

Abstract
Inhibiting individual histone deacetylases (HDAC) is emerging as well-tolerated anticancer strategy compared with pan-HDAC inhibitors. Through pre-clinical studies, we demonstrated that the sensitivity to the leading HDAC6 inhibitor (HDAC6i) ricolinstat can be predicted by a computational network-based algorithm (HDAC6-score). Analysis of ~3,000 human breast cancers (BCs) showed that ~30% of them could benefice from HDAC6i therapy. Thus, we designed a phase Ib dose-escalation clinical trial to evaluate the activity of ricolinostat plus nab-paclitaxel in metastatic BC patients (NCT02632071). Study results showed that the two agents can be safely combined, that clinical activity is identified in patients with HR+/HER2− disease, and that the HDAC6-score has potential as predictive biomarker. Analysis of other tumor types also identified multiple cohorts with predicted sensitivity to HDAC6is. Mechanistically, we have linked the anticancer activity of HDAC6i/s to their ability to induce c-Myc hyperacetylation (ac-K148) promoting its proteasome-mediated degradation in sensitive cancer cells.
Keywords: HDAC6, ricolinostat, network activity, breast cancer
Editor summary:
Silva and colleagues develop a network-based HDAC6 score which could predict sensitivity to the HDAC6 inhibitor ricolinstat in preclinical models, as well as patients with HR+/HER2− breast cancer that received ricolinstat in a phase Ib clinical trial.
Homeostasis of cancer cells presents different oncogene and non-oncogene dependencies compared to non-transformed cells. Therapies aimed at targeting these dependencies represent more selective and less toxic anticancer strategies than standard chemotherapy1. Inhibition of histone deacetylases (HDACs) using pan-inhibitors has proven anticancer activity, especially in hematopoietic malignancies 2. However, toxicity associated with pleiotropic inhibition of multiple HDAC family members has limited their clinical use2. Thus, the interest has turned towards more selective inhibitors, targeting specific HDACs.
HDAC6 is a class-IIB deacetylase responsible for deacetylating a variety of substrates that is emerging as a promising therapeutic target. Anticancer activity of ricolinostat alone or in combination with additional drugs has been recently reported in pre-clinical models of multiple myeloma (MM)3, pancreatic and ovarian cancer4, esophageal cancer5, melanoma6, and lymphoma7. Recently, we reported that HDAC6 function is essential for maintaining the viability of an aggressive BC subtype called inflammatory breast cancer (IBC, ~1-4% of all BC8) and demonstrated that the leading HDAC6 inhibitor ricolinostat3 induces IBC cell death in vitro and in vivo9. HDAC6 is rarely amplified or mutated and the expression level of HDAC6 is similar between IBCs and non-IBC. Remarkably, in our previous studies, we used a transcriptomic-based system biology analysis aimed at measuring protein activity9 and found marked higher HDAC6 activity in IBCs. In brief, we first used the ARACNe algorithm10 to identify candidate HDAC6 transcriptional targets (i.e., transcripts whose expression is affected by HDAC6, HDAC6 regulon) from microarray-based transcriptome profiles of the TCGA breast cancer cohort (BRCA-TCGA11). Then, we integrated the expression of all transcripts of the HDAC6 regulon in a single score, termed as HDAC6 score, by summarizing their expression values. Importantly, from these analyses, the HDAC6 score emerged as a candidate biomarker for the effective identification of tumors where HDAC6 behaves as a master regulator of tumor cell state, thus presenting a non-oncogene dependency essential for cancer cell viability9.
We thus reasoned that other BCs in addition to IBCs may present the same dependency and that the HDAC6 score can be used as a biomarker to identify patient populations that can benefit from HDAC6 targeted therapy. In the new study reported here, we used the HDAC6 score to analyze all the primary and metastatic tumors included in the TCGA11, METABRIC12, and MBC13 data sets (~3,000 primary and ~270 metastatic BCs). Interestingly, we found that a group of BCs (~30% of all BCs) that were enriched in hormone receptor-positive pathological and molecular characteristics (HR+) present an HDAC6 score predictive of potential response to HDAC6 inhibitors. Next, we used a variety of experimental models that include in vitro cultures as well as in vivo mouse models to confirm the correlation between the HDAC6 score and the anticancer response to HDAC6 inhibition in BC cells. Based on these results, we designed a phase Ib clinical trial in partnership with Acetylon/Celgene to investigate ricolinostat, the leading HDAC6 inhibitor (HDAC6i)3 plus nab-paclitaxel as BC therapy (clinical trial ID#: NCT02632071). Paclitaxel was included based on pre-clinical data showing synergistic activity with ricolinostat, as well as for consistency with the standard of care treatment in BC. Notably, we observed that ricolinostat plus nab-paclitaxel can be safely combined, and that clinical activity is identified specifically in patients with HR+/HER2− disease. Furthermore, retrospective analysis that includes the NY CLIA-certified OncoTarget test, confirmed that the HDAC6 score could successfully stratify patients based on clinical outcomes. To complement these studies, we also expanded these studies to all cancer cohorts in TCGA, using tumor type-specific HDAC6 scores, and confirmed their predictive values by dose-response studies in vitro.
Although some HDAC targets have been identified14–16, the full substrate repertoire is far from being fully characterized. To define the anticancer mechanism of HDAC6i/s we combined comprehensive proteomic with biochemical targeted studies and identified that c-Myc is a substrate for HDAC6, and that HDAC6-mediated acetylation promotes its stability. HDAC6 inhibition by genetic or pharmacological approaches induces hyperacetylation and proteasome-mediated degradation of c-Myc leading to reduced cell fitness.
Overall, our studies support the coupling of ricolinostat in combination with cytotoxic treatment with HDAC6 score for treating multiple human cancers and support a mechanistic model based on c-Myc regulation explaining its anticancer activity.
RESULTS
Next-generation HDAC6 score predicts response to HDAC6i
Since our previous studies revealed that dependency on the HDAC6 function was linked to a high HDAC6 score in IBCs, we decided to explore its utility as a biomarker of HDAC6 inhibitor sensitivity. To accomplish this goal, we first assessed the fraction of breast tumor samples that would be predicted as HDAC6i based on the HDAC6 score analysis across both the BRCA-TCGA11 and METABRIC12 data sets (~3,000 primary tumors) as well as the set of BC lines available at the cancer cell line encyclopedia (CCLE; 47 lines)17 (Extended Data Fig. 1a).
While the original HDAC6 regulon used to generate the HDAC6 score was based on microarray-based gene expression profiling of 359 BRCA-TCGA samples9, the inclusion of additional datasets including a more heterogeneous set of tumors and different profiling technologies (i.e., different gene expression microarray platforms and RNA-seq) required the adaptation of the original HDAC6 score. Thus, we revised the original regulon used to compute the HDAC6 score—i.e., its repertoire of positively regulated and repressed transcriptional targets—to make it consistent with these different technologies and tumor models. For this, we used the SJARACNe18, an algorithm designed to reverse-engineer gene regulatory networks from a large number of transcriptomic profiles (n > 100). This generated a refined HDAC6 regulon specific to breast cancer based on the integrative analysis of all available gene expression profiles from the BRCA-TCGA (RNA-seq, n=1,221) and METABRIC (microarray, n=1,904) cohorts. We also improved the HDAC6 score calculation by using the NetBID (data-driven network-based Bayesian inