

Abstract
Macroautophagy (hereafter referred to as autophagy) is an essential quality-control pathway in neurons, which face unique functional and morphological challenges in maintaining the integrity of organelles and the proteome. To overcome these challenges, neurons have developed compartment-specific pathways for autophagy. In this review, we discuss the organization of the autophagy pathway, from autophagosome biogenesis, trafficking, to clearance, in the neuron. We dissect the compartment-specific mechanisms and functions of autophagy in axons, dendrites, and the soma. Furthermore, we highlight examples of how steps along the autophagy pathway are impaired in the context of aging and neurodegenerative disease, which underscore the critical importance of autophagy in maintaining neuronal function and survival.
Autophagy is a key neuroprotective pathway
Autophagy is an evolutionarily conserved lysosomal pathway that maintains cellular homeostasis by degrading and recycling aged or damaged cellular components [1–3]. During autophagy, cargo (e.g. proteins and organelles) that are targeted for degradation are engulfed by an isolation membrane that fuses with itself to form a closed, double-membraned autophagosome (Figure 1). Autophagosomes then mature into degradative compartments through a stepwise fusion with endolysosomal organelles [1–3]. The first key step involves fusion with organelles containing endosomal signatures to form amphisomes. The second key step is a terminal fusion event with degradative lysosomes to form autolysosomes that enable resident lysosomal hydrolases to digest the contents originating from the autophagosome. The term autophagic vacuoles (AVs) encompasses any LC3-positive compartment along this spectrum, including autophagosomes, amphisomes, and autolysosomes [4,5]. At the end of this process, degradation products are then exported from the lysosome and recycled to fuel new biosynthetic reactions. In this way, autophagy can control the composition and integrity of the cellular proteome and organelles.
Figure 1. Schematic of the autophagy-lysosomal pathway.
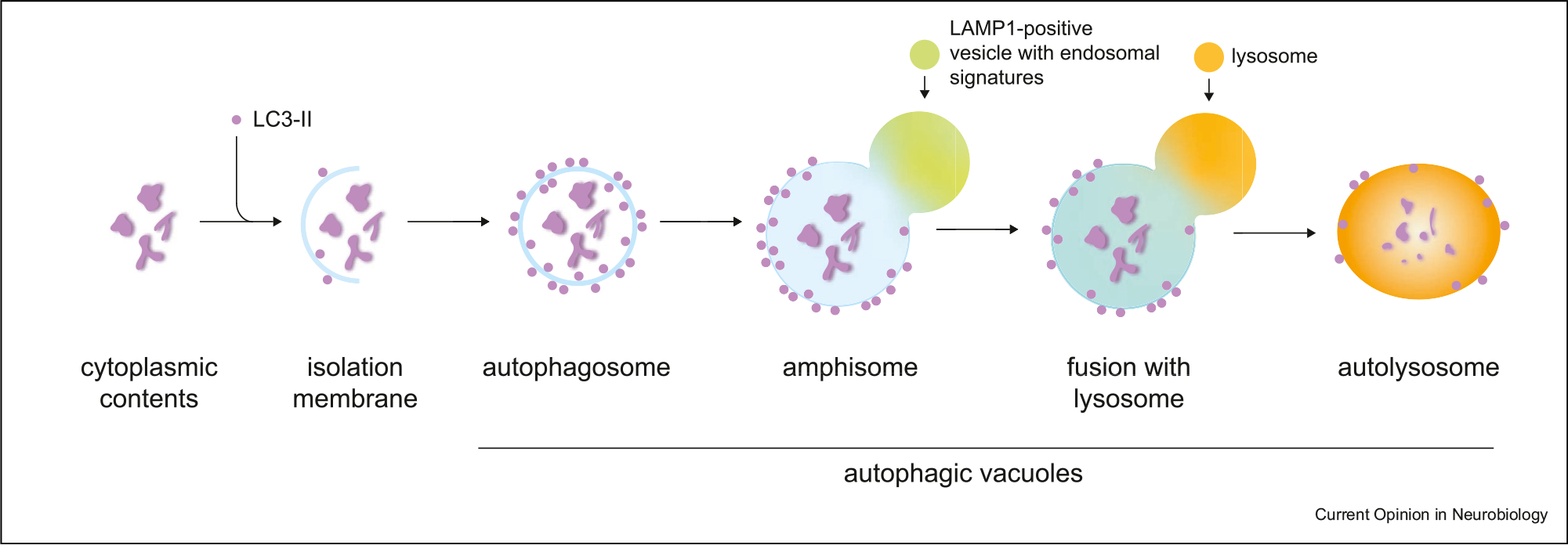
Cytoplasmic contents targeted for degradation are engulfed by an isolation membrane coated with LC3-II, a lipidated form of LC3. The membrane fuses with itself to form a double-membrane autophagosome. Autophagosomes mature into degradative autolysosomes through a series of fusion events with endolysosomal organelles. Contents of the autophagosome are digested by lysosomal hydrolases and breakdown products are recycled to fuel new biosynthetic reactions.
Neurons face unique challenges in regulating the quality and composition of their proteome and organelles. Neurons must establish and maintain a network of synaptic connections for nearly a century of time! A single neuron can make ~7000 synaptic connections (e.g. a human neocortical neuron) [6] and fire action potentials at rates up to 50 impulses per second (e.g. an inhibitory Purkinje cell in the rat cerebellum) [7,8]. Maintaining functionality for this extended lifetime renders neurons vulnerable to accumulating damage to their proteome and organelles. Since neurons are post-mitotic, they cannot simply dilute out proteotoxins by cell division. Thus, neurons are dependent on quality–control pathways to preserve the integrity and function of the neuronal proteome and organelles. Moreover, robust degradative pathways are also needed to remodel synapses to shape neuronal circuits throughout a lifetime.
Autophagy is essential for the proper connectivity, function, and survival of the nervous system. Loss of autophagy can elicit defects during neurodevelopment in axon outgrowth and guidance [9–12], dendritic growth and branching [13], synaptic development [11,14], and synaptic pruning [15]. Loss of autophagy can also elicit degeneration of established neuronal networks and neuromuscular junctions [12,16–22]. In fact, mutations in key autophagy genes are linked to neurodevelopmental and neurodegenerative disorders in humans [23–25]. Moreover, global loss of autophagy and acute reduction of autophagy specifically in the hippocampus are sufficient to cause deficits in learning and memory in mouse models, underscoring critical roles for autophagy in synaptic function and plasticity [21,26–28]. Stimulation of autophagy can rescue age-related cognitive decline in mice [26]. Combined, overwhelming evidence indicates autophagy is a key neuroprotective pathway, and the diversity of functions of autophagy in regulating neuronal physiology varies with context and neuronal subtype.
How is autophagy organized and regulated in neurons to enable neuronal function and survival? Neurons have a highly branched dendritic network that receives electrochemical inputs, and a single axon to transmit electrical information across distances that can reach up to 1 m. This complex morphology poses a challenge to organelle trafficking and requires an efficient method for delivering autophagosomes across this exceptionally polarized structure. These morphological and functional challenges necessitate unique compartment-specific regulation of the autophagy pathway [29]. In this review, we discuss the mechanisms and regulation of autophagy in each compartment of the neuron (i.e. axon versus soma versus dendrites) from autophagosome biogenesis, trafficking, to clearance. We also highlight examples that illustrate how defects in various stages of autophagy are associated with aging and neurodegeneration.
Axonal autophagy is a vectorial pathway that delivers cargo from the distal axon to the soma
Biogenesis
In axons, the majority of autophagosomes are produced in distal regions of the axon at presynaptic sites (Figure 2a, b) [11,30–35]. Formation of neuronal autophagosomes uses core proteins conserved from yeast (e.g. Atg13, a member of the initiation complex, and Atg5, a protein involved in elongation of the isolation membrane) that are recruited to specific subdomains of the endoplasmic reticulum (ER) in an ordered fashion [35,36] (Figure 2b). Autophagosome biogenesis in the distal axon is also dependent on the delivery of the transmembrane core protein ATG9, a lipid scramblase that facilitates expansion of the isolation membrane, in vesicles derived from the trans-Golgi network (TGN) [11,37–39]. Blocking delivery of ATG9 from the soma to the distal axon reduces autophagosome production and early stages of maturation [11,37,40]. At presynaptic sites, ATG-9 undergoes exocytosis and endocytosis in response to synaptic activity, a process that couples autophagosome formation with activity [39]. Thus, neurons use the same core machinery as yeast to generate autophagosomes, but this process is regulated by proteins enriched in presynaptic terminals. For example, the presynaptic scaffolding protein Bassoon negatively regulates autophagy by sequestering ATG5 [41]. Knockdown of Bassoon increased the number of autophagosomes in the axon and soma, but not in dendrites [41]. Endophilin A, a key player in synaptic vesicle endocytosis, induces autophagy by generating highly curved membranes for the assembly of autophagosome biogenesis factors [33]. Endophilin B may play a similar role in autophagosome production in presynaptic terminals [42]. Synaptojanin, a lipid phosphatase involved in synaptic vesicle trafficking, is also required for autophagosome formation in presynaptic terminals [43]. Evidence also suggests unexpected roles for the dynein adaptor Rab-interacting lysosomal protein (RILP) in promoting autophagosome biogenesis in axons of cortical neurons [44]. The RILP N-terminal domain binds to ATG5 and the RILP C-terminal domain appears to facilitate the closure of isolation membranes to form autophagosomes [44].
Figure 2. Overview of the compartment-specific organization of the autophagy-lysosomal pathway in neurons.
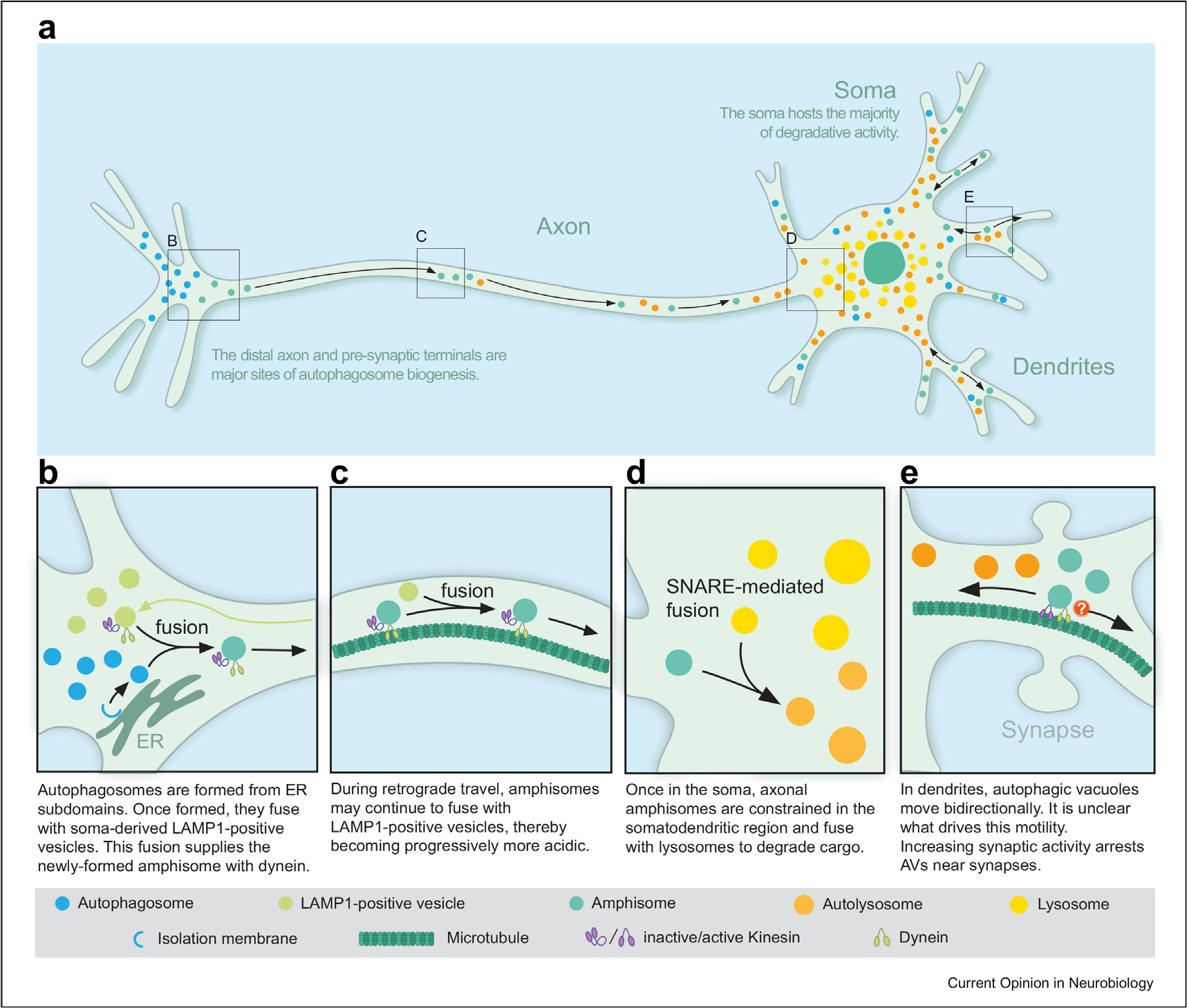
(a). Axonal autophagosomes form distally (b) and travel retrogradely toward the soma (c), where the majority of degradative activity is housed. There, autophagic vacuoles fuse with mature lysosomes (d) to degrade and recycle cargo. In dendrites, AVs exhibit bidirectional motility, and station at or near synapses in models of enhanced synaptic activity (e).
Trafficking
Following formation, axonal autophagosomes initially move bidirectionally in the distal axon but then transition to primarily unidirectional retrograde transport to the soma (Figure 2a) [31,32,45,46]. This switch in motility is triggered by a change in maturation state involving fusion of autophagosomes with an organelle population positive for late endosomal/lysosomal markers (Figure 2b) [31,45,47]. This fusion event is proposed to supply dynein to activate retrograde transport (Figure 2b, c); blocking fusion decreases the retrograde motility of autophagosomes, and autophagosomes accumulate in the distal axon [47]. In fact, ~95% of LC3-positive organelles in the mid-axon are co-positive for LAMP1 (marker for late endosomes and lysosomes) suggesting that LC3-positive organelles are autophagosomes for only a limited time before they mature into amphisomes [31,48]. Interestingly, the LAMP1-positive compartments that fuse with distal autophagosomes appear to be derived from the soma [49–51]. In fact, specifically blocking anterograde transport of soma-derived LAMP1-positive organelles inhibits the egress of autophagosomes from the distal axon [49,50]. Since LAMP1 can label a heterogeneous population of organelles [52,53], the precise nature of these soma-derived organelles is under investigation. Some evidence suggests that this population may include proteolytically active lysosomes [49,50]. In contrast, a more recent study suggests that this population may include TGN-derived transport carriers that deliver lysosomal components and degradative enzymes to organelles in the distal axon, but do not yet possess proteolytic activity [51]. Interestingly, many of these anterograde LAMP1 carriers were tubular in nature [51]. Recent evidence shows that LAMP-positive tubules originate from degradative lysosomes in the pre-axonal region through contacts with the ER [54]. However, these tubules may not be degradative [54,55]. Taken together, these data suggest an inside-out trafficking mechanism to clear autophagosomes from the distal axon, where soma-derived anterograde LAMP1-positive organelles facilitate the retrograde transport of autophagosomes formed in the distal axon.
As autophagosomes journey from the distal axon to the cell soma, they mature into autolysosomes. Retrograde GFP-LC3 carriers which are predominantly amphisomes have endocytic signatures and are co-positive for LAMP1, Rab7, and are partially acidified as measured with Lysotracker, an acidotropic dye that accumulates in organelles with pH ≤ 6 [31,45,47,51]. Using dyes reported to fluoresce with cathepsin B/D activity, recent studies indicate that axonal amphisomes may possess some proteolytic capability [50,51]. Axonal amphisomes are positive for various cargoes including cytoplasmic proteins and organelles such as mitochondria [31,56]. Recent proteomic analysis of brain-derived autophagosomes identified synaptic proteins and mitochondrial fragments as the major cargoes engulfed within neuronal autophagosomes [56]. Thus, axonal autophagic cargoes may start to break down en route to the cell soma. However, axons have a limited degradative capacity relative to the soma, which houses the vast majority of degradative lysosomes in the neuron [51–53,57]. Indeed, use of the acid-sensitive mCherry-GFP-LC3 reporter indicates that fully acidified autolysosomes (mCherry-positive, GFP-negative LC3 organelles) are concentrated in the soma [31,45,48,51]. Complete maturation of an axonal AV into a degradative organelle occurs upon entering the soma, possibly due to fusion events with proteolytically competent lysosomes resident in the soma (Figure 2d). Upon entry into the soma, axonal AVs (specifically the less mature species labeled with GFP-LC3) are restricted to the somatodendritic domain and prevented from reentering the axon, which may facilitate encounters with degradative lysosomes enriched in the soma of the neuron [30]. Therefore, autophagosomes travel the distance of the axon to deliver cargo to the soma for terminal degradation. Importantly, this pathway for axonal autophagy of distal formation followed by retrograde transport linked with organelle maturation is observed not only in primary neurons but is conserved in intact nervous systems in vivo, indicating a foundational pathway for basal autophagy to maintain axonal homeostasis [34,58–60]. Conditions of cellular stress, however, may elicit more localized autophagy within the axon [61].
Many accessory proteins have been identified to regulate the dynein-driven retrograde transport of AVs in the axon, including JIP1, HAP1-Huntingtin, JIP3, and RILP [34,44,62,63]. Loss of these accessory proteins reduces retrograde transport of AVs and disrupts their maturation into degradation-competent organelles [34,44,62,63], thereby linking organelle dynamics with their function. Why are so many different dynein effectors needed? Different dynein effectors may regulate the retrograde transport of AVs at distinct stages of maturation [64]. JIP1 regulates the initiation of retrograde transport in the distal axon, HAP1 functions in the mid-axon, and JIP3 functions in the mid/proximal axon to preferentially impact the retrograde motility of mature autolysosomes [64]. In addition to dynein, the anterograde motor kinesin is also bound to AVs [31]. Thus, disruptions in retrograde motility can also be caused by activation of bound kinesin, recruitment of kinesin motors, or both [62,65,66]. For example, hyperactivation of LRRK2 by a mutation associated with Parkinson’s disease increases JIP4 on AVs, which activates bound kinesin motors [65]. Consequently, dynein and kinesin engage in a tug-of-war that reduces the processivity of AVs in the axon, and their maturation into degradative autolysosomes [65]. Kinesin motors can also be recruited to autophagosomes depending on the phosphorylation state of LC3B [66]. Phosphorylation of LC3B at threonine 50 (T50) facilitates retrograde transport, whereas dephosphorylation at T50 engages anterograde transport via recruitment of the kinesin adaptor FYCO1 [66]. Across these studies, a unifying theme is that disruptions in the retrograde transport of AVs in the axon lead to defects in their maturation. Given that many of these proteins have been implicated in neurodegenerative disease, these findings provide a mechanistic basis for linking defects in autophagy with neuronal dysfunction and death.
The soma houses the vast majority of proteolytically active lysosomes in the neuron
Degradative lysosomes are concentrated in the neuronal soma [48,51–53,57] (Figure 2a). This finding is based on the distribution and activity of lysosomal hydrolases in the neuron [51–53,57]. Thus, the cell soma is the primary site of terminal degradation of various cargoes originating in dendrites and axons [53]. In fact, blocking lysosome function with Bafilomycin A1 results in the accumulation of GFP-LC3-positive organelles predominantly in the soma, indicating the soma as the final site of autophagosome clearance [30]. A recent study has shown that loss of WDR91, a Rab7 effector, leads to uncontrolled lysosomal fusion resulting in enlarged lysosomes with reduced degradative capacity [67]. Consequently, immature AVs accumulate in the soma and fail to degrade autophagic cargo [67]. These events are sufficient to drive neurodegeneration [67]. Thus, axonal autophagosomes travel the distance of the axon to reach mature lysosomes in the soma for effective degradation, which may enable efficient recycling of contents to primary sites of biosynthesis. In addition to autophagosomes originating in the axon, autophagosomes can also be formed locally within the soma [30,35]. Selective engulfment of damaged mitochondria into autophagosomes, via the mitophagy receptor optineurin, in neurons occurs mostly in the soma and is less frequently observed in axons and dendrites [68]. Surprisingly, these organelles persist in a non-acidified state for hours to days, suggesting that the degradation of damaged mitochondria may be slow and inefficient in neurons, representing a point of vulnerability [68].
Dendritic autophagy is a more localized process
Comparatively less is known about AV trafficking and clearance in dendrites versus axons. Interestingly, dendrites appear to contain few immature AVs as detected by the acid-sensitive GFP-LC3 marker [35], and appear to contain more mature AVs [48]. These AVs are likely derived from the soma, as autophagosome biogenesis is less frequent in dendrites under basal conditions [35]. Similar to axons, ~90% of LC3-positive organelles in dendrites are positive for LAMP1, suggesting that the majority of AVs in dendrites are amphisomes or autolysosomes [48]. To distinguish a population of AVs that are degradative autolysosomes, Kulkarni et al. used DQ-BSA, an endocytic substrate that fluoresces only upon proteolytic cleavage [48]. Kulkarni et al. found that dendrites have a greater percentage of DQ-BSA labeled degradative autolysosomes as compared with axons [48]. Further, AVs in dendrites fuse with newly endocytosed cargo to a greater extent than AVs in axons [48]. Thus, compared with axons, dendrites have a greater percentage of DQ-BSA labeled degradative autolysosomes due to compartment-specific differences in how the autophagy pathway intersects with the endolysosomal pathway [48].
