

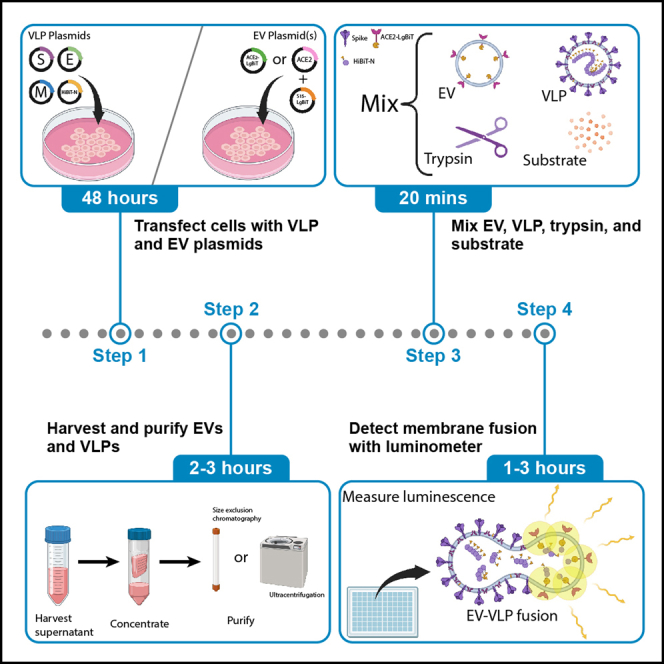
Summary
Here we present a protocol to measure coronavirus-mediated membrane fusion, an essential event in coronavirus cell entry. The approach uses nanoluciferase (Nluc) “HiBiT”-tagged corona virus-like particles (VLPs) and Nluc “LgBiT”-containing extracellular vesicles (EVs) as proxies for virus and cell, respectively. VLP-EV membrane fusion allows HiBiT and LgBiT to combine into measurable Nluc, which signifies virus fusion with target cell membranes. We highlight assay utility with methods to assess coronavirus-mediated fusion and its inhibition by antibodies and antiviral agents.
For complete details on the use and execution of this protocol, please refer to Qing et al. (2021),1 Qing et al. (2022),2 and Marcink et al. (2022).3
Subject areas: Cell Biology, Microbiology, Molecular Biology
Graphical abstract
Highlights
-
•
Quick, biosafe, high-throughput assay to measure coronavirus membrane fusion
-
•
Optimized for detection of SARS-CoV-2 and can be expanded to diverse coronaviruses
-
•
Effective at quantifying coronavirus neutralization by antibodies and fusion inhibitors
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here we present a protocol to measure coronavirus-mediated membrane fusion, an essential event in coronavirus cell entry. The approach uses nanoluciferase (Nluc) “HiBiT”-tagged corona virus-like particles (VLPs) and Nluc “LgBiT”-containing extracellular vesicles (EVs) as proxies for virus and cell, respectively. VLP-EV membrane fusion allows HiBiT and LgBiT to combine into measurable Nluc, which signifies virus fusion with target cell membranes. We highlight assay utility with methods to assess coronavirus-mediated fusion and its inhibition by antibodies and antiviral agents.
Before you begin
Introduction
Coronaviruses are enveloped RNA viruses infecting humans and animals. Animal coronavirus emergence into the human population can cause severe pandemic disease. Several assay platforms are required to understand coronavirus emergence, transmission, replication, and virulence. These include assays that quickly and easily measure coronavirus entry into host cells. Appropriate coronavirus entry assays should be able to isolate the entry step from other infection events, identify coronavirus variations impacting the entry steps, dissect the dynamics of the virus-cell interactions during entry, and characterize the action mechanisms of various antiviral agents blocking specific cell entry stages. Here we outline a coronavirus entry assay that meets these expectations. The assay is rapid, sensitive, modular, applicable to divergent coronaviruses, cell-free, and does not employ biohazardous replication-competent viruses. While we have optimized the assay for SARS-CoV-2, it is readily adaptable for general evaluation of coronavirus fusion.
The assay measures the membrane fusion between corona “virus-like particles” (VLPs) and extracellular vesicles (EVs). VLPs are structurally similar to authentic viruses; their assembly and egress from producer cells as well as their entry into target cells reflect aspects of the authentic coronavirus infection process. VLPs are produced by co-expressing four coronavirus structural proteins [S (spike), E (envelope), M (membrane), and N (nucleocapsid)].4 EVs are membrane-bound particles secreted from cells; a subset of EVs are generated by budding from the plasma membrane and thus contain plasma membrane proteins.5 EVs that incorporate coronavirus receptors are used as target membranes in viral fusion assays. VLP-EV fusion reflects authentic coronavirus-cell fusion in its dependence on viral S proteins, cell receptor proteins, and S-cleaving proteases.
