

Abstract
Aims
Adult polyglucosan body disease (APBD) is a progressive neurogenetic disorder caused by 1,4-alpha-glucan branching enzyme 1 (GBE1) mutation with an accumulation of polyglucosan bodies (PBs) in the central and peripheral nervous systems as a pathological hallmark. Here we report two siblings in a family with a GBE1 mutation with prominent frontotemporal lobar degeneration with TAR DNA-binding protein 43 (FTLD-TDP) and ageing-related tau astrogliopathy (ARTAG) co-pathologies with PBs in the central nervous system.
Methods
Whole-genome sequencing (WGS) followed by Sanger sequencing (SS) was performed on three affected and two unaffected siblings in a pedigree diagnosed with familial frontotemporal dementia. Out of the affected siblings, autopsies were conducted on 2 cases, and brain samples were used for biochemical and histological analyses. Brain sections were stained with haematoxylin and eosin, and immunostained with antibodies against ubiquitin, tau, amyloid β, α-synuclein, TDP-43, and fused in sarcoma (FUS).
Results
A novel single nucleotide deletion in GBE1, c.1280delG, was identified, which is predicted to result in a reading frameshift, p.Gly427Glufs*9. This variant segregated with disease in the family, is absent from population databases, and is predicted to cause loss-of-function, a known genetic mechanism for APBD. The affected siblings showed a greater than 50% decrease in GBE protein levels. Immunohistochemical analysis revealed widespread FTLD-TDP (type A) and ARTAG pathologies as well as PBs in the brains of two affected siblings for whom an autopsy was performed.
Conclusions
This is the first report of a family with several individuals with a FTD clinical phenotype and underlying co-pathologies of APBD, FTLD-TDP, and ARTAG with a segregating GBE1 loss-of-function mutation in affected siblings. The finding of co-pathologies of APBD and FTLD-TDP suggests these processes may share a disease mechanism resulting from this GBE1 mutation.
Keywords: GBE1, adult polyglucosan body disease, corpora amylacea, FTLD-TDP, ARTAG
Graphical Abstract
A family with FTD clinical phenotype with a novel GBE1 mutation c.1280delG was identified, which is predicted to result in a reading frameshift, p.Gly427Glufs*9.
Two of affected siblings showed a reduction in GBE protein expression levels with abundant FTLD-TDP Type A pathology in the grey matter, abundant PBs, and ARTAG in the subpial matter and the white matter.
The finding of co-pathologies of APBD and FTLD-TDP suggests these processes may share a disease mechanism resulting from this GBE1 loss-of-function mutation.
Introduction
Adult polyglucosan body disease (APBD) is a rare neurogenetic disorder characterized by adult-onset, progressive upper and lower motor neuropathy, sensory impairment, and neurogenic bladder. As the disease progresses, about half of the patients develop cognitive impairment.1 The majority of patients with APBD are of Ashkenazi Jewish ancestry and have a common p.Tyr329Ser mutation in the 1,4-alpha-glucan branching enzyme 1 (GBE1) gene with a reduction of GBE activity.2, 3 Other mutations in GBE1, however, have also been detected in other populations.1, 4–8
APBD is characterized by the intracellular accumulation of polyglucosan bodies (PBs), also called corpora amylacea, in the central and peripheral nervous systems and other tissues. PBs are composed of poorly branched, aggregated, and insoluble glycogen caused by GBE deficiency. In the brain, most PBs are observed in the cerebral white matter, accompanied by demyelination and gliosis.9
Frontotemporal lobar degeneration (FTLD) is characterized by atrophy in the frontal and temporal lobes of the brain. FTLD is divided into three main histopathological subtypes, namely, 1) FTLD-tau, 2) FTLD with TDP-43 inclusions (FTLD-TDP), and 3) FTLD with fused in sarcoma inclusions (FTLD-FUS). FTLD-TDP is characterized by inclusions that are ubiquitin- and TDP-43-positive but tau and FUS-negative.10 Based on histopathological differences, FTLD-TDP is subdivided into five subtypes: 1) Type A, with small neurites and neuronal cytoplasmic inclusions in the superficial cortical layers; 2) Type B, comprised of neuronal and glial cytoplasmic inclusions in both superficial and deep cortical layers; 3) Type C, demonstrating long neuritic profiles found in the superficial cortical laminae; 4) Type D, with neuronal intranuclear inclusions and dystrophic neurites, and 5) Type E, showing a wide neuroanatomic distribution comprised of granulofilamentous neuronal inclusions, abundant grains, and oligodendroglial inclusions.10–12
Two different cases of co-existing APBD and FTLD-TDP have been reported without identification of a causative gene mutation.13, 14 In addition, an example of co-existing APBD and FTLD-FUS, but not FTLD-TDP, has been identified with an intronic mutation in GBE1 (IVS5+2C>T).14 However, there are no reports of a GBE1 mutation resulting in both APBD and FTLD-TDP pathology.
Ageing-related tau astrogliopathy (ARTAG) describes a pathological accumulation of abnormally phosphorylated tau protein in astrocytes with the morphological subtypes of 1) thorn-shaped astrocytes (TSA) and 2) granular/fuzzy astrocytes (GFA).15 ARTAG occurs in the brain during normal ageing as well as in most neurodegenerative diseases, including FTLD.15 The presence of TDP-43 pathology is frequently associated with subpial ARTAG in the middle temporal lobe but not in the white matter.16
Here we report on the finding of a family with a GBE1 mutation in three siblings with frontotemporal dementia (FTD), two of which have autopsies showing abundant FTLD-TDP Type A pathology in the grey matter, abundant PBs, and ARTAG in the subpial matter and the white matter.
Materials and Methods
Patients
Members of a kindred affected by FTD were consented and enrolled in research studies at the University of Pennsylvania (Penn) Frontotemporal Degeneration Center or Alzheimer’s Disease Research Center. Autopsies on two deceased individuals were performed at the Penn Center for Neurodegenerative Disease Research (CNDR) following informed consent from the next-of-kin at the time of death.17
Ethical statement
Consent was obtained according to the Declaration of Helsinki and approved by the Penn Institutional Review Board.
Genetic analysis
Genomic DNA was extracted from blood or brain tissue using QuickGene-610L (Autogen Inc., MA) or QIAamp DNA Mini kits (Qiagen). Whole genome sequencing of DNA from three affected cases and two unaffected siblings was performed at the New York Genome Center using Illumina TruSeq PCR-free library preparation kits and HiSeqX sequencer with 2 × 150 bp cycles. Sequencing reads were aligned to human genome build hg38, and the sequence variant call format files were analysed in Geneticist Assistant software (Soft Genetics). For germline variants, variants with poor quality and false positives were filtered by variant allele frequency (< 25%) and the variant quality score recalibration (VQSR) scores. The effect of variants was analysed by 1) allele frequency in the population database (Genome Aggregation Database, GnomAD)18 as well as in neurological healthy controls in our Penn cohort, and 2) combined prediction scores (range 0–12) calculated from four functional predictors (PolyPhen2, SIFT, MutationTaster, MutationAssessor) and two meta-predictors (CADD and REVEL).19, 20 The evaluated rare variants with deleterious prediction scores were manually assessed for functional/clinical impacts and segregation/inheritance pattern using publicly available databases including ClinVar and Varsome and analysis software including Alamut (Sofia Genetics) and Geneticist Assistant (Soft Genetics). The identified variant GBE1(NM_000158.4):c.1280delG (p.Gly427Glufs*9) was confirmed by Sanger sequencing on ABI3730 Genetic Analyzer at the DNA sequencing facility at the University of Pennsylvania.
Western blotting
The procedure for western blotting applied in this study has been reported previously.21, 22 Briefly, 100 mg of frozen tissues of frontal cortical grey matters from affected siblings (II-1 and II-5) and control cases were homogenized using a Dounce homogenizer in ten volumes (v/w) high-salt buffer (10 mM Tris-HCl, pH 7.4, 0.8 M NaCl, 1 mM EDTA, and 2 mM dithiothreitol [DTT]) with 1% Triton X-100, proteinase inhibitor cocktail, phosphatase inhibitor, and PMSF. After the sonication at 20 pulses, the samples were centrifuged at 45,000 rpm (100,000 × g) for 30 min at 4 °C, and the supernatant was collected. The protein concentrations were determined using BCA assay, and the samples with 10–20 μg of proteins were loaded on SDS-PAGE. The samples were transferred to nitrocellulose membranes and blocked in Odyssey blocking buffer (LI-COR Biosciences) before being immunoblotted with specific primary antibodies for GBE (1:1,000, ab180596 [EP11113], abcam) and GAPDH (1:10,000, R-RGM2, Advanced Immunochemical). The blots were further incubated with IRDye-labelled secondary antibodies and scanned using an ODY-2816 imager.
Histological and immunohistochemical staining
Eighteen regions (amygdala, hippocampus, entorhinal cortex, cingulate gyrus, middle frontal gyrus, angular gyrus, superior/middle temporal gyrus, occipital cortex, motor cortex, caudate, putamen, globus pallidus, thalamus, midbrain, pons, medulla, cerebellum, and spinal cord) were examined by neuropathologists. The neuropathological assessment was performed as reported previously.23, 24 Briefly, brain sections were fixed in 70% ethanol in 150 mM NaCl and underwent staining with haematoxylin and eosin (H&E), the Klüver-Barrera (KB) method, Thioflavin S, and Periodic acid–Schiff (PAS). For immunohistochemistry, the sections were stained with antibodies raised against ubiquitin (1:40,000, MAB1510, Millipore), FUS (1:40,000, 11570–1-AP, Protein Tech Group, Inc), phosphorylated tau (PHF1, 1:1000, a gift from Dr Peter Davies), phosphorylated TDP-43 (pS409/410, 1:500, a contribution from Dr Manuela Neumann and Elisabeth Kremmer), amyloid β (Aβ) (NAB228, 1:20,000, developed at CNDR), and pathological conformation of α-synuclein (Syn303, 1:16,000, generated at CNDR). Antigen retrieval using 88% formic acid was used for tau, TDP-43, and α-synuclein antibodies. Antigen retrieval using a citrate-based unmasking solution (pH 6.0, Vector, H-3300) was used for ubiquitin and FUS antibodies. Primary antibody binding is visualized with the avidin-biotin complex detection method (VECTASTAIN ABC kit; Vector Laboratories, Burlingame, CA) with diamino-benzidine peroxidase substrate (ImmPACT, Vector Laboratories) as the chromogen.
The images were captured using Observer 7 (Zeiss), TCS SP8 WLL Confocal with STED 3X (Leica), or PANNORAMIC 250 (3DHISTECH) imaging systems, and analysed using HALO (Indica Labs). The neuropathological changes were rated using a semi-quantitative scale (0, no changes; 0.5, rare; 1, mild; 2, moderate; 3, severe) as described previously.23, 24 The average rating scores of 2 cases for each region were applied to a heatmap using HSB colour models (0, H0°S0%B75%; 0.5, H90°S100%B100%; 1, H60°S100%B100%; 2, H30°S100%B100%; 3, H0°S100%B100%) in the software (Adobe Illustrator CC). A gradient smoothing method was used for the boundaries between assessed regions.
Results
Clinical Description
Six siblings in the pedigree (Figure 1A and Table 1) were enrolled in research studies. To better preserve anonymity, ages have been approximated, and sex has been masked. There was no evidence that the neurologic disease in this pedigree was sex-linked. Individuals II-1 and II-5 (proband) underwent clinical examination and autopsy. II-3 was clinically diagnosed as behavioural variant of FTD (bvFTD) at the age of ~75 years by a neurologist but did not undergo an autopsy when they died at the age of ~85years.25 Siblings II-2, II-4, and II-6 were asymptomatic. No DNA was collected from II-4 before death.
Figure 1. Pedigree with FTD and Genetic analysis.
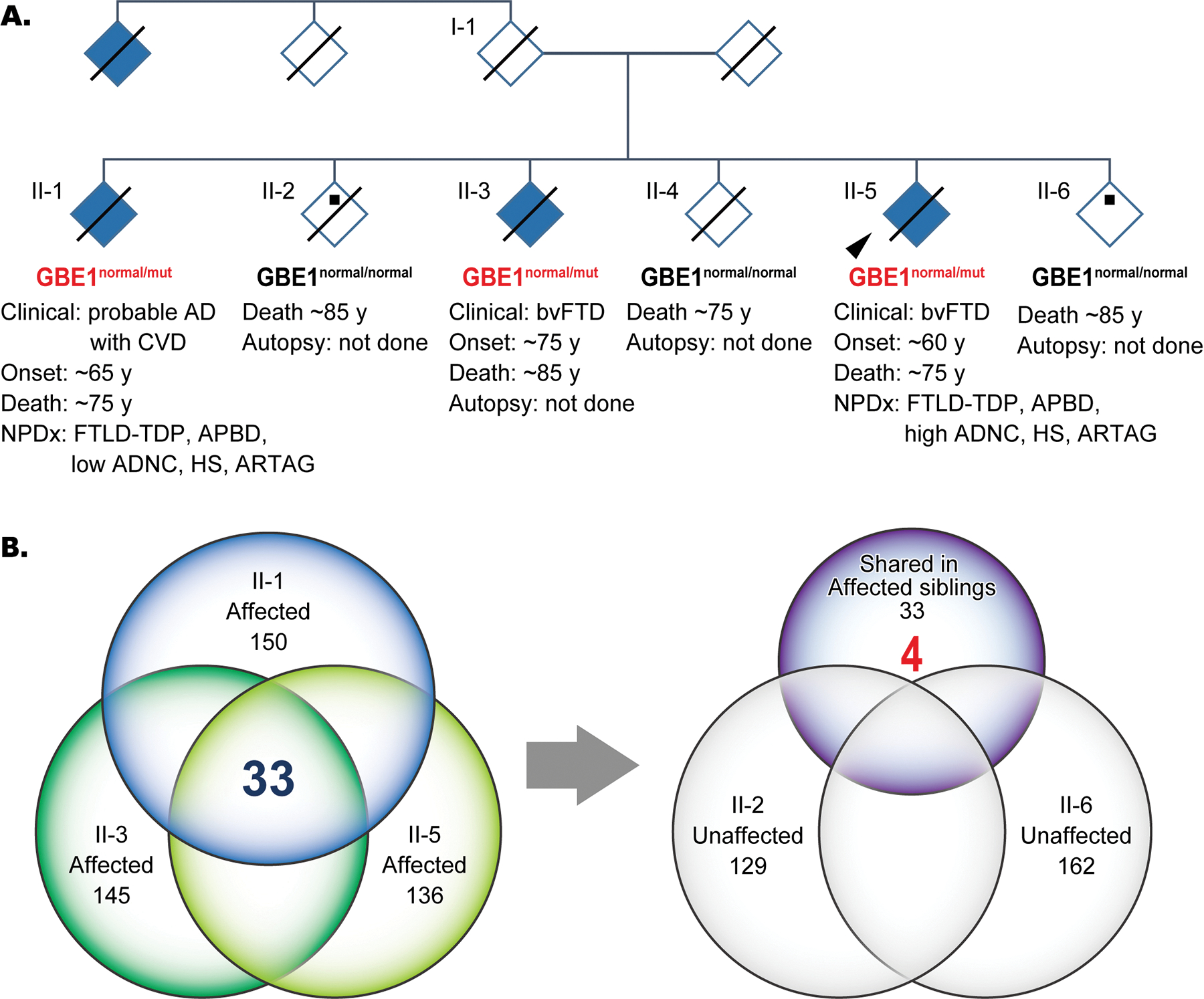
(A) Pedigree diagnosed with FTD. An autopsy was performed on two affected siblings (II-1 and -5). Arrowhead shows proband. Black dots and solid lines indicate cases with available DNA and those deceased, respectively. Whole genome sequencing was performed with samples from 3 affected siblings (II-1, -3, -5) and two unaffected siblings (II-2 and -6). The heterozygous status of the GBE1 mutation is shown in red. (B) Venn diagrams of numbers of rare variants identified from whole genome sequencing among the affected and unaffected siblings. Thirty-three variants that were shared among the three affected siblings were filtered against the variants identified in unaffected siblings, which resulted in 4 variants. Abbreviations: ADNC, Alzheimer’s disease neuropathologic change; APBD, adult polyglucosan body disease; ARTAG, ageing-related tau astrogliopathy; bvFTD, behavioural variant of frontotemporal dementia; CVD, cerebrovascular disease; FTLD-TDP, frontotemporal lobar degeneration with TDP-43 inclusions; mut, mutant; PDD, Parkinson’s disease dementia
Table 1.
Patient demographics.
| Case | Affected/Unaffected | Age at onset | Age at death | Clinical Dx | Autopsy | Neuropathological findings |
|---|---|---|---|---|---|---|
|
| ||||||
| II-1 | Affected | ~65 | ~75 | bvFTD probable AD with CVD | Yes | FTLD-TDP, APBD, low ADNC, HS, ARTAG |
| II-2 | Unaffected | - | ~85 | - | No | - |
| II-3 | Affected | ~75 | ~85 | bvFTD | No | - |
| II-4 | Unaffected | - | ~75 | - | No | - |
| II-5 | Affected (Proband) | ~60 | ~75 | bvFTD | Yes | FTLD-TDP, APBD, high ADNC, HS, ARTAG |
| II-6 | Unaffected | - | ~85 | - | No | - |
Abbreviations; AD, Alzheimer’s disease; ADNC, Alzheimer’s disease neuropathologic change; APBD, adult polyglucosan body disease; ARTAG, Aging-related tau astrogliopathy; bvFTD, behavioural variant frontotemporal dementia; HS, hippocampal sclerosis.
II-1, a Caucasian, was seen at the Penn FTD Center at the age of ~75 years for a second opinion about dementia that started at the age of ~65 years. They complained of increased memory problems, progressive difficulty in walking, and loss of bowel function over several years. They were clinically diagnosed with possible Alzheimer’s disease and small vessel disease.
Neurological examination showed no cranial nerve impairment. They had a slight resting tremor on the right and mild limb muscle weakness. Gait was wide-based and positive for the Romberg test. They were intact to light touch, pinprick vibration, and proprioception.
They were alert and oriented at the first visit, but recognition was poor. The mini-mental examination was 20/30. The patient had poor performance on an oral alternation pattern and continually perseverated. Although repetition was good, there were significant problems copying a figure as well as copying a known sequence. They named both high and low frequency items and were able to read spelt words irregularly. They died at the age of ~75 years.
II-5 is a Caucasian who was seen in the Penn FTD Center at the age of ~60 years, complaining of memory and thinking problems that began 2 years before visiting the centre. They showed progressive cognitive decline with behavioural abnormalities as well as profound apraxia, which was diagnosed as bvFTD.
Neurological examination showed mild rigidity to activation in the right arm and slight hypomimia but no Parkinson-like tremor, bradykinesia, or myoclonus. When ~65 years of age, they scored 14/28 on the severe impairment battery, 5/30 on a mini mental examination, and 8/15 on the geriatric depression rating scale. They died at the age of ~75 years.
GBE1 variant identified
To search for a genetic cause for the familial FTD in this family, whole genome sequencing (WGS) was performed on DNA from the three affected siblings (II-1, II-3, II-5) and the two unaffected siblings (II-2 and II-6) (Figure 1A). Variants from WGS were filtered and evaluated according to our variant analysis workflow (see Methods). Among the 150, 145, and 136 rare variants identified from symptomatic siblings II-1, -II-3, and II-5, respectively, 33 variants were shared by the three. Of these 33 variants, only four heterozygous variants were also absent in the two asymptomatic siblings (Figure 1B and Supplementary Table S1). Among these four variants, a novel frameshift variant in GBE1(NM_000158.4):c.1280delG (p.Gly427Glufs*9) was the strongest candidate for pathogenicity. Aside from familial segregation with APBD and FTD, criteria for pathogenicity include the fact that it is a heterozygous null variant in a gene where autosomal dominant loss-of-function is a known disease mechanism, it is novel and absent from all population databases. Because of the co-segregation of FTD with APBD, the variant is also a strong candidate gene for being associated with the FTD phenotype.26 We next performed functional studies to evaluate this pathogenic mechanism.
Western blotting
Western blotting was performed using frozen frontal cortical tissues to evaluate the protein expression levels of GBE in affected siblings II-1 and II-5. For age-matched control, we used two unremarkable adult brains and three primary age-related tauopathy cases (also known as PART), all of which showed no remarkable evidence of PBs, tauopathy, synucleinopathy, or TDP-43 proteinopathies in the frontal lobes (Supplementary table S2). As expected, for the heterozygous frameshift variant of GBE, the GBE protein expression levels were decreased by 50% or more in the affected siblings compared with control cases (40% in II-1 and 17.6% in II-5, respectively). Results are the average of 5 technical replicates. (Figures 2A and 2B).
Figure 2. Western blot analysis for GBE1 protein expression levels.