Unlike the long-range unidirectional motility exhibited by amphisomes in the axon, AVs in dendrites undergo short-range bidirectional motility [30,35,48] (Figure 2e). This bidirectional transport could be achieved via dynein navigating the dendritic microtubule cytoskeleton, which is mixed in polarity, in contrast to the uniform polarity in axons [69]. Alternatively, this bidirectional transport could be achieved via a competition between bound dynein and kinesin motors [70]. Strikingly, the motility of dendritic AVs is controlled by synaptic activity [48]. Stimulation of synaptic activity reduces AV motility, whereas silencing synaptic activity increases AV motility [48] (Figure 2e). These activity-dependent effects on dendritic AV motility are local, reversible, and position AVs near synapses [48]. Most strikingly, synaptic activity increases the percentage of dendritic AVs that are autolysosomes, thus increasing their degradative function [48]. Importantly, this activity-dependent decrease in AV motility and increase in AV degradative function occurs specifically in dendrites and not in axons [48]. These findings are consistent with Goo et al. that show an activity-dependent localization of LAMP1-positive compartments to dendritic spines that may promote degradation of AMPARs [71]. Thus, this process may locally control the composition of the synaptic proteome, either through degradation of specific postsynaptic components or by providing fuel for local biosynthesis at synapses. The balance of these activities may help to regulate synaptic function and plasticity to impact learning and memory.
Molecular functions of autophagy in pre- versus postsynaptic compartments
The compartment-specific pathways for autophagy suggest that autophagy serves distinct functions in the axon and somatodendritic regions. Neurons regulate the neuronal proteome in response to synaptic activity [72,73]. At the synapse, autophagy may function to turn over the local proteome and organelles. In fact, various models of learning increase levels of autophagy in the hippocampus [26–28], and this activity-induced autophagy promotes long-term memory [21,26–28]. Stimulation of neuronal activity increases autophagosome biogenesis in presynaptic terminals [33,34,39,43] and their retrograde flux toward the soma [74]. Synaptic activity can also induce autophagosome formation in postsynaptic compartments [75]. However, some reports suggest that under specific contexts, autophagy is suppressed to facilitate learning and memory [76,77]. Thus, levels of autophagy may be finely tuned to enable learning and memory under various paradigms.
What are the precise functions and cargoes for autophagy at synapses? Autophagy has been shown to negatively regulate presynaptic neurotransmission by controlling the axonal ER [78]. Kuijpers et al. find that conditional knockout of Atg5 in excitatory neurons of the mouse cortex and hippocampus increased excitatory neurotransmission compared to wild-type controls, and led to neuronal death without altering synapse number or density [78]. Mechanistically, loss of autophagy resulted in the accumulation of tubular ER specifically in axons and not in dendrites, leading to an elevated release of calcium from ER stores and increased exocytosis of synaptic vesicles [78]. Autophagy has also been shown to negatively regulate presynaptic neurotransmission by controlling the synaptic vesicle pool size and release probability [76,79]. Hernandez et al. find that conditional knockout of Atg7 in dopaminergic neurons increased dopamine release compared to wild-type mice [79]. Conversely, modestly stimulating autophagy with rapamycin reduced the synaptic vesicle pool, raising the possibility that synaptic vesicles may be substrates for autophagic degradation in presynaptic terminals [79]. Consistent with this model, Gu et al. find that conditional knockout of Atg5 in CA3 hippocampal neurons increased the number of synaptic vesicles in the readily releasable pool and their rate of release [76]. Moreover, several groups also find that autophagosomes contain synaptic vesicle proteins [41,56,80–82] and components of the presynaptic active zone [34,56].
Dendritic autophagy degrades many postsynaptic proteins including AMPARs [75,83], and scaffolding proteins of dendritic spines such as PSD-95, PICK1, SAP97, and SHANK3 [56,75,77]. Studies in hippocampal neurons indicate that autophagy can impact both the strengthening of a synapse through long-term potentiation (LTP) [21,26,77] and the weakening of a synapse through long-term depression (LTD) [75,84,85]. For example, Glatigny et al. find that chemical induction of LTP increases autophagy and this activity-induced autophagy is required for the formation of new dendritic spines [26]. Nikoletopoulou et al. report that autophagy negatively regulates LTP potentially via degradation of postsynaptic scaffolding proteins [77]. Kallergi et al. find that induction of LTD by activation of NMDA receptors or metabotropic glutamate receptors triggers autophagosome formation in dendrites, and dendritic autophagy, in turn, promotes LTD [75]. Moreover, Compans et al. suggest that autophagy facilitates NMDAR-mediated LTD via degradation of PSD-95 to allow for lateral diffusion of AMPARs away from nanodomains that face presynaptic release sites [85]. Thus, autophagy is emerging as a key modulator of synaptic plasticity with critical roles on both sides of the synapse.
Defects in autophagosome biogenesis, trafficking, and clearance in aging and neurodegenerative disease
Defects in biogenesis
Various stages in the life of an autophagosome, from formation to clearance, can be altered in the context of aging and neurodegenerative disease. A recent study by Stavoe et al. shows that neurons cultured from aged mice exhibit a reduction in the rate of autophagosome biogenesis as evidenced by an increase in isolation membranes stalled at an early stage of development that fail to recruit LC3 [86]. Interestingly, overexpression of WIPI2B, a protein important for autophagosome formation, is sufficient to restore rates of autophagosome biogenesis in aged neurons (16–17 months) to levels observed in younger mice (3 months) [86]. Autophagosome biogenesis depends on tightly regulated WIPI2B phosphorylation and de-phosphorylation events, which become impaired during aging [86]. Further research should elucidate the posttranslational modifiers of WIPI2B and how their activity and localization might be misregulated in aged neurons. Reduced autophagosome formation in age-related neurodegenerative diseases may also be attributed to changes in the transcriptional regulation of autophagy proteins [87]. In fact, postmortem analysis of the parahippocampal gyrus from brains of Alzheimer’s disease (AD) patients showed a decreased expression of genes enriched in autophagosome biogenesis including those that encode ATG5, ATG12, ATG16, GABARAPL1, LC3, and the autophagy kinase complexes BECN1-PIK3C3 and ULK1/2-FIP200 [87].