The VLP and EV system has utilities that make it an attractive addition to existing assays that measure coronavirus entry. First, the VLPs and EVs can be assembled with any combination of coronavirus S or cellular S-binding receptor, making for a modular assay system. Second, the EVs are devoid of fusion-activating proteases, making it so that titrated proteases (typically trypsin) can measure differences in proteolytic activation of viral fusion.1,2 Third, the EVs and VLPs can be prepared as larger stocks, then aliquoted and frozen for future assays. The fusion assays themselves can be completed within 3 h in high-throughput fashion, in non-sterile BSL1 benchtop settings.
The assay depends on small nanoluciferase fragments (HiBiT) appended to coronavirus N proteins, and complementary large nanoluciferase fragments (LgBiT) on coronavirus receptor cytoplasmic tails. Alternatively, LgBiT can be tethered to EV membranes via lipophilic tags. Fusion of the VLP and EV membrane allows the union of HiBiT and LgBiT fragments to generate functional nanoluciferase enzyme activities, measured as readouts of viral membrane fusion.
Production of SARS-CoV-2 virus-like particles (VLPs)
Timing: ≈4 days
VLPs are produced by co-transfecting plasmids encoding coronavirus S, E, M, and N into “producer” cells. Conditioned media are then harvested and VLPs purified for subsequent assays. The N plasmids encode a modified N-terminal HiBiT-tagged form.
-
1.
The day before transfection (day 1): Seed ≈6 × 106 HEK293T cells in c-DMEM into each of two 145 cm2 tissue culture plates; note that number and size of plates can be scaled up or down.
-
2.
The day of transfection (day 2): At ∼ 80% cell confluence, replace media with fresh, pre-warmed c-DMEM approximately 30–60 min before transfection.
-
3.Obtain required plasmids for two transfection conditions: (a) complete VLPs (+S) and (b) “bald” VLPs (-S). See key resources table for plasmid sources. Plasmid maps are available upon request.
-
a.+S VLP transfection plasmids:
-
i.pcDNA3.1-SARS2-S-D614G.
-
ii.pcDNA3.1-SARS2-E.
-
iii.pcDNA3.1-SARS2-M.
-
iv.pcDNA3.1-SARS2- HiBiT-N.
-
i.
-
b.-S VLP transfection plasmids:
-
i.pcDNA3.1- empty vector.
-
ii.pcDNA3.1-SARS2-E.
-
iii.pcDNA3.1-SARS2-M.
-
iv.pcDNA3.1-SARS2- HiBiT-N.
-
i.
-
a.
-
4.Transfect cells using LipoD293 reagent:
-
a.Add 4 μg of each plasmid to 500 μL of serum free DMEM (total 16 μg plasmid per condition).
-
b.Add 48 μL of LipoD to 500 μL of serum free DMEM for each condition.
-
c.Immediately pipette the diluted LipoD293 into the diluted plasmid mixture. Not the reverse order.
-
d.Incubate plasmid/LipoD293 mixture for 10 min at room temperature.
-
e.Add 1 mL of LipoD/plasmid mixture dropwise to 145 cm2 dishes of 293T cells.
-
a.
-
5.
≈16 h post transfection (day 3): Replace media with pre-warmed serum free DMEM (20 mL per plate).
-
6.48 h post transfection (day 4): Collect and clarify the media from each transfected plate.
-
a.Centrifuge media 300 × g for 10 min at 4°C.; discard pellets.
-
b.Centrifuge media 3,000 × g for 10 min at 4°C.; discard pellets.
-
a.
-
7.
Store clarified media at 4°C.
-
8.
Add clarified media to 100k Amicon 15 mL filters.
-
9.
Centrifuge at 2,500 × g at 4°C until media is concentrated at least ≈40×.
-
10.
Collect concentrated supernatant from filter and bring up volume to 40× concentration with serum free media (if 20 mL starting supernatant bring concentrated volume to 500 μL).
-
11.
Aliquot concentrated supernatant and store at −80°C until use.
Production of ACE2+ LgBiT+ EVs
Note: EV and VLP production can be performed in parallel.
EVs are produced by transfecting plasmids encoding coronavirus receptors into EV “producer” cells. The plasmids encode receptors that are modified with C-terminal LgBiT appendages. Alternatively, plasmids encoding untagged receptors are co-transfected with plasmids encoding LgBiT that is N-terminally tagged with a 15-residue lipophilic “S15” peptide.6 EVs that bud from plasma membranes incorporate the plasmid-encoded proteins.
-
12.
The day before transfection (day 1): Seed ≈6 × 106 HEK293T cells in c-DMEM into each of two 145 cm2 tissue culture plates; note that number and size of plates can be scaled up or down.
-
13.
The day of transfection (day 2): At ∼ 80% cell confluence, replace media with fresh, pre-warmed c-DMEM approximately 30–60 min before transfection.
-
14.Obtain and thaw plasmids required for transfection. See key resources table for plasmid sources. Plasmid maps are available upon request.
-
a.For receptor covalently attached to LgBiT use plasmid:
-
i.pcDNA3.1-ACE2-LgBiT.
-
i.
-
b.For receptor separated from LgBiT (but still able to co-incorporate into EVs) use plasmids:
-
i.pcDNA3.1-S15-LgBiT.