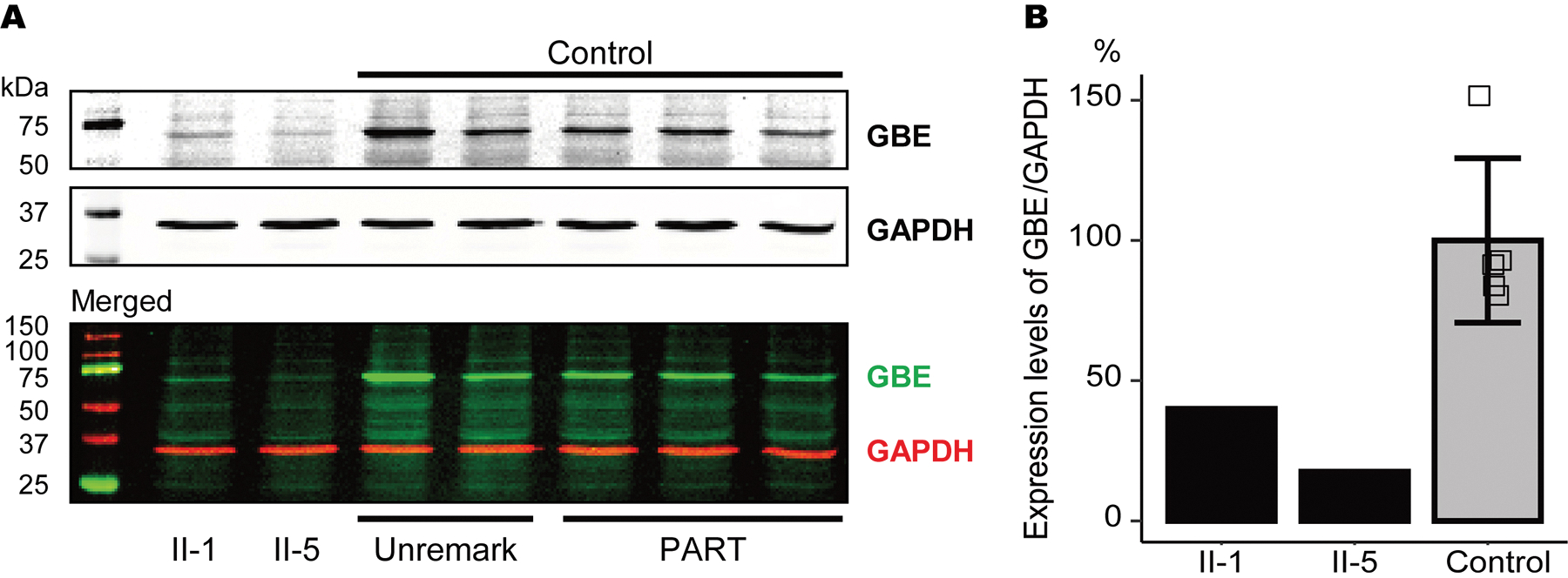
(A) Representative images of western blot analysis using GBE and GAPDH antibodies. (B) Relative expression levels of GBE/GAPDH in II-1, II-5, and control cases. The GBE protein expression levels were reduced in affected sibling cases compared with control cases. Vertical bars represent mean ± SD. Abbreviations: PART, primary age-related tauopathy; Unremark, unremarkable adult brain.
Histological and immunohistochemical analysis
For II-1, an autopsy was performed 19 hours after death. Mild brain atrophy was observed in the frontotemporal regions. The brain weight was 1011 grams. Neuron loss was severe and accompanied by gliosis in the hippocampal regions and moderate in the frontotemporal regions. For II-5, an autopsy was performed 10.5 hours after death. Severe atrophy was observed throughout the brain. The brain weight was 810 grams. Neuron loss was severe and accompanied by gliosis in the hippocampal regions and moderate in the neocortical regions.
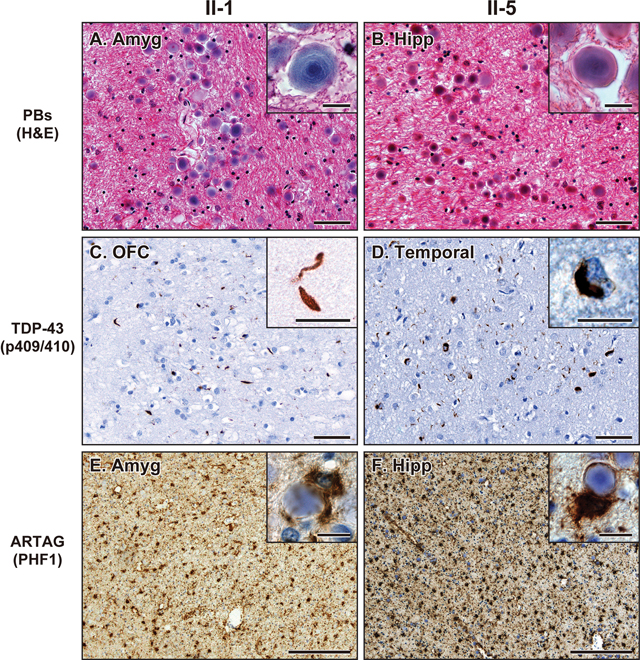
Consistent with APBD with GBE1 mutation,1–8 both II-1 and II-5 showed abundant PBs in the subpial matter and the deep white matter of frontal, temporal, parietal, and occipital lobes and cerebellum as well as some parts of the brainstem and the spinal cord (Figure 3A, 3B and 4A). PBs were especially abundant in the subpial matter as well as subependymal and perivascular areas of the white matter (Figures 3A, 3B, and 4A). TDP-43 neuronal cytoplasmic inclusions (NCI) and dystrophic neurites (DN) were observed in the neocortical, allocortical, and subcortical grey matter and partially in the grey matter of the brainstem and spinal cord (Figure 3C, 3D and 4B). The TDP-43 pathologies were especially severe in the frontotemporal cortices. In the neocortical area, the TDP-43 NCI and DN were concentrated in the superficial cortical layers, which is consistent with TDP-43 type A.27 ARTAG was present as dense accumulations of TSA in the white matter and subpial, subependymal, and perivascular areas of the frontotemporal lobes as well as the amygdala and hippocampus (Figure 3E, 3F, and 4C). A small amount of GFA was partially observed in the temporal cortices. The TSA and PBs were closely distributed in the subpial matter and white matter. Some TSA enveloped PBs in their foot processes (insets in Figure 3F). Ubiquitin staining highlighted the TDP-43 NCI and DN, subpial PBs, and a subset of the white matter PBs. FUS and α-synuclein positive pathologies were not observed. Both II-1 and II-5 exhibited hippocampal sclerosis and severe white matter attenuation (Supplementary Figure S1). II-1 had mild Aβ plaque burden limited in the neocortical regions, and neurofibrillary tangles and neuropil threads were limited to the hippocampus resulting in a low Alzheimer’s disease neuropathologic change (ADNC) stage. II-5 had abundant Aβ plaques, neurofibrillary tangles, neuropil threads, and neuritic plaques throughout the brain resulting in a high ADNC.28
Figure 3. Neuropathological findings in the cases with GBE1 mutation.