Defects in autophagosome formation can also manifest through loss of function of the adaptor protein complex AP-4 which causes hereditary spastic paraplegia [37,40]. AP-4 deficiency arrests ATG9 in the TGN, thereby reducing autophagosome formation in the distal axon [37,40]. Moreover, C9ORF72, a Rab GEF that is implicated in ALS pathology, has been found to promote autophagosome biogenesis in primary neurons via interactions with the autophagy initiation complex, ULK1-FIP200-ATG13-ATG101 [88–90]. Loss of C9ORF72 impairs autophagy in primary neurons and synergizes with another ALS-linked protein Ataxin-2 to cause lower-motor neuron degeneration [89], suggesting defects in autophagosome biogenesis in ALS. However, C9ORF72 may suppress autophagosome biogenesis in nonneural cells [91]. Importantly, the GGGGCC (G4C2) hexanucleotide repeat expansion in C9orf72 that causes ALS and FTD impairs autophagic cargo degradation in Drosophila motor neurons [92]. This phenotype is partially caused by the loss of nuclear localization of the Drosophila homolog of TFEB, a transcriptional regulator of autophagosome and lysosome biogenesis [92]. G4C2-mediated neurodegeneration can be rescued by knocking down expression of the Drosophila homolog of the autophagy receptor p62, linking defective autophagy with neuronal death [92].
Additionally, mutations in autophagy receptors that recruit specific ubiquitinated cargoes to the growing autophagosome via an LC3-interacting region (e.g. p62 or optineurin) have been linked to neurodegeneration [93–96]. Moreover, mutations in proteins that regulate mitophagy such as kinases TBK1 and PINK1, and the ubiquitin E3 ligase parkin are connected to neurodegenerative diseases [93,97]. A recent study shows that ALS-linked mutations in TBK1, a kinase that phosphorylates optineurin to promote mitophagy [98], can lead to an accumulation of mitochondria with altered morphology and membrane potential, and stalled mitophagy, in the neuronal soma [99]. These findings indicate that a failure to effectively load cargo into autophagosomes may underlie the progression of neurodegenerative disease. In total, a variety of mechanisms can reduce the capacity of neurons to generate autophagosomes or engulf proteotoxins, thereby disrupting proteostasis networks and contributing to neuronal death.
Defects in trafficking and clearance
AV trafficking and clearance are also disrupted in models of neurodegenerative disease. Models of AD exhibit an accumulation of defective AVs and endolysosomal organelles in axonal swellings [45,57]. This phenotype can be due at least in part to deficits in anterograde trafficking of soma-derived LAMP1-positive vesicles that are required for the retrograde motility of axonal AVs [100,101]. Furthermore, defects in retrograde transport of AVs in AD can also be attributed to impaired AV-motor interactions, as Aβ 1–42, the pathogenic Aβ oligomer species linked to AD, blocked the recruitment of dynein motors to amphisomes [102]. Consequently, the failure to clear axonal AVs augments AD pathology by retaining axonal BACE1, the enzyme that converts APP to Aβ, which increases Aβ production and synaptic deficits [103,104]. Loss of the dynein effector JIP3 is also sufficient to accumulate LAMP1/Rab7 co-positive organelles, which likely includes amphisomes, in axon swellings, and increase axonal BACE1 and Aβ production [105]. Volpicelli et al. find that addition of preformed fibrils of α-synuclein to primary hippocampal neurons alters retrograde axonal transport of Rab7-positive endosomes and GFP-LC3-positive AVs, and reduces their maturation into degradative autolysosomes [106]. Lastly, the terminal stages of autophagy have also been reported to be disrupted in neurodegenerative disease; overexpression of α-synuclein reduces autophagosome-lysosome fusion via decreased expression of the SNARE protein SNAP29 on AVs [107]. Co-expression of SNAP29 with α-synuclein rescued the fusion defect [107]. Interestingly, defects in autophagy promote cell-to-cell spread of pathology via secretion of α-synuclein and tau into the extracellular space [108–110]. Taken together, defects throughout the autophagy pathway are associated with the progression of neurodegenerative diseases, underscoring the importance of the highly compartmentalized nature and functions of autophagy in neuroprotection.
Conclusions and future directions
Autophagosome biogenesis and trafficking in neurons are highly spatially organized. Autophagosomes in the axon are created predominantly in the axon terminal, and travel retrogradely along microtubules to the soma via fusion with soma-derived LAMP1-positive vesicles. This fusion supplies autophagosomes with the adaptors needed to associate with dynein and begin their retrograde trafficking to the soma. Once in the soma, neuronal autophagosomes fuse with perinuclear lysosomes to degrade their cargo. This retrograde pathway for axonal autophagy is regulated by the activity state of the neuron. Synaptic activity stimulates autophagosome formation in axon terminals and subsequent movement to the soma. In contrast, AVs in dendrites exhibit bidirectional motility and synaptic activity arrests AVs near synapses to localize degradative function at postsynaptic compartments. Lastly, defects in autophagosome dynamics, from biogenesis to trafficking and fusion with lysosomes, can impair autophagic function and cause neuronal degeneration.
Many exciting avenues of exploration in the field of neuronal autophagy need further investigation. We need to understand the diversity of the functions of autophagy in the neuron based on neuronal subtype, developmental stage, and context. For example, what are the roles and dynamics of constitutive, basal autophagy as compared to autophagy induced by cellular stress or neuronal activity, in the context of all compartments of the neuron? Particular focus needs to be applied to autophagy in dendrites where much less is understood. For example, the molecular mechanisms by which synaptic activity controls AV motility and maturation into degradative organelles in dendrites need to be explored further. Moreover, it remains unclear what proportion of autophagic output contributes to new biosynthesis, as a form of creation by destruction, in postsynaptic compartments that may impact learning and memory. Future studies need to precisely define the intersection between autophagy and endolysosomal trafficking to establish how these two critical pathways that regulate the proteome are organized in neurons. Lastly, how autophagy is affected in neurodegenerative disease and how it might be manipulated to promote neuroprotective outcomes warrants further investigation. By developing a comprehensive understanding of autophagy in neuronal function, we may find solutions to mitigate neuronal dysfunction and degeneration.
Acknowledgments
This work was supported by NIH grant R01NS110716 and associated Research Supplement to Promote Diversity in Health-Related Research to SM. We thank Maeve Coughlan, Max Stempel, and Vineet Kulkarni for constructive feedback on the manuscript.
Footnotes
Conflict of interest statement
Nothing declared.