-
ii.pcDNA3.1-hACE2-C9.
-
i.
-
a.
-
15.Transfect cells using LipoD293 reagent:
-
a.Dilute a total of 16 μg plasmid in 500 μL serum free DMEM per dish.
-
i.For pcDNA3.1-ACE2-LgBiT plasmid use 16 μg per dish.
-
ii.For -- pcDNA3.1-S15-LgBiT and pcDNA3.1-S15-LgBiT plasmids use 8 μg of each plasmid per dish (16 μg total).
-
i.
-
b.Add 48 μL of LipoD to 500 μL of serum free DMEM for each condition.
-
c.Immediately pipette the diluted LipoD293 into the diluted plasmid mixture. Not the reverse order.
-
d.Incubate plasmid/LipoD293 mixture for 10 min at room temperature.
-
e.Add 1 mL of LipoD/plasmid mixture dropwise to 145 cm2 dishes of 293T cells.
-
a.
-
16.
≈16 h post transfection (day 3): Replace media on the transfected plates with pre-warmed serum free DMEM (20 mL per plate).
-
17.
48 h post transfection (day 4): Collect the supernatant from each transfected plate.
-
18.Clarify the collected supernatant through centrifugation.
-
a.Centrifuge supernatant 300 × g for 10 min at 4°C.; discard pellet.
-
b.Centrifuge supernatant 3,000 × g for 10 min at 4°C.; discard pellet.
-
a.
-
19.
Store clarified supernatant at 4°C.
-
20.
Add clarified supernatant to 100k Amicon 15 mL filters.
-
21.
Centrifuge at 2,500 × g at 4°C until supernatant is concentrated at least ≈160×.
-
22.
Collect concentrated supernatant from filter and bring up volume to at least 160× concentration with serum free media (if 80 mL starting supernatant bring concentrated volume to 500 μL).
-
23.
Store at −80°C or purify further.
Purification of EVs and VLPs
EVs and VLPs are purified to remove extravesicular LgBiT and HiBiT fragments that generate background signals in the VLP-EV fusion assays. Size exclusion chromatography (SEC) or equilibrium density gradient centrifugation is used to purify EVs and VLPs. The methods below describe the SEC method.
-
24.
Wash a qEVoriginal/35 nm SEC column (IZON) with ≈20 mL PBS at room temperature.
-
25.
Load 500 μL of the concentrated supernatant into the SEC column.
-
26.
Elute the sample by gravity (∼ 1-mL/min) in PBS according to manufacturer instructions.
-
27.
Collect 16 × 500 μL fractions.
-
28.Dispense 5 μL of each fraction into wells of a white-walled 96 well plate.
-
a.Less of each fraction can be used if RLU generated from reactions is too high to be read accurately with luminometer.
-
a.
-
29.Add 35 μL per well of master mix containing the following:
-
a.0.2 μL per well of commercial purified HiBiT or LgBiT (Promega).
-
i.Use HiBiT to complement the LgBiT in EVs or LgBiT to complement the HiBiT in the VLPs.Note: The added HiBiT/LgBiT must be in excess in order to measure signal from sample. Using 0.2 μL per well of commercial purified HiBiT/LgBiT as described should provide excess reagent. Lower amounts can be used if care is taken that sample is still the limiting factor.
-
i.
-
b.0.2 μL per well of Nano-Glo substrate (Promega).
-
c.34.6 μL per well of 1× Passive Lysis Buffer (PLB, Promega).
-
a.
-
30.
Incubate plate 10 min at room temperature.
-
31.Read fractions in a room temperature luminometer.
-
a.We measure with a 0.5 s integration time using a GloMax® Explorer Multimode Microplate Reader (Cat. GM3510).
-
a.
-
32.Save early-eluting peak fractions (typically fractions 7–9).
-
a.Particles within the size range of EVs and VLPs will elute between fractions 7–9 (3.5–4.5 mL elution volume, see Figure 1).
-
a.
Note: complementation signals in later eluting fractions (after fraction 10) come from HiBiT or LgBiT – containing proteins that are not associated with VLPs or EVs. There should be a clear separation between the VLP-EV peak fractions and the later eluting free proteins. If not, then sequential rounds of SEC will be required to remove the contaminating non-vesicular LgBiT or HiBiT proteins that will generate background signals in the fusion assays.
-
33.
Add sodium azide (final 0.02%) to the purified EV – VLP samples for preservation.
-
34.Store SEC purified EVs and VLPs at 4°C.
-
a.SEC purified EVs are stable for at least 2–3 months at 4°C with minimal degradation.
-
b.SEC purified VLPs are stable for around 1 week at 4°C.
-
c.SEC purified EVs and VLPs in PBS elution buffer are functionally inactivated by freeze-thaw cycles. However, DMEM, sucrose, BSA and serum has cryopreservative properties. We typically add an equal volume of 2× DMEM to the EV and VLP samples before aliquoting and freezing at −80°C. Frozen samples are stable for months and possibly longer.
-
a.
