temperature under hydrogen atmosphere (in a double walled balloon (balloon inside similar size balloon)). After 14 h, the balloon with hydrogen gas was detached and more water (0.3 mL) and EtOH (0.5 mL) was added to rinse back into solution any reaction content that was splashed onto the inside surface of vial. The vial was then flushed with hydrogen gas and the reaction was allowed to continue under hydrogen gas atmosphere (in balloon). After an additional 7 hours, the reaction mixture was filtered by passing through celite plug while rinsing using water (5 mL) and MeOH (5.0 mL). A low stream of pressurized nitrogen gas was blown over the filtrate overnight to get rid of the solvents, then approximately 5 mL of water was added and the solution was then lyophilized to give 7.5 mg of crude material that was purified by passing through sephadex LH-20 while using water (HPLC grade) as the mobile phase to give 6.4 mg of 2. NMR showed presence of an impurity. The product was then repurified by passing through bio-gel P2 while using water (HPLC grade) as the mobile phase to give 2.8 mg of clean 2 (white solid, 41% yield).
Supplementary Material
Acknowledgements
The authors acknowledge the National Institutes of Health (R15AI140226) for generous support of this research. The authors thank Ms. Connie David and Dr. Fabrizio Donnarumma (LSU) for assistance with high-resolution and other mass spectrometry experiments. The authors thank Dr. Thomas Weldeghiorghis (LSU) for assistance with, and helpful conversations about, NMR. The authors also thank Dr. Evgeny Vinogradov (NRC-Canada) for generously providing NMR data for the original O-glycan isolate.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting information for this article is given via a link at the end of the document.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- [1].Huang W, Yao Y, Long Q, Yang X, Sun W, Liu C, Jin X, Li Y, Chu X, Chen B, Ma Y, PLOS One 2014, 9, e100727–e100739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Garcia-Quintanilla M, Pulido MR, McConnell MJ, Curr. Pharm. Biotechnol. 2013, 14, 897–902. [DOI] [PubMed] [Google Scholar]
- [3].Lazureanu V, Porosnicu M, Gandac C, Moisil T, Baditoiu L Laza R, Musta V, Crisan A, Marinescu A-R, BMC Infect. Dis. 2016, 16(Suppl.), 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vila J,; Pachón J, Expert Opin. Pharmacother. 2008, 9, 587–599. [DOI] [PubMed] [Google Scholar]
- [5].Wisplinghoff H, Edmond MB, Pfaller MA, Jones RN, Wenzel RP, Seifert H, Clin. Infect. Dis. 2000, 31, 690–697. [DOI] [PubMed] [Google Scholar]
- [6].De Oliveira DMP, Forde BM, Kidd TJ, Harris PNA, Schembri MA, Beatson SA, Paterson DL, Walker MJ, Clin. Microbiol. Rev. 2020, 33, e00181–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Luo G, Lin L, Ibrahim AS, Baquir B, Pantapalangkoor P, Bonomo RA, Doi Y, Adams MA, Russo TA, Spellberg B, PLOS One 2012, 7, e29446–e29456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dijkshoorn L, Nemec A, Seifert H, Nat. Rev. Microbiol. 2007, 5, 939–951. [DOI] [PubMed] [Google Scholar]
- [9].Pachon J, McConnell MJ, Vaccine 2014, 32, 2534–2536. [DOI] [PubMed] [Google Scholar]
- [10].Gellings PS, Wilkins AA, Morici LA, Pathogens 2020, 9, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Giguère D, Carbohydr. Res. 2015, 418, 29–43. [DOI] [PubMed] [Google Scholar]
- [12].Vinogradov EV, Duus JØ, Brade H, Holst O, Eur. J. Biochem. 2002, 269, 422–430. [DOI] [PubMed] [Google Scholar]
- [13].Vinogradov EV, Peterson BO, Thomas-Oates JE, Duus J, Brade H, Holst O, J. Biol. Chem. 1998, 273, 28122–28131. [DOI] [PubMed] [Google Scholar]