References
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Mizushima N: A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol 2018, 20:521–527. [DOI] [PubMed] [Google Scholar]
- 2.Nakatogawa H: Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol 2020, 21:439–458. [DOI] [PubMed] [Google Scholar]
- 3.Melia TJ, Lystad AH, Simonsen A: Autophagosome biogenesis: from membrane growth to closure. J Cell Biol 2020, 219:e202002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T: LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000, 19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo-Arozena A, et al. : Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17:1–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pakkenberg B, Pelvig D, Marner L, Bundgaard MJ, Gundersen HJ, Nyengaard JR, Regeur L: Aging and the human neocortex. Exp Gerontol 2003, 38:95–99. [DOI] [PubMed] [Google Scholar]
- 7.Harris JJ, Attwell D: The energetics of CNS white matter. J Neurosci 2012, 32:356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LeDoux MS, Lorden JF: Abnormal spontaneous and harmaline-stimulated Purkinje cell activity in the awake genetically dystonic rat. Exp Brain Res 2002, 145:457–467. [DOI] [PubMed] [Google Scholar]
- 9.Dragich JM, Kuwajima T, Hirose-Ikeda M, Yoon MS, Eenjes E, Bosco JR, Fox LM, Lystad AH, Oo TF, Yarygina O, et al. : Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. Elife 2016, 5:e14810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kannan M, Bayam E, Wagner C, Rinaldi B, Kretz PF, Tilly P, Roos M, McGillewie L, Bar S, Minocha S, et al. : WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc Natl Acad Sci U S A 2017, 114:E9308–E9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stavoe AK, Hill SE, Hall DH, Colon-Ramos DA: KIF1A/UNC-104 transports ATG-9 to regulate neurodevelopment and autophagy at synapses. Dev Cell 2016, 38:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaguchi J, Suzuki C, Nanao T, Kakuta S, Ozawa K, Tanida I, Saitoh T, Sunabori T, Komatsu M, Tanaka K, et al. : Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy 2018, 14:764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark SG, Graybeal LL, Bhattacharjee S, Thomas C, Bhattacharya S, Cox DN: Basal autophagy is required for promoting dendritic terminal branching in Drosophila sensory neurons. PLoS One 2018, 13, e0206743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen W, Ganetzky B: Autophagy promotes synapse development in Drosophila. J Cell Biol 2009, 187:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, et al. : Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83:1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. : Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441:885–889. [DOI] [PubMed] [Google Scholar]
- 17.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. : Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441:880–884. [DOI] [PubMed] [Google Scholar]
- 18.Komatsu M, Wang QJ, Holstein GR, Friedrich VL Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z: Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A 2007, 104:14489–14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang CC, Wang C, Peng X, Gan B, Guan JL: Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem 2010, 285:3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M: Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 2007, 3:591–596. [DOI] [PubMed] [Google Scholar]
- 21.Zhao YG, Sun L, Miao G, Ji C, Zhao H, Sun H, Miao L, Yoshii SR, Mizushima N, Wang X, et al. : The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015, 11:881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudnick ND,Griffey CJ,Guarnieri P,Gerbino V,Wang X,Piersaint JA, Tapia JC, Rich MM, Maniatis T: Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc Natl Acad Sci U S A 2017, 114:E8294–E8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maday S: Mechanisms of neuronal homeostasis: autophagy in the axon. Brain Res 2016, 1649:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim M, Sandford E, Gatica D, Qiu Y, Liu X, Zheng Y, Schulman BA, Xu J, Semple I, Ro SH, et al. : Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. Elife 2016, 5:e12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collier JJ, Guissart C, Olahova M, Sasorith S, Piron-Prunier F, Suomi F, Zhang D, Martinez-Lopez N, Leboucq N, Bahr A, et al. : Developmental consequences of defective ATG7-mediated autophagy in humans. N Engl J Med 2021, 384:2406–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.*. Glatigny M, Moriceau S, Rivagorda M, Ramos-Brossier M, Nascimbeni AC, Lante F, Shanley MR, Boudarene N, Rousseaud A, Friedman AK, et al. : Autophagy is required for memory formation and reverses age-related memory decline. Curr Biol 2019, 29:435–448. Here, the authors show that activity-induced autophagy in the hippocampus is required to form novel memories by promoting structural and functional synaptic plasticity. Moreover, the authors show that stimulating autophagy can reverse age-related cognitive decline in mice.
- 27.**. Pandey K, Yu XW, Steinmetz A, Alberini CM: Autophagy coupled to translation is required for long-term memory. Autophagy 2021, 17:1614–1635. In this paper, the authors show that learning induces autophagy in the hippocampus and this learning-induced autophagy is not required for learning or short-term memory, but rather for long-term memory. This study underscores the importance of synaptic autophagy as an essential mechanism of long-term memory.
- 28.Hylin MJ, Zhao J, Tangavelou K, Rozas NS, Hood KN, MacGowan JS, Moore AN, Dash PK: A role for autophagy in long-term spatial memory formation in male rodents. J Neurosci Res 2018, 96:416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulkarni VV, Maday S: Compartment-specific dynamics and functions of autophagy in neurons. Dev Neurobiol 2018, 78:298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maday S, Holzbaur EL: Compartment-specific regulation of autophagy in primary neurons. J Neurosci 2016, 36:5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maday S, Wallace KE, Holzbaur EL: Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 2012, 196:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hollenbeck PJ: Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol 1993, 121:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernandez-Diaz S, Swerts J, Schoovaerts N, Vilain S, Gounko NV, Vints K, et al. : A LRRK2-dependent EndophilinA phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 2016, 92:829–844. [DOI] [PubMed] [Google Scholar]
- 34.Hill SE, Kauffman KJ, Krout M, Richmond JE, Melia TJ, Colon-Ramos DA: Maturation and clearance of autophagosomes in neurons depends on a specific cysteine protease isoform, ATG-4.2. Dev Cell 2019, 49:251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maday S, Holzbaur EL: Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 2014, 30:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koyama-Honda I, Itakura E, Fujiwara TK, Mizushima N: Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 2013, 9:1491–1499. [DOI] [PubMed] [Google Scholar]
- 37.Ivankovic D, Drew J, Lesept F, White IJ, Lopez Domenech G, Tooze SA, Kittler JT: Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome. Autophagy 2020, 16:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matoba K, Kotani T, Tsutsumi A, Tsuji T, Mori T, Noshiro D, Sugita Y, Nomura N, Iwata S, Ohsumi Y, et al. : Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat Struct Mol Biol 2020, 27:1185–1193. [DOI] [PubMed] [Google Scholar]
- 39.*. Yang S, Park D, Manning L, Hill SE, Cao M, Xuan Z, Gonzalez I, Dong Y, Clark B, Shao L, et al. : Presynaptic autophagy is coupled to the synaptic vesicle cycle via ATG-9. Neuron 2022, 110:824–840. The authors show that ATG-9 is present on vesicles that undergo exo-endocytosis at presynaptic terminals, and mislocalization of ATG-9 leads to defects in activity-induced presynaptic autophagy. This paper identifies a mechanism that couples autophagosome biogenesis with synaptic activity.
- 40.De Pace R, Skirzewski M, Damme M, Mattera R, Mercurio J, Foster AM, Cuitino L, Jarnik M, Hoffmann V, Morris HD, et al. : Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet 2018, 14, e1007363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okerlund ND, Schneider K, Leal-Ortiz S, Montenegro-Venegas C, Kim SA, Garner LC, Waites CL, Gundelfinger ED, Reimer RJ, Garner CC: Bassoon controls presynaptic autophagy through Atg5. Neuron 2017, 93:897–913. [DOI] [PubMed] [Google Scholar]
- 42.Hernandez-Diaz S, Ghimire S, Sanchez-Mirasierra I, Montecinos-Oliva C, Swerts J, Kuenen S, Verstreken P, Soukup SF: Endophilin-B regulates autophagy during synapse development and neurodegeneration. Neurobiol Dis 2022, 163:105595. [DOI] [PubMed] [Google Scholar]
- 43.Vanhauwaert R, Kuenen S, Masius R, Bademosi A, Manetsberger J, Schoovaerts N, Bounti L, Gontcharenko S, Swerts J, Vilain S, et al. : The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J 2017, 36:1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khobrekar NV, Quintremil S, Dantas TJ, Vallee RB: The dynein adaptor RILP controls neuronal autophagosome biogenesis, transport, and clearance. Dev Cell 2020, 53:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee S, Sato Y, Nixon RA: Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci 2011, 31:7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yue Z: Regulation of neuronal autophagy in axon: implication of autophagy in axonal function and dysfunction/degeneration. Autophagy 2007, 3:139–141. [DOI] [PubMed] [Google Scholar]
- 47.Cheng XT, Zhou B, Lin MY, Cai Q, Sheng ZH: Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol 2015, 209:377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.**. Kulkarni VV, Anand A, Herr JB, Miranda C, Vogel MC, Maday S: Synaptic activity controls autophagic vacuole motility and function in dendrites. J Cell Biol 2021, 220:e202002084. In this paper, the authors show that synaptic activity decreases the motility and increases the degradative function of AVs within dendrites but not axons. This work defines how the activity state of the neuron dynamically regulates autophagy, and highlights the compartment-specific nature of intracellular trafficking pathways for degradation in neurons.