Note: EVs and VLPs can also be purified and concentrated by equilibrium density gradient ultracentrifugation. Concentration with 100k Amicon filters is not required. Load EV/VLP-containing supernatants into appropriate ultracentrifuge tubes (i.e., Beckman SW28 tubes). Underlay with 20% and 50% (w/w) sucrose prepared in DMEM. Spin at 200,000 × g for 2 h at 4°C in a swing-out rotor. EVs and VLPs will band between 20% and 50% sucrose layers and are collected by fractionation at air-gradient interfaces. Fractions containing EVs and VLPs are identified by BiT complementation as described above.
Figure 1.

SEC elution profiles for LgBiT-containing EVs
S15-LgBiT EV (+/- hACE2) - containing supernatants were harvested, concentrated, and purified by SEC. Fractions of eluate were collected in PBS. 10 μL of each 0.5-mL fraction were complemented with HiBiT in presence of nanoluciferase substrate and passive lysis buffer. The signals at fractions 7–9 include peak EV eluates. Error bars represent standard error of the mean (SEM).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| COVID-19 convalescent sera samples | Dr. Susan Uprichard (Loyola University Chicago) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DrkBiT peptide ≥90% purity (VSGWALFKKIS) | Genscript (Fast Peptide Synthesis) | N/A |
| HiBiT control protein | Promega | Cat#N3010 |
| LgBiT protein (included in Nano-Glo HiBiT Extracellular Detection System kit) | Promega | Cat#N401A |
| Passive Lysis 5× Buffer | Promega | Cat#E1941 |
| Trypsin from bovine pancreas, TPCK treated | Sigma-Aldrich | Cat#T1426-50MG |
| LipoD293 | SignaGen | Cat#SL100668 |
| Corning® 10× Phosphate-Buffered Saline (PBS), pH 7.4 ± 0.1 | Fisher Scientific | Cat#46-013-CM |
| Corning™ DMEM with L-Glutamine, 4.5 g/L Glucose and Sodium Pyruvate | Fisher Scientific | Cat#MT10013CV |
| Penicillin-Streptomycin solution | Fisher Scientific | Cat#SV30010 |
| Corning™ HEPES, Liquid | Fisher Scientific | Cat#MT25060CI |
| MEM Non-Essential Amino Acids Solution (100×) | Fisher Scientific | Cat#11-140-050 |
| Cytiva L-Glutamine | Fisher Scientific | Cat#SH30034.01 |
| Fetal bovine serum - premium, heat inactivated | R&D Systems | Cat#S11150H |
| Critical commercial assays | ||
| Nano-Glo HiBiT Extracellular Detection System | Promega | Cat#N2420 |
| Nano-Glo Luciferase Assay System | Promega | Cat#N1110 |
| Experimental models: Cell lines | ||
| HEK-293T | Laboratory of Edward Campbell (Loyola University Chicago) | N/A |
| Recombinant DNA | ||
| pcDNA3.1-SARS2-S-D614G | (Qing et al.1) | N/A |
| pcDNA3.1-SARS2-E | (Kumar et al.4) | N/A |
| pcDNA3.1-SARS2-M | (Kumar et al.4) | N/A |
| pcDNA3.1-SARS2- HiBiT-N | (Kumar et al.4) | N/A |
| pcDNA3.1- empty vector | Invitrogen | Cat#V79020 |
| pcDNA3.1-hACE2-C9 | Dr. Michael Farzan (Scripps Research Institute) | N/A |
| pcDNA3.1-S15-LgBiT | (Marcink et al.3) | N/A |
| pcDNA3.1-ACE2-LgBiT | (Kumar et al.4) | N/A |
| pCMV6-Entry-huDPP4 (MERS receptor) | Origene (Barlan et al.7) | N/A |
| pcDNA3.1- CEACAM-Long (MHV receptor) | (Boscarino et al.8) | N/A |
| pCAGGS-MHV-A59 S | (Boscarino et al.8) | N/A |
| pCAGGS-MHV-A59 E | (Boscarino et al.8) | N/A |
| pCAGGS-MHV-A59 HiBiT-N | (Kumar et al.4) | N/A |
| pCAGGS-MHV-A59 M | (Boscarino et al.8) | N/A |
| pcDNA3.1- MERS-EMC S-C9 | (Barlan et al.7) | N/A |
| pcDNA3.1- MERS-EMC E | (Qing et al., 20209) | N/A |
| pcDNA3.1- MERS-EMC HiBiT-N | (Kumar et al.4) | N/A |
| pcDNA3.1- MERS-EMC M | (Qing et al., 20209) | N/A |
| Software and algorithms | ||
| Prism | GraphPad | N/A |
| Other | ||
| qEVoriginal / 35nm Legacy Column (SEC columns) | Izon | Cat#SP5 |
| GloMax® Explorer Multimode Microplate Reader | Promega | Cat#GM3510 |
| Corning™ 96-Well Solid White Polystyrene Microplates | Fisher Scientific | Cat#07-200-589 |
| Amicon® Ultra-15 Centrifugal Filter Unit (100 kDa cutoff) | Millipore | Cat#UFC910024 |
| Automatic fraction collector (AFC, V1) | IZON | N/A |
Materials and equipment
Luminometer
A luminometer is required to detect the luminescent signal released from the fusion reactions. The optimal temperature for these reactions is 37°C. Therefore, a temperature controlled luminometer is ideal. We use the GloMax® Explorer Multimode Microplate Reader (Promega, Cat#GM3510). This luminometer can be programmed to maintain a desired temperature and perform automatic readings at defined timepoints.
Size exclusion columns
The purification of the EV and VLP samples can increase the signal:background ratio of the assay by removing extravesicular HiBiT/LgBiT. We typically use size exclusion chromatography for purification as it is quick, gentle, and effective. We use qEVoriginal / 35nm Legacy Columns (IZON, Cat#SP5). An alternative to using these columns is purification using equilibrium density gradient centrifugation as described above.
Complete Dulbecco’s Modified Eagle Media (c-DMEM)
| Reagent | Final concentration | Amount |
|---|---|---|
| Corning™ DMEM with L-Glutamine, 4.5 g/L Glucose and Sodium Pyruvate | N/A | 430 mL |
| Fetal Bovine Serum - Premium, Heat Inactivated | 10% | 50 mL |
| HEPES buffer (1 M) | 10 mM | 5 mL |
| Penicillin-Streptomycin solution (100×) | 1× | 5 mL |
| L-Glutamine (200 mM) | 2 mM | 5 mL |
| MEM Non-Essential Amino Acids Solution (100×) | 1× | 5 mL |
| Total | N/A | 500 mL |
Serum-free Dulbecco’s Modified Eagle Media (DMEM)
| Reagent | Final concentration | Amount |
|---|---|---|
| Corning™ DMEM with L-Glutamine, 4.5 g/L Glucose and Sodium Pyruvate | N/A | 480 mL |
| HEPES buffer (1 M) | 10 mM | 5 mL |
| Penicillin-Streptomycin solution (100×) | 1× | 5 mL |
| L-Glutamine (200 mM) | 2 mM | 5 mL |
| MEM Non-Essential Amino Acids Solution (100×) | 1× | 5 mL |
| Total | N/A | 500 mL |
Note: All media should be stored at 4°C and used within a month of preparation. SEC columns should be stored according to manufacturer instructions.
Step-by-step method details
VLP-EV fusion assay
VLP-EV fusion is initiated by adding activating proteases (Figures 2 and 3). We use trypsin as the activating protease because it is known to activate several coronaviruses for cell entry,10,11 and has a similar cleavage specificity to TMPRSS2.12 Furimazine turnover is used to measure the HiBiT-LgBiT complementation that reads out VLP-EV fusion kinetics. Assay signals are interpreted relative to control conditions in which VLPs lack S proteins or EVs lack receptors.
-
1.
Place a white walled 96 well plate on ice.
CRITICAL: Keep plate and all reagents on ice until fusion reaction is ready to start. The fusion reaction is temperature sensitive.
-
2.Obtain and thaw required reagents:
-
a.1 mg/mL trypsin (TPCK treated, Sigma).
-
b.Nano-Glo HiBiT extracellular detection substrate (Promega).
-
c.Concentrated VLPs (+/- S) prepared in earlier steps.
-
i.Unconcentrated supernatant can be used but may require larger reaction volumes.
-
i.
-
d.Concentrated EVs prepared in earlier steps.
-
i.Unconcentrated supernatant can be used but may require larger reaction volumes.
-
i.
-
e.1× PBS.
-
a.
Optional: Inhibit extravesicular LgBiT in EVs with DrkBiT peptide by mixing the DrkBiT peptide with an aliquot of EVs diluted in 25 µL PBS. (10 µM final DrkBiT concentration). Incubate EV/DrkBiT mix at 37°C for 1 hour.
Note: DrkBiT is a small peptide competitive inhibitor of LgBiT. DrkBiT can be added to inhibit extravesicular LgBiT in fusion assays. This is typically not required if using sufficiently pure EVs. If using unpurified EVs, DrkBiT should be used to minimize background signals.
-
3.Dispense 25 μL diluted EVs in white walled 96 well plate.
-
a.Dilute in 1× PBS to 25 μL per well.
-
b.Amount of EVs added depends on concentration; 1 μL of 100× concentrated EV per well is a good starting point.
-
a.
-
4.On ice, prepare at least two parallel master mixes, one containing VLPs that contain spike and one containing VLPs that lack spike.
-
a.Nano-Glo extracellular detection substrate: 0.5 μL per well.
-
b.VLP (amount added depends on concentration):
-
i.1 μL of 40× concentrated VLP per well is a good starting point.
-
i.
-
c.Trypsin to a final concentration of 50 ng/μL.
-
d.PBS up to 25 μL volume per well.
-
a.
-
5.
Dispense 25 μL of master mixes into the wells containing EVs (final volume 50 μL).
-
6.
Bring the plate to 37°C.
-
7.Read luminescence at frequent time intervals for 1–3 h.