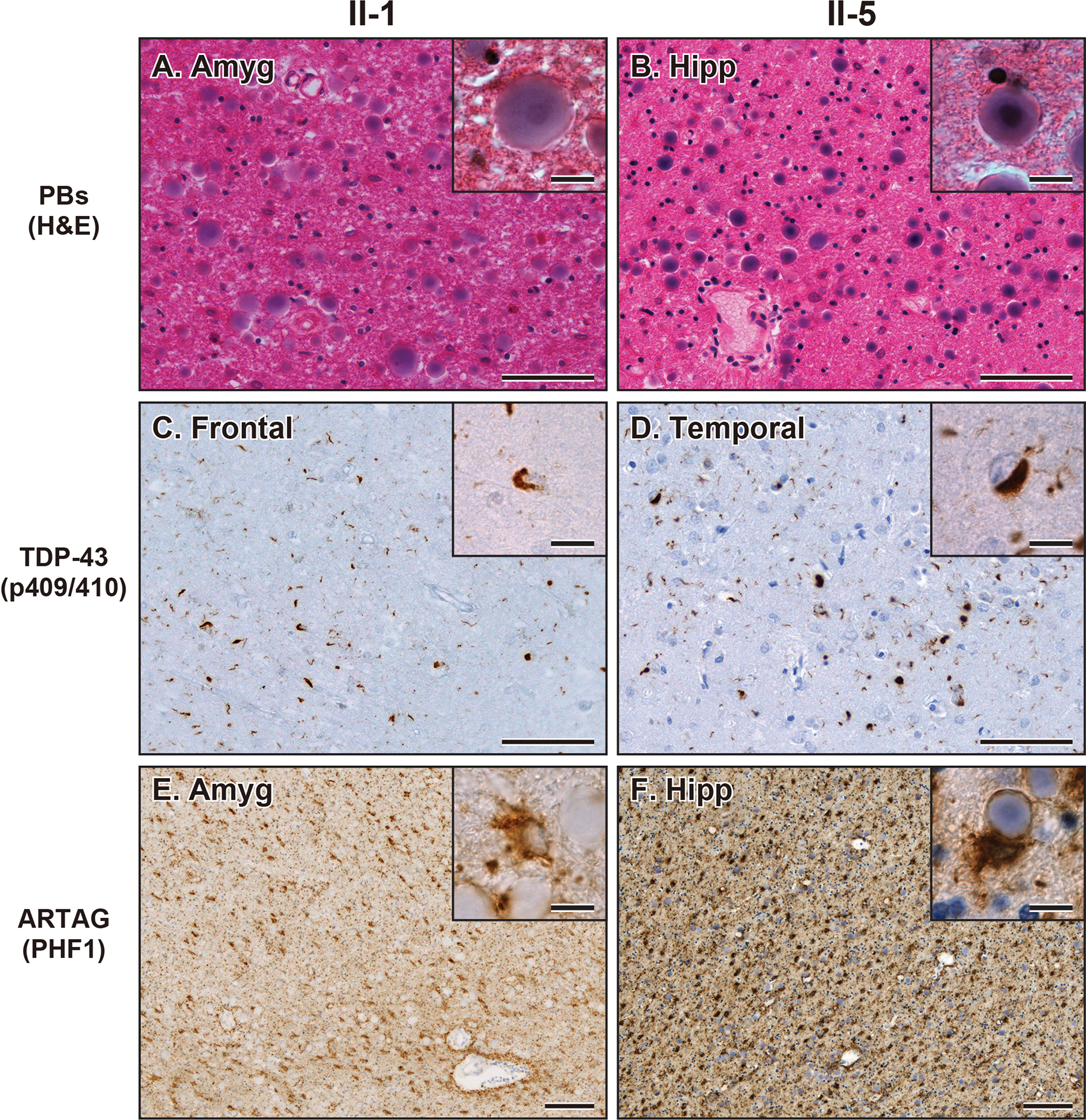
(A, B) Representative images of H&E staining showing PBs in the amygdala in case II-1 (A) and hippocampus in case II-5 (B). Bars represent 50 μm and 10 μm (insets). (C, D) Representative images of TDP-43 (p409/410) staining showing NCI and DN in the frontal cortex in case II-1 (C) and temporal cortex in case II-5 (D). Bars represent 50 μm and 10 μm (insets). (E, F) Representative images of PHF1 staining showing ARTAG in the amygdala in case II-1 (E) and hippocampus in case II-5 (F). Insets show PBs adjacent to ARTAG. Bars represent 200 μm and 10 μm (insets). Abbreviations: Amyg, amygdala; Hipp, hippocampus; Temp, temporal cortex.
Figure 4. Distribution of PBs, TDP-43 NCI and DN, and ARTAG in the CNS with GBE1 mutation.
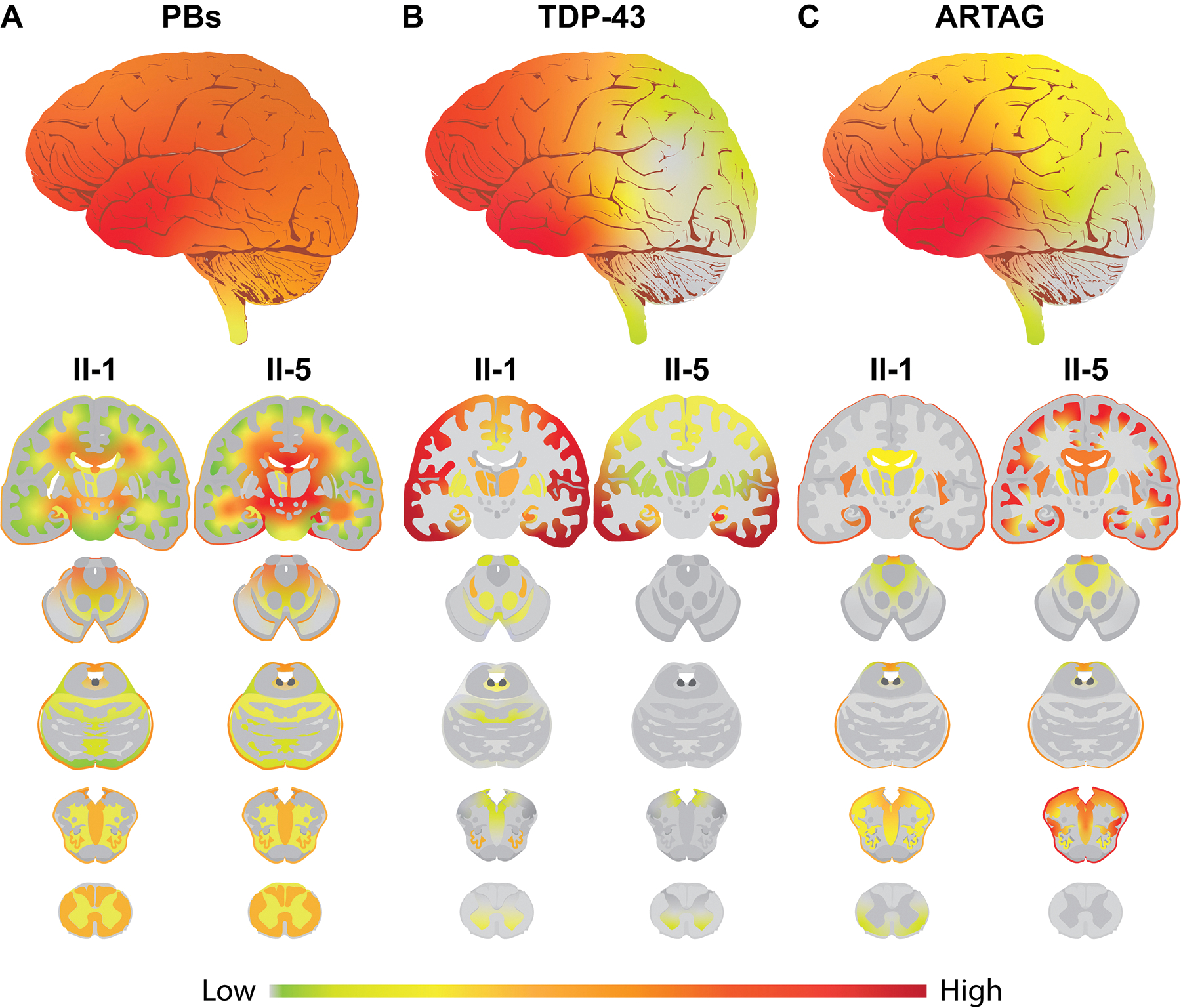
(A) Heatmap of PB pathology. The upper whole brain image represents the average of two cases with interpolated smoothing. Lower sagittal and coronal images represent PB distribution in II-1 and II-5. PBs are observed in the subpial matter and the deep white matter of frontal, temporal, parietal, and occipital lobes, and the cerebellum as well as some parts of the brainstem and the spinal cord. II-5 shows more abundant PB pathology than II-1. (B) Heatmap of TDP-43 pathology. The upper whole brain image represents the average of two cases with interpolated smoothing. Lower sagittal and coronal images represent TDP-43 distribution in II-1 and II-5. TDP-43 NCI and DN are observed in the neocortical, allocortical, and subcortical grey matter and partially in the grey matter of the brainstem and spinal cord. The TDP-43 pathologies are especially abundant in the frontotemporal cortices. II-1 shows more broadly distributed pathology than II-5. (C) Heatmap of ARTAG pathology. The upper whole brain image represents the average of two cases with interpolated smoothing. Lower sagittal and coronal images represent ARTAG distribution in II-1 and II-5. ARTAG is abundantly observed in the subpial matter and the white matter of frontotemporal lobes, amygdala, and hippocampus and partially observed in the subpial matter and the white matter of the brainstem and spinal cord. II-5 shows more abundant and broadly distributed ARTAG than II-1. A gradient smoothing method was used for the boundaries between each assessed legion.
Although the morphologies of PBs, FTLD-TDP, and ARTAG were identical between the two cases, there are some differences in abundance and distribution between II-1 and II-5. For example, PBs and ARTAG were more abundant in II-5 than in II-1, whereas TDP-43 pathology was more widely distributed in II-1 than II-5 (Figure 4A–C and Supplementary Table S3).
Discussion