- 49.Farias GG, Guardia CM, De Pace R, Britt DJ, Bonifacino JS: BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc Natl Acad Sci U S A 2017, 114: E2955–E2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farfel-Becker T, Roney JC, Cheng XT, Li S, Cuddy SR, Sheng ZH: Neuronal soma-derived degradative lysosomes are continuously delivered to distal axons to maintain local degradation capacity. Cell Rep 2019, 28:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.**. Lie PPY, Yang DS, Stavrides P, Goulbourne CN, Zheng P, Mohan PS, Cataldo AM, Nixon RA: Post-Golgi carriers, not lysosomes, confer lysosomal properties to pre-degradative organelles in normal and dystrophic axons. Cell Rep 2021, 35:109034. In this paper, the authors comprehensively map and define endolysosomal and autophagic organelles in each compartment of the neuron. They propose that LAMP1-positive vesicles that pickup distal AVs for retrograde transport are transport carriers derived from the TGN, rather than degradative lysosomes.
- 52.Cheng XT, Xie YX, Zhou B, Huang N, Farfel-Becker T, Sheng ZH: Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. J Cell Biol 2018, 217:3127–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yap CC, Digilio L, McMahon LP, Garcia ADR, Winckler B: Degradation of dendritic cargos requires Rab7-dependent transport to somatic lysosomes. J Cell Biol 2018, 217: 3141–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.**. Ozkan N, Koppers M, van Soest I, van Harten A, Jurriens D, Liv N, Klumperman J, Kapitein LC, Hoogenraad CC, Farias GG: ER - lysosome contacts at a pre-axonal region regulate axonal lysosome availability. Nat Commun 2021, 12:4493. The authors show that ER-lysosome contacts at the pre-axonal region regulate the availability of late endosomes/lysosomes in the axon. This study identifies a new mechanism whereby ER-lysosome contacts facilitate lysosome tubulation, fission, and subsequent translocation into the axon.
- 55.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. : Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465:942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldsmith J, Ordureau A, Harper JW, Holzbaur ELF: Brain-derived autophagosome profiling reveals the engulfment of nucleoid-enriched mitochondrial fragments by basal autophagy in neurons. Neuron 2022, 110:967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, De Camilli P, Ferguson SM: Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci U S A 2015, 112:E3699–E3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neisch AL, Neufeld TP, Hays TS: A STRIPAK complex mediates axonal transport of autophagosomes and dense core vesicles through PP2A regulation. J Cell Biol 2017, 216:441–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sung H, Tandarich LC, Nguyen K, Hollenbeck PJ: Compartmentalized regulation of parkin-mediated mitochondrial quality control in the Drosophila nervous system in vivo. J Neurosci 2016, 36:7375–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jin EJ, Kiral FR, Ozel MN, Burchardt LS, Osterland M, Epstein D, Wolfenberg H, Prohaska S, Hiesinger PR: Live observation of two parallel membrane degradation pathways at axon terminals. Curr Biol 2018, 28:1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL: Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 2014, 206:655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu MM, Nirschl JJ, Holzbaur ELF: LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev Cell 2014, 29:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wong YC, Holzbaur EL: The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci 2014, 34:1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.*. Cason SE, Carman PJ, Van Duyne C, Goldsmith J, Dominguez R, Holzbaur ELF: Sequential dynein effectors regulate axonal autophagosome motility in a maturation-dependent pathway. J Cell Biol 2021, 220:e202010179. This study defines a role for the dynein effectors JIP1, Htt/HAP1, and JIP3 at various stages of autophagosomal transport in axons, in a manner dependent on the distribution and organelle maturation state in the axon. They propose an integrated model for how the multitude of dynein effectors may collaborate to facilitate retrograde transport of AVs, in a maturation-dependent manner.
- 65.Boecker CA, Goldsmith J, Dou D, Cajka GG, Holzbaur ELF: Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr Biol 2021, 31:2140–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nieto-Torres JL, Shanahan SL, Chassefeyre R, Chaiamarit T, Zaretski S, Landeras-Bueno S, Verhelle A, Encalada SE, Hansen M: LC3B phosphorylation regulates FYCO1 binding and directional transport of autophagousomes. curr Biol 2021, 31;3440–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xing R, Zhou H, Jian Y, Li L, Wang M, Liu N, Yin Q, Liang Z, Guo W, Yang C: The Rab7 effector WDR91 promotes autophagy-lysosome degradation in neurons by regulating lysosome fusion. J Cell Biol 2021, 220:e202007061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evans CS, Holzbaur EL: Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. Elife 2020, 9:e50260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baas PW, Black MM, Banker GA: Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J Cell Biol 1989, 109:3085–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hendricks AG, Perlson E, Ross JL, Schroeder HW 3rd, Tokito M, Holzbaur EL: Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport. Curr Biol 2010, 20:697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goo MS, Sancho L, Slepak N, Boassa D, Deerinck TJ, Ellisman MH, Bloodgood BL, Patrick GN: Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J Cell Biol 2017, 216:2499–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hafner AS, Donlin-Asp PG, Leitch B, Herzog E, Schuman EM: Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science 2019, 364. [DOI] [PubMed] [Google Scholar]
- 73.Dorrbaum AR, Alvarez-Castelao B, Nassim-Assir B, Langer JD, Schuman EM: Proteome dynamics during homeostatic scaling in cultured neurons. Elife 2020, 9:e52939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang T, Martin S, Papadopulos A, Harper CB, Mavlyutov TA, Niranjan D, Glass NR, Cooper-White JJ, Sibarita JB, Choquet D, et al. : Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type a. J Neurosci 2015, 35:6179–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kallergi E, Daskalaki AD, Kolaxi A, Camus C, Ioannou E, Mercaldo V, Haberkant P, Stein F, Sidiropoulou K, Dalezios Y, et al. : Dendritic autophagy degrades postsynaptic proteins and is required for long-term synaptic depression in mice. Nat Commun 2022, 13:680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gu Q, Jiao S, Duan K, Wang YX, Petralia RS, Li Z: The BAD-BAX-caspase-3 cascade modulates synaptic vesicle pools via autophagy. J Neurosci 2021, 41:1174–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nikoletopoulou V, Sidiropoulou K, Kallergi E, Dalezios Y, Tavernarakis N: Modulation of autophagy by BDNF underlies synaptic plasticity. Cell Metabol 2017, 26:230–242. [DOI] [PubMed] [Google Scholar]
- 78.*. Kuijpers M, Kochlamazashvili G, Stumpf A, Puchkov D, Swaminathan A, Lucht MT, Krause E, Maritzen T, Schmitz D, Haucke V: Neuronal autophagy regulates presynaptic neurotransmission by controlling the axonal endoplasmic reticulum. Neuron 2021, 109:299–313. Here, the authors show that loss of ATG5 results in an accumulation of ER in the distal axon, and calcium release from the ER which stimulates synaptic vesicle exocytosis. This paper establishes autophagy as a key regulator of presynaptic neurotransmission via regulation of axonal ER.