-
a.Fusion is interpreted as signal above background (background defined as RLU from condition with VLPs lacking spike).
-
a.
Figure 2.

Receptor and protease dependence in VLP-EV fusion assays
SEC purified S15-LgBiT EVs (+/- ACE2) were pre-incubated with DrkBiT before adding substrate, VLP and either trypsin (50 ng/μL) or PBS. Readings were taken every 2 min in a 37°C luminometer. Background is defined as RLU generated by parallel fusion assays in which VLPs lacked S proteins. Backgrounds were subtracted from each condition and graphed. Error bars represent standard error of the mean (SEM) and a line of best fit (LOWESS curve) was generated with GraphPad Prism software.
Figure 3.

Trypsin sensitivities of VLP-EV fusion assays
SEC purified ACE2 containing EVs were incubated with SARS-2 VLPs, substrate, and DrkBiT at 37°C for 1 h. After incubation, trypsin was added at indicated final concentrations. The reactions were read in a 37°C luminometer every 2 min. The data shown were taken from the 60 min-post trypsin time point. Error bars represent standard error of the mean (SEM).
Using the VLP-EV fusion assay to measure activities of antiviral agents
VLP-EV fusion is inhibited by antibodies and other antiviral inhibitors. The VLP-EV fusion assay accurately measures serum neutralization titers from convalescent or vaccinated patients, matching those determined by plaque-reduction neutralization titer (PRNT) assays involving authentic SARS-CoV-2 and other infectious coronaviruses (Figure 4). The VLP-EV fusion assay also accurately measures the activity of antiviral peptide fusion inhibitors as recently described in Marcink et al.3
-
8.
Place a white-walled 96 well plate on ice.
-
9.Obtain and thaw required reagents:
-
a.1 mg/mL trypsin (TPCK treated, Sigma).
-
b.Nano-Glo HiBiT extracellular detection substrate (Promega).
-
c.Concentrated +S and -S VLPs prepared in earlier steps.
-
d.EVs prepared in earlier steps.
-
e.PBS.
-
f.PBS containing 0.5% BSA.
-
g.Serum to be tested.
-
a.
-
10.Serially dilute sera samples in PBS containing 0.5% BSA per well.
-
a.Typically, half-log or log dilutions.
-
a.
-
11.Dilute VLP (+/- S), into 5 μL PBS per well.
-
a.Amount of VLP added may vary depending on yield. 1 μL of VLP per well may be a good starting point.
-
a.
-
12.Add the 5 μL diluted VLPs per well of the cold white walled 96 well plate.
-
a.Parallel of spike and bald VLPs for each serum sample dilutions.
-
a.
-
13.
Add 5 μL of serum sample dilutions per well to VLP containing wells.
-
14.
Incubate the plate at 37°C for 30 min.
-
15.
Bring plate back to ice.
-
16.Add a 40 μL master mix to each well composed of the following:
-
a.Nano-Glo HiBiT extracellular detection substrate: 0.5 μL per well.
-
b.ACE2 containing EV: amount added depends on yield.
-
c.Trypsin: to 50 ng/ μL final concentration in the total 50 μL reaction per well.
-
d.PBS: to 40 μL.
-
a.
-
17.
Incubate 30 min on ice.
-
18.
Shift to 37°C and read luminescence at frequent time intervals for 1–3 h.
Figure 4.

Comparison of antibody neutralization titers obtained by PRNT and VLP-EV fusion assays
PRNT assay: Infectious SARS-CoV-2 viruses were incubated with COVID-19 convalescent patient sera in half-log dilutions at 37°C for 30 min, inoculated onto Vero cells, and plaques enumerated two days later. The sera dilution factors generating 50% reduction of enumerated plaques were stated as Plaque Reduction Neutralization Titer (PRNT) values. VLP-EV fusion assay: SARS-2 VLPs were incubated with COVID-19 convalescent patient sera in half-log dilutions at 37°C for 30 min. After incubation, a mix of ACE2-containing EVs, substrate, and trypsin (50 ng/μL) were added to each well. The reactions were incubated on ice for 30 min. The reactions were then brought to 37°C and readings were recorded with a luminometer. IC50 values were calculated in GraphPad prism using the 60 min timepoint values.
Broadening the VLP-EV fusion assay to several coronaviruses
This VLP-EV fusion platform can be adapted to coronaviruses beyond SARS-CoV-2. We have used the assay format to evaluate mouse hepatitis coronavirus (MHV) and Middle East Respiratory Syndrome coronavirus (MERS-CoV) fusion with respective receptor-bearing EVs (see Figure 5).
-
19.Produce VLPs using the designated protocols, with the following changes:
-
a.Replace SARS-CoV-2 S, E, M, and HiBiT-N plasmids with plasmids encoding the S, E, M, and HiBiT-N proteins of the coronavirus of interest.
-
a.
-
20.Produce and purify EVs using the designated protocols, with the following changes:
-
a.Instead of co-transfecting ACE2 and S15-LgBiT plasmids, swap ACE2 with the corresponding coronavirus receptor (hDPP4 for MERS-CoV; mCEACAM for MHV).