Herein we report on identifying a heterozygous loss-of-function mutation in GBE1 that segregates clinically with FTD and pathologically in the central nervous system with abundant PBs, FTLD-TDP type A, and ARTAG in a family for which several individuals, both affected and unaffected, underwent sequence analysis. Aside from segregating with the disease, the identified GBE1 c.1280delG mutation is novel and absent in population databases. Taken together, these findings support classification as a pathogenic variant. Furthermore, from a functional perspective, GBE protein levels were decreased in the brain tissue of two of the affected siblings, suggesting that the heterozygous frameshift reduced GBE protein expression.
GBE1 in humans encodes GBE. Mutations in GBE1 have been shown to reduce GBE enzymatic activity and are associated with glycogen storage disease type IV (also known as Andersen’s disease) in newborns and APBD in adults. In APBD cases, diffuse PBs composed of accumulated abnormally branched glycogen are observed in the central and peripheral nervous systems. In our study, cases II-1 and II-5 showed abundant PBs in the brains and spinal cords, which was consistent with APBD with GBE1 mutation.
In addition, the patients had abundant FTLD-TDP NCI and DN (TDP-43 type A) in the central nervous system, which was consistent with the clinical diagnosis of bvFTD.29 Two cases of coexisting APBD and FTLD-TDP have been reported.13, 14 Because genetic analyses were not conducted in those, no causative gene could be linked to the patients with APBD and FTLD-TDP. A case of APBD with clinical FTD had also been reported before FTLD-TDP and FTLD-FUS were described.29 This is the first report of familial FTD cases with a GBE1 mutation showing APBD and FTLD-TDP.
Bit-Ivan et al. reported a case of APBD with GBE1 haploinsufficiency with concomitant FTLD-FUS, which had no TDP-43 pathology 14 while our cases of APBD and FTLD-TDP with GBE1 mutation had no FUS aggregation. Although the enzymatic activities of TDP-43 and FUS are similar, they regulate splicing of mainly distinct RNA targets and show different pathogenic mechanisms in FTLD.30 How GBE1 mutations could be differentially associated with FTLD-FUS and FTLD-TDP is unknown. Interestingly, Bit-Ivan et al. investigated 49 FTLD-TDP and three FTLD-FUS cases and found two FTLD-TDP cases having focal clusters of cortical PBs that were of higher density than the occasional cortical corpora amylacea, which is morphologically and ultrastructurally identical to PBs.8 Because corpora amylacea are frequently observed in ageing brains,31 APBD may be missed, ignored, or simply reported as ageing corpora amylacea in FTLD-TDP and FTLD-FUS cases.
ARTAG was also widespread in the current cases. While ARTAG is frequently observed in the ageing brain,14 the amount of ARTAG in these cases was dramatically increased, suggesting that ARTAG is associated with PBs. Notably, the distributions of ARTAG and PBs are quite similar: both are abundant in the subpial matter, the subependymal and perivascular regions, and the white matter. That being said, ARTAG is frequently clustered in the gyral white matter, while PBs are more common in deep white matter. Nonetheless, the two pathologies were closely related, and some PBs were enwrapped by or included in the processes of TSA (Figure 2E).