- 79.Hernandez D, Torres CA, Setlik W, Cebrian C, Mosharov EV, Tang G, Cheng HC, Kholodilov N, Yarygina O, Burke RE, et al. : Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012, 74:277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoffmann-Conaway S, Brockmann MM, Schneider K, Annamneedi A, Rahman KA, Bruns C, Textoris-Taube K, Trimbuch T, Smalla KH, Rosenmund C, et al. : Parkin contributes to synaptic vesicle autophagy in Bassoon-deficient mice. Elife 2020, 9:e56590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hoffmann S, Orlando M, Andrzejak E, Bruns C, Trimbuch T, Rosenmund C, Garner CC, Ackermann F: Light-activated ROS production induces synaptic autophagy. J Neurosci 2019, 39:2163–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Luningschror P, Binotti B, Dombert B, Heimann P, Perez-Lara A, Slotta C, Thau-Habermann N, RvC C, Karl F, Damme M, et al. : Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease. Nat Commun 2017, 8:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K: Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci 2012, 32:10413–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shen H, Zhu H, Panja D, Gu Q, Li Z: Autophagy controls the induction and developmental decline of NMDAR-LTD through endocytic recycling. Nat Commun 2020, 11:2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Compans B, Camus C, Kallergi E, Sposini S, Martineau M, Butler C, Kechkar A, Klaassen RV, Retailleau N, Sejnowski TJ, et al. : NMDAR-dependent long-term depression is associated with increased short term plasticity through autophagy mediated loss of PSD-95. Nat Commun 2021, 12:2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stavoe AK, Gopal PP, Gubas A, Tooze SA, Holzbaur EL: Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. Elife 2019, 8:e44219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lachance V, Wang Q, Sweet E, Choi I, Cai CZ, Zhuang XX, Zhang Y, Jiang JL, Blitzer RD, Bozdagi-Gunal O, et al. : Autophagy protein NRBF2 has reduced expression in Alzheimer’s brains and modulates memory and amyloid-beta homeostasis in mice. Mol Neurodegener 2019, 14:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ, et al. : The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 2016, 35:1656–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, et al. : Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J 2016, 35:1276–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ho WY, Tai YK, Chang JC, Liang J, Tyan SH, Chen S, Guan JL, Zhou H, Shen HM, Koo E, et al. : The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy 2019, 15:827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ugolino J, Ji YJ, Conchina K, Chu J, Nirujogi RS, Pandey A, Brady NR, Hamacher-Brady A, Wang J: Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet 2016, 12, e1006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.**. Cunningham KM, Maulding K, Ruan K, Senturk M, Grima JC, Sung H, Zuo Z, Song H, Gao J, Dubey S, et al. : TFEB/Mitf links impaired nuclear import to autophagolysosomal dysfunction in C9-ALS. Elife 2020, 9:e59419. Here, the authors elucidate a pathogenic cascade by which the G4C2 hexanucleotide repeat expansion in C9orf72 disrupts nuclear import of the Drosophila homolog of TFEB, leading to reduced autophagosome biogenesis, dysfunctional lysosomes, and neurodegeneration. This study provides a mechanistic link between defective autophagy and disease pathogenesis in models of ALS.
- 93.Deng Z, Purtell K, Lachance V, Wold MS, Chen S, Yue Z: Autophagy receptors and neurodegenerative diseases. Trends Cell Biol 2017, 27:491–504. [DOI] [PubMed] [Google Scholar]
- 94.Wong YC, Holzbaur EL: Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A 2014, 111:E4439–E4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Deng Z, Lim J, Wang Q, Purtell K, Wu S, Palomo GM, Tan H, Manfredi G, Zhao Y, Peng J, et al. : ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 2020, 16:917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goode A, Butler K, Long J, Cavey J, Scott D, Shaw B, Sollenberger J, Gell C, Johansen T, Oldham NJ, et al. : Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12:1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ge P, Dawson VL, Dawson TM: PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. Mol Neurodegener 2020, 15:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. : Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 2016, 113:4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harding O, Evans CS, Ye J, Cheung J, Maniatis T, Holzbaur ELF: ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc Natl Acad Sci U S A 2021, 118, e2025053118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Snouwaert JN, Church RJ, Jania L, Nguyen M, Wheeler ML, Saintsing A, Mieczkowski P, Manuel de Villena FP, Armao D, Moy SS, et al. : A mutation in the Borcs7 subunit of the lysosome regulatory BORC complex results in motor deficits and dystrophic axonopathy in mice. Cell Rep 2018, 24:1254–1265. [DOI] [PubMed] [Google Scholar]
- 101.**. Roney JC, Li S, Farfel-Becker T, Huang N, Sun T, Xie Y, Cheng XT, Lin MY, Platt FM, Sheng ZH: Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type C. Dev Cell 2021, 56:1452–1468. The authors show that alterations in lipid content of lysosomal organelles in Niemann-Pick disease type C alter association with molecular motors, thereby reducing anterograde delivery into the axon and accumulation of axonal AVs. This paper establishes a pathological mechanism by which a neurodegenerative lysosomal storage disorder impairs the trafficking of autophagic and endolysosomal organelles and axonal homeostasis.
- 102.Tammineni P, Ye X, Feng T, Aikal D, Cai Q: Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. Elife 2017, 6:e21776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ye X, Feng T, Tammineni P, Chang Q, Jeong YY, Margolis DJ, Cai H, Kusnecov A, Cai Q: Regulation of synaptic amyloid-beta generation through BACE1 retrograde transport in a mouse model of alzheimer’s disease. J Neurosci 2017, 37:2639–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feng T, Tammineni P, Agrawal C, Jeong YY, Cai Q: Autophagy-mediated regulation of BACE1 protein trafficking and degradation. J Biol Chem 2017, 292:1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gowrishankar S, Wu Y, Ferguson SM: Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J Cell Biol 2017, 216:3291–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VM: Formation of alpha-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell 2014, 25:4010–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang Q, Gao P, Arzberger T, Hollerhage M, Herms J, Hoglinger G, Koeglsperger T: Alpha-Synuclein defects autophagy by impairing SNAP29-mediated autophagosome-lysosome fusion. Cell Death Dis 2021, 12:854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen X, Li Y, Wang C, Tang Y, Mok SA, Tsai RM, Rojas JC, Karydas A, Miller BL, Boxer AL, et al. : Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol Neurodegener 2020, 15:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ: Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp Mol Med 2013, 45:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Poehler AM, Xiang W, Spitzer P, May VE, Meixner H, Rockenstein E, Chutna O, Outeiro TF, Winkler J, Masliah E, et al. : Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10:2171–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]