-
a.
-
21.Perform the VLP-EV fusion reactions using designated protocols, with the following changes:
-
a.Volumes of EV and VLP required for each reaction may vary from the assays described for SARS-CoV-2.
-
a.
Figure 5.

Expanding VLP-EV fusion assays to diverse coronaviruses
(A and B) S15-LgBiT EVs containing either DPP4 (A) or CEACAM (B) were harvested and purified by SEC. 5 μL of each 0.5-mL fraction were complemented with HiBiT in presence of substrate and passive lysis buffer. The signals at fractions 7–9 include peak EV eluates. Error bars represent standard error of the mean (SEM).
(C) MERS VLPs were mixed with SEC-purified DPP4 EVs.
(D) MHV VLPs were mixed with SEC-purifed CEACAM EVs. VLPs and EVs were pre-incubated for 30 min at 37°C. Trypsin (50 ng/μL) and nanoluciferase substrate were then added, and luminescence readings were taken every 2 min in a 37°C luminometer. Background is defined as RLU generated by parallel fusion assays in which VLPs lacked S proteins. Error bars represent standard error of the mean (SEM).
Expected outcomes
Expected outcome of the VLP-EV fusion assay are illustrated in Figure 2. Background is defined as the RLU measured from EVs mixed with VLPs lacking spike. Expect signal:background ratios of >10 fold. Maximum fusion signals typically peak around 60 min after initiating the reactions with trypsin. The reactions are expected to be entirely dependent on trypsin or alternative proteases that cleave S proteins at the S2′ position.2 Trypsin concentrations required to activate SARS-CoV-2 VLPs (D614G) are around 5–50 ng/ μL (Figure 3). VLPs with variant spikes vary widely in the amounts of trypsin required for activation.1,2 Trypsin concentrations above 50 ng/μL may be inhibitory to fusion.
Limitations
The VLP-EV fusion assay cannot register virus-cell entry pathways. Most coronaviruses endocytose before fusion, and this is not revealed by the in vitro VLP-EV assays. The VLP-EV assays are poorly responsive to the pH dependence of fusion activation because nanoluciferase activities are reduced by acidic conditions. The VLP-EV assays are robust after trypsin cleavage of S proteins. Many coronaviruses are activated naturally by trypsin but others are more dependent on TMPRSS2 or cathepsin.11,13,14,15,16,17,18 TMPRSS2 and cathepsin weakly activate fusion in the VLP-EV assays. Our assay also depends on transfection of plasmids to produce the required EVs and thus does not use EVs from natural SARS-CoV-2 target cells.
Troubleshooting
Problem 1
High background values.
Potential solution
This problem usually derives from contaminating free HiBiT or free LgBiT in the VLP or EV preparations. To reduce background, ensure that DrkBiT is being used at sufficient concentrations. 10 μM DrkBiT is usually a high enough concentration to inhibit virtually all extravesicular LgBiT in our hands, but higher concentrations may be necessary if samples are heavily contaminated with extravesicular LgBiT. Optimally, ensure the DrkBiT is incubated with EV samples at 37°C for 1 h before adding HiBiT containing VLPs to ensure all available LgBiT is already bound to DrkBiT before HiBiT has a chance to bind.
Problem 2
Low signal values.
Potential solution
Ensure adequate amounts of EVs and VLPs are added per reaction. Increasing amounts of EVs and/or VLPs per reaction will increase signal strength.
Problem 3
Low EV/VLP yield.
Potential solution
The typical yield of EVs and VLPs from the production protocol above is sufficient to perform 500–1,000 reactions. Low yield may result from unhealthy or overconfluent cells. Ensure cells are healthy and are 70%–80% confluent at time of transfection. Ensure media changes are done gently and with pre-warmed media to prevent cell detachment. To preserve cell viability, the transfection mix should be removed from the cells 4–6 h post transfection and then replaced with fresh c-DMEM overnight. Harvest conditioned c-DMEM and replace with fresh c-DMEM every 24 h to ensure cell viabilities.
Problem 4
EVs/VLPs destroyed by freeze thaw.
Potential solution
EVs and VLPs can be frozen at −80°C if unpurified. Aliquot frozen material to minimize freeze thaws. If EVs or VLPs are purified with SEC, add 0.02% sodium azide for preservation and store at 4°C. EVs are stable for months at 4°C while VLPs are only stable for days. SEC purified EVs/VLPs will be destroyed by freeze thaw unless DMEM is added. Add DMEM (serum free) to a final concentration of 1× to the sample. Serum, sucrose or BSA will also cryopreserve the samples.
Problem 5
Sera neutralization titers are inconsistent.
Potential solution
Ensure 0.5% BSA is used in serum diluent to keep protein levels similar across serial dilutions.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tom Gallagher (tgallag@luc.edu).
Materials availability
Materials generated in this study are available upon request.