The most common clinical findings of APBD are neurogenic bladder and sensory-motor deficits; mild cognitive decline may affect up to half of the patients as the disease progresses.1 In the present case reports, II-1 started to have difficulty in walking and loss of bowel function at ~65 years old (yo), but they were able to walk at ~75 yo. II-5 had no complaint of walking difficulty or bowel dysfunction at ~60 yo, when they started to have cognitive impairment. Considering that the average age of onset of neurogenic bladder and motor dysfunction in APBD has been reported to be 50–55 years and around 50% of APBD patients become wheelchair dependent at ~65 yo,1 the current novel GBE1 c.1280delG mutation may cause sensory-motor symptoms that are milder and slower than typical APBD. In the present cases, the main symptom of the patients was cognitive decline. FTLD-TDP type A pathology in both cases, and a high level of ADNC in II-5, may have contributed to the severe cognitive impairment. The FTLD-TDP pathology in II-1 and II-5 was identical to previously reported FTLD-TDP type A pathology. Consistent with that, three affected siblings out of six have been diagnosed with bvFTD. Given that the ages at onset were compatible with that of typical FTLD-TDP type A patients,32 it is unclear whether co-pathologies of APBD, FTLD-TDP, ARTAG, and ADNC have exacerbated cognitive dysfunction or not.
One limitation of this study is that the association between GBE1 mutation and FTLD-TDP is still enigmatic. The association of autosomal dominant mutations in GBE1 with APBD is well documented,1–8 but FTLD-TDP is not always found in GBE1 mutation cases. Likewise, not all FTLD-TDP cases have APBD.14 Therefore, whether FTLD-TDP is primary, secondary, or unrelated to the identified GBE1 mutation is unclear. A second limitation is that although total GBE1 levels were significantly reduced in the patients with the GBE1 mutation, the enzyme activity of these patients could not be determined due to technical difficulty in the measurement of GBE1 enzyme activity using post-mortem brain tissue. Given the marked decrease in GBE1 protein levels in patients with the GBE1 mutation and in silico evidence suggesting it causes a loss-of-function, it seems reasonable to assume that GBE1 enzyme activity will be decreased. The third limitation is that the interaction between PBs and ARTAG is still unclear. PBs in APBD cases have been reported to be in the processes of neurons and astrocytes.33 Electron microscopic analysis will be performed in the future to determine the main cell preferences of those PBs: whether those PBs are in the processes of ARTAG or the neuronal axons enwrapped by the processes of ARTAG.
In conclusion, we are the first to report a case of familial FTD showing abundant PBs, FTLD-TDP, and ARTAG with a novel GBE1 mutation. Because some cases with both APBD and clinical FTD have been reported,13, 14, 22 coexisting APBD and FTLD-TDP/FTLD-FUS might be more common than expected, and therefore, GBE1 should be included among the genes considered for familial FTD.
Supplementary Material
Key Points:
We identified a family with FTD clinical phenotype with a novel GBE1 mutation c.1280delG, which is predicted to result in a reading frameshift, p.Gly427Glufs*9.
Two of the affected siblings with the GBE1 mutation showed a reduction in GBE protein expression levels with abundant FTLD-TDP Type A pathology in the grey matter, abundant PBs, and ARTAG in the subpial matter and the white matter.
The finding of co-pathologies of APBD and FTLD-TDP suggests these processes may share a disease mechanism resulting from this GBE1 loss-of-function mutation.
Acknowledgements
We are grateful to the patients and their families for participation in this research study. This study was funded by the National Institutes of Health (NIH) (P30AG072979, P01AG066597, R01NS109260, and U19AG062418) and the ALS Association to the New York Genome Center ALS Consortium (15-LGCA-234).
We thank Manuela Neumann and Elisabeth Kremmer for providing the phosphorylation-specific TDP-43 antibody TAR5P-1D3. We thank Theresa Schuck and Junle Chen for their technical support. We thank all colleagues in CNDR who helped with this study, as well as all the patients and their families.
Abbreviations
- Aβ
amyloid β
- APBD
Adult polyglucosan body disease
- ARTAG
Aging-related tau astrogliopathy
- bvFTD
behavioural variant of FTD
- DN
dystrophic neurites
- FTD
frontotemporal dementia
- FTLD
Frontotemporal lobar degeneration
- FTLD-TDP
FTLD with TDP-43 inclusions
- FTLD-FUS
FTLD with fused in sarcoma inclusions
- GBE1
1,4-alpha-glucan branching enzyme 1
- GFA
granular/fuzzy astrocytes
- H&E
haematoxylin and eosin
- HS
hippocampal sclerosis
- NCI
neuronal cytoplasmic inclusions
- PAS
Periodic acid–Schiff
- PBs
polyglucosan bodies
- TSA
thorn-shaped astrocytes
- WGS
whole genome sequencing
References
- 1.Mochel F, Schiffmann R, Steenweg ME, Akman HO, Wallace M, Sedel F, Laforêt P, Levy R, Powers MJ, Demeret S, Maisonobe T, Froissart R, Nobrega B, Fogel BL, Natowicz MR, Lubetzki C, Durr A, Brice A, Rosenmann H, Barash V, Kakhlon O, Gomori MJ, Knaap MSvd, Lossos A. Adult polyglucosan body disease: Natural History and Key Magnetic Resonance Imaging Findings. Annals of neurology 2012; 72: 433–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akman HO, Kakhlon O, Coku J, Peverelli L, Rosenmann H, Rozenstein-Tsalkovich L, Turnbull J, Meiner V, Chama L, Lerer I, Shpitzen S, Leitersdorf E, Paradas C, Wallace M, Schiffmann R, DiMauro S, Lossos A, Minassian BA. Deep Intronic GBE1 Mutation in Manifesting Heterozygous Patients With Adult Polyglucosan Body Disease. Jama Neurol 2015; 72: 441–5 [DOI] [PubMed] [Google Scholar]
- 3.Lossos A, Meiner Z, Barash V, Soffer D, Schlesinger I, Abramsky O, Argov Z, Shpitzen S, Meiner V. Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. Annals of neurology 1998; 44: 867–72 [DOI] [PubMed] [Google Scholar]
- 4.Harigaya Y, Matsukawa T, Fujita Y, Mizushima K, Ishiura H, Mitsui J, Morishita S, Shoji M, Ikeda Y, Tsuji S. Novel GBE1 mutation in a Japanese family with adult polyglucosan body disease. Neurology Genetics 2017; 3 (2): e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akman OH, Kurt Y, Wallace M, Schiffmann R, DiMauro S. Compound heterozygosis of a splice site and the common Ashkenazi Jewish mutation in GBE1 causes adult onset polyglucosan body disease. (P2.026). Neurology 2015; 84: P 2.026 [Google Scholar]
- 6.Sampaolo S, Esposito T, Gianfrancesco F, Napolitano F, Lombardi L, Lucà R, Roperto F, Iorio GD. A novel GBE1 mutation and features of polyglucosan bodies autophagy in Adult Polyglucosan Body Disease. Neuromuscular Disorders 2015; 25: 247–52 [DOI] [PubMed] [Google Scholar]
- 7.Ziemssen F, Sindern E, Schröder JM, Shin YS, Zange J, Kilimann MW, Malin JP, Vorgerd M. Novel missense mutations in the glycogen-branching enzyme gene in adult polyglucosan body disease. Annals of neurology 2000; 47: 536–40 [PubMed] [Google Scholar]