- [14].Iwashkiw JA, Seper A, Weber BS, Scott NE, Vinogradov E, Stratilo C, Reiz B, Cordwell SJ, Whittal R, Schild S, Feldman MF, PLoS Pathogens 2012, 8, e1002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Verez-Bencomo V, Fernández-Santana V, Hardy E, Toledo ME, Rodriguez MC, Heynngnezz L, Rodriguez A, Baly A, Herrera L, Izquierdo M, Villar A, Valdés Y, Cosme K, Deler ML, Montane M, Garcia E, Ramos A, Aguilar A, Medina E, Toraño G, Sosa I, Hernandez I, Martinez R, Muzachio A, Carmenates A, Costa L, Cardoso F, Campa C, Diaz M, Roy R, Science 2004, 305, 522–525. [DOI] [PubMed] [Google Scholar]
- [16].Verheul AFM, Boons GJPH, Van der Marel GA, Van Boom JH, Jennings HJ, Snippe H, Verhoef J, Hoogerhout P, Poolman JT, Infect. Immun. 1991, 59, 3566–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boltje TJ, Zhong W, Park J, Wolfert MA, Chen W, Boons G-J, J. Am. Chem. Soc. 2012, 134, 14255–14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Adamo R, Nilo A, Castagner B, Boutureira O, Berti F, Bernardes GJL, Chem. Sci. 2013, 4, 2995–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Anish C, Schumann B, Pereira CL, Seeberger PH, Chem. Biol. 2014, 21, 8–50. [DOI] [PubMed] [Google Scholar]
- [20].Arihara R, Kakita K, Yamada K, Nakamura S, Hashimoto S, J. Org. Chem. 2015, 80, 4278–4288. [DOI] [PubMed] [Google Scholar]
- [21].Kenyon JJ, Hall RM, PLoS One 2013, 8, e62160–e62171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang S, Seeberger PH, Chem. Eur. J. 2021, 27, 17444–17451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Njeri DK, Ragains JR, Org. Lett. 2022, 24, 3461–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhou X-Y, Li L-X, Zhang Z, Duan S-C, Huang Y-W, Luo Y-Y, Mu X-D, Chen Z-W, Qin Y, Hu J, Yin J, Yang J-W, Angew. Chem. Int. Ed. 2022, 61, e202204420. [DOI] [PubMed] [Google Scholar]
- [25].Yang F-L, Lou T-C, Kuo S-C, Wu W-L, Chern J, Lee Y-T, Chen S-T, Zou W, Lin N-T, Wu S-H, Vaccine 2017, 35, 1440–1447. [DOI] [PubMed] [Google Scholar]
- [26].Qin C, Schumann B, Zou X, Pereira CL, Tian G, Hu J, Seeberger PH, Yin J, J. Am. Chem. Soc. 2018,140, 3120–3127. [DOI] [PubMed] [Google Scholar]
- [27].Mende M, Nieger M, Bräse S, Chem. – Eur. J. 2017, 23, 12283–12296. [DOI] [PubMed] [Google Scholar]
- [28].Hagen B, van Dijk JHM, Zhang Q, Overkleeft HS, van der Marel GJ, Codée JDC, Org. Lett. 2017, 19, 2514–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Puri K, Kulkarni SS, Org. Lett. 2021, 23, 7083–7087. [DOI] [PubMed] [Google Scholar]
- [30].Isoda Y, Kitamura K, Takahashi S, Nokami T, Itoh T, ChemElectroChem 2019, 6, 4149–4152. [Google Scholar]
- [31].Qiao Y, Ge W, Jia L, Hou X, Wang Y, Pedersen CM, Chem. Comm. 2016, 52, 11418–11421. [DOI] [PubMed] [Google Scholar]
- [32].Njeri DK, Pertuit CJ, Ragains JR, Org. Biomol. Chem. 2020, 18, 2405–2409. [DOI] [PubMed] [Google Scholar]
- [33].Sianturi J, Priegue P, Hu J, Yin J, Seeberger PH, Angew. Chem. Int. Ed. 2022, 10.1002/anie.202209556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ramadan S, Yang W, Zhang Z, Huang X, Org. Lett. 2017, 19, 4838–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Meng S, Zhong W, Yao W, Li Z, Org. Lett. 2020, 22, 2981–2986. [DOI] [PubMed] [Google Scholar]
- [36].Wang C-C, Lee J-C, Luo S-Y, Kulkarni SS, Huang Y-W, Lee C-C, Chang K-L, Hung S-C, Nature 2007, 446, 896–899. [DOI] [PubMed] [Google Scholar]
- [37].Tian G, Qin C, Liu Z, Shen D, Zou X, Fu J, Hu J, Seeberger PH, Yin J, Chem. Comm. 2020, 56, 344–347. [DOI] [PubMed] [Google Scholar]
- [38].Fu J, Laval S, Yu B, J. Org. Chem. 2018, 83, 7076–7084. [DOI] [PubMed] [Google Scholar]
- [39].Chao C-S, Li C-W, Chen M-C, Chang S-S, Mong K-KT, Chem. Eur. J. 2009, 15, 10972–10982. [DOI] [PubMed] [Google Scholar]
- [40].Chao C-S, Chen M-C, Lin S-C, Mong K-KT, Carbohydr. Res. 2008, 343, 957–964. [DOI] [PubMed] [Google Scholar]
- [41].Noti C, de Paz JL, Polito L, Seeberger PH, Chem. Eur. J. 2006, 12, 8664–8686. [DOI] [PubMed] [Google Scholar]
- [42].Tamura K, Mizukami H, Maeda K, Watanabe H, Uneyama K, J. Org. Chem. 1993, 58, 32. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.