Acknowledgments
The study was supported by NIH grant 5P01 AI060699. We thank Susan Uprichard, Loyola University Chicago, for providing convalescent SARS-CoV-2 sera. We thank Abby Odle and Stanley Perlman, University of Iowa, for providing SARS-CoV-2 plaque reduction neutralization data.
Author contributions
T.K. performed experiments and wrote the original draft. T.K., E.Q., G.M.H., and A.W. performed supporting experiments and commented on the manuscript. T.K. and T.G. reviewed and edited the manuscript. T.G. supervised the study.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Thomas Kicmal, Email: tkicmal@luc.edu.
Tom Gallagher, Email: tgallag@luc.edu.
Data and code availability
All datasets generated during this study are available upon request.
References
- 1.Qing E., Kicmal T., Kumar B., Hawkins G.M., Timm E., Perlman S., Gallagher T. Dynamics of SARS-CoV-2 spike proteins in cell entry: control elements in the amino-terminal domains. mBio. 2021;12:e0159021. doi: 10.1128/mBio.01590-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qing E., Li P., Cooper L., Schulz S., Jäck H.M., Rong L., Perlman S., Gallagher T. Inter-domain communication in SARS-CoV-2 spike proteins controls protease-triggered cell entry. Cell Rep. 2022;39 doi: 10.1016/j.celrep.2022.110786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marcink T.C., Kicmal T., Armbruster E., Zhang Z., Zipursky G., Golub K.L., Idris M., Khao J., Drew-Bear J., McGill G., et al. Intermediates in SARS-CoV-2 spike–mediated cell entry. Sci. Adv. 2022;8:eabo3153. doi: 10.1126/sciadv.abo3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar B., Hawkins G.M., Kicmal T., Qing E., Timm E., Gallagher T. Assembly and entry of severe acute respiratory syndrome coronavirus 2 (SARS-CoV2): evaluation using virus-like particles. Cells. 2021;10:853. doi: 10.3390/cells10040853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doyle L.M., Wang M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. 2019;8:727. doi: 10.3390/cells8070727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell E.M., Perez O., Melar M., Hope T.J. Labeling HIV-1 virions with two fluorescent proteins allows identification of virions that have productively entered the target cell. Virology. 2007;360:286–293. doi: 10.1016/j.virol.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barlan A., Zhao J., Sarkar M.K., Li K., McCray P.B., Jr., Perlman S., Gallagher T. Receptor variation and susceptibility to Middle East respiratory syndrome coronavirus infection. J. Virol. 2014;88:4953–4961. doi: 10.1128/JVI.00161-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boscarino J.A., Logan H.L., Lacny J.J., Gallagher T.M. Envelope protein palmitoylations are crucial for murine coronavirus assembly. J. Virol. 2008;82:2989–2999. doi: 10.1128/JVI.01906-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qing E., Hantak M., Perlman S., Gallagher T. Distinct roles for sialoside and protein receptors in coronavirus infection. mBio. 2020;11 doi: 10.1128/mBio.02764-19. e02764–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menachery V.D., Dinnon K.H., 3rd, Yount B.L., Jr., McAnarney E.T., Gralinski L.E., Hale A., Graham R.L., Scobey T., Anthony S.J., Wang L., et al. Trypsin treatment unlocks barrier for zoonotic bat coronavirus infection. J. Virol. 2020;94 doi: 10.1128/JVI.01774-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffmann M., Hofmann-Winkler H., Pöhlmann S. Springer International Publishing; 2018. Priming Time: How Cellular Proteases Arm Coronavirus Spike Proteins; pp. 71–98. [DOI] [Google Scholar]
- 12.Lu M., Uchil P.D., Li W., Zheng D., Terry D.S., Gorman J., Shi W., Zhang B., Zhou T., Ding S., et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. Cell Host Microbe. 2020;28:880–891.e8. doi: 10.1016/j.chom.2020.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeda M. Proteolytic activation of SARS-CoV-2 spike protein. Microbiol. Immunol. 2022;66:15–23. doi: 10.1111/1348-0421.12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleine-Weber H., Elzayat M.T., Hoffmann M., Pöhlmann S. Functional analysis of potential cleavage sites in the MERS-coronavirus spike protein. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-34859-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heurich A., Hofmann-Winkler H., Gierer S., Liepold T., Jahn O., Pöhlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014;88:1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaudhry M.Z., Eschke K., Hoffmann M., Grashoff M., Abassi L., Kim Y., Brunotte L., Ludwig S., Kröger A., Klawonn F., et al. Rapid SARS-CoV-2 adaptation to available cellular proteases. J. Virol. 2022;96 doi: 10.1128/jvi.02186-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glowacka I., Bertram S., Müller M.A., Allen P., Soilleux E., Pfefferle S., Steffen I., Tsegaye T.S., He Y., Gnirss K., et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011;85:4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao M.M., Yang W.L., Yang F.Y., Zhang L., Huang W.J., Hou W., Fan C.F., Jin R.H., Feng Y.M., Wang Y.C., Yang J.K. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021;6:134. doi: 10.1038/s41392-021-00558-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets generated during this study are available upon request.