- 8.Hussain A, Armistead J, Gushulak L, Kruck C, Pind S, Triggs-Raine B, Natowicz MR. The adult polyglucosan body disease mutation GBE1 c.1076A>C occurs at high frequency in persons of Ashkenazi Jewish background. Biochem Bioph Res Co 2012; 426: 286–8 [DOI] [PubMed] [Google Scholar]
- 9.Gray F, Gherardi R, Marshall A, Janota I, Poirier J. Adult polyglucosan body disease (APBD). Journal of neuropathology and experimental neurology 1988; 47: 459–74 [DOI] [PubMed] [Google Scholar]
- 10.Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, Lee VM, Trojanowski JQ. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta neuropathologica 2015; 129: 469–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM. A harmonized classification system for FTLD-TDP pathology. Acta neuropathologica 2011; 122: 111–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee EB, Porta S, Michael Baer G, Xu Y, Suh E, Kwong LK, Elman L, Grossman M, Lee VM, Irwin DJ, Van Deerlin VM, Trojanowski JQ. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta neuropathologica 2017; 134: 65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farmer JG, Crain BJ, Harris BT, Turner SR. Coexisting adult polyglucosan body disease with frontotemporal lobar degeneration with transactivation response DNA-binding protein-43 (TDP-43)-positive neuronal inclusions. NeuroII-2013; 19: 67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bit-Ivan EN, Lee KH, Gitelman D, Weintraub S, Mesulam M, Rademakers R, Isaacs AM, Hatanpaa KJ, White CL, Mao Q, Akman O, DiMauro S, Bigio EH. Adult polyglucosan body disease with GBE1 haploinsufficiency and concomitant frontotemporal lobar degeneration. Neuropathology and Applied Neurobiology 2014; 40: 778–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, Cairns NJ, Crary JF, Duyckaerts C, Ghetti B, Halliday GM, Ironside JW, Love S, Mackenzie IR, Munoz DG, Murray ME, Nelson PT, Takahashi H, Trojanowski JQ, Ansorge O, Arzberger T, Baborie A, Beach TG, Bieniek KF, Bigio EH, Bodi I, Dugger BN, Feany M, Gelpi E, Gentleman SM, Giaccone G, Hatanpaa KJ, Heale R, Hof PR, Hofer M, Hortobágyi T, Jellinger K, Jicha GA, Ince P, Kofler J, Kövari E, Kril JJ, Mann DM, Matej R, McKee AC, McLean C, Milenkovic I, Montine TJ, Murayama S, Lee EB, Rahimi J, Rodriguez RD, Rozemüller A, Schneider JA, Schultz C, Seeley W, Seilhean D, Smith C, Tagliavini F, Takao M, Thal DR, Toledo JB, Tolnay M, Troncoso JC, Vinters HV, Weis S, Wharton SB, White CL 3rd, Wisniewski T, Woulfe JM, Yamada M, Dickson DW. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta neuropathologica 2016; 131: 87–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovacs GG, Robinson JL, Xie SX, Lee EB, Grossman M, Wolk DA, Irwin DJ, Weintraub D, Kim CF, Schuck T, Yousef A, Wagner ST, Suh E, Van Deerlin VM, Lee VM, Trojanowski JQ. Evaluating the Patterns of Aging-Related Tau Astrogliopathy Unravels Novel Insights Into Brain Aging and Neurodegenerative Diseases. Journal of neuropathology and experimental neurology 2017; 76: 270–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farmer JMM-WE, Lee VM-Y, Trojanowski JQ. An immortal legacy: how donation of human tissues impacts research and drives advances in diagnosis and therapy. Amherst, NY: Prometheus Books. 2008 [Google Scholar]
- 18.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME; Genome Aggregation Database Consortium, Neale BM, Daly MJ, MacArthur DG. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; 581(7809):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groß C, de Ridder D, Reinders M. Predicting variant deleteriousness in non-human species: applying the CADD approach in mouse. BMC Bioinformatics. 2018; 19(1): 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, Cussenot O, Cancel-Tassin G, Giles GG, MacInnis RJ, Maier C, Hsieh CL, Wiklund F, Catalona WJ, Foulkes WD, Mandal D, Eeles RA, Kote-Jarai Z, Bustamante CD, Schaid DJ, Hastie T, Ostrander EA, Bailey-Wilson JE, Radivojac P, Thibodeau SN, Whittemore AS, Sieh W. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016; 99(4): 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, Gathagan RJ, Iba M, McBride JD, Trojanowski JQ, Lee VM. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med. 2016; 213(12):2635–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, Zhang B, Gathagan RJ, Yue C, Dengler C, Stieber A, Nitla M, Coulter DA, Abel T, Brunden KR, Trojanowski JQ, Lee VM. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med. 2018; 24(1): 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL, Xie SX, McBride J, Wood EM, Schuck T, Irwin DJ, Gross RG, Hurtig H, McCluskey L, Elman L, Karlawish J, Schellenberg G, Chen-Plotkin A, Wolk D, Grossman M, Arnold SE, Shaw LM, Lee VM, Trojanowski JQ. A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 2014; 10: 477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uemura MT, Robinson JL, Cousins KAQ, Tropea TF, Kargilis DC, McBride JD, Suh E, Xie SX, Xu Y, Porta S, Uemura N, Van Deerlin VM, Wolk DA, Irwin DJ, Brunden KR, Lee VM, Lee EB, Trojanowski JQ. Distinct characteristics of limbic-predominant age-related TDP-43 encephalopathy in Lewy body disease. Acta Neuropathol. 2022; 143(1): 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bott NT, Radke A, Stephens ML, Kramer JH. Frontotemporal dementia: diagnosis, deficits and management. Neurodegener Dis Manag 2014; 4: 439–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann M, Lee EB, Mackenzie IR. Frontotemporal Lobar Degeneration TDP-43-Immunoreactive Pathological Subtypes: Clinical and Mechanistic Significance. Adv Exp Med Biol 2021; 1281: 201–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 2012; 8: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boulan-Predseil P, Vital A, Brochet B, Darriet D, Henry P, Vital C. Dementia of frontal lobe type due to adult polyglucosan body disease. Journal of Neurology 1995; 242: 512–6 [DOI] [PubMed] [Google Scholar]
- 30.Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem 2016; 138 Suppl 1: 95–111 [DOI] [PubMed] [Google Scholar]
- 31.Snow AD, Mar H, Nochlin D, Raskind M, Wight TN. Corpora Amylacea in Aging and Alzheimer’s Brain: Immunolocalization of Chondroitin Sulfate and Heparan Sulfate Proteoglycans. In Amyloid and Amyloidosis Eds. Isobe T, Araki S, Uchino F, Kito S, Tsubura E. Boston, MA: Springer US. 1988: 561–6 [Google Scholar]
- 32.Gefen T, Ahmadian SS, Mao Q, Kim G, Seckin M, Bonakdarpour B, Ramos EM, Coppola G, Rademakers R, Rogalski E, Rademaker A, Weintraub S, Mesulam MM, Geula C, Bigio EH. Combined Pathologies in FTLD-TDP Types A and C. J Neuropathol Exp Neurol. 2018. May 1;77(5):405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robitaille Y, Carpenter S, Karpati G, DiMauro SD. A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora’s disease and normal ageing. Brain 1980; 103: 315–36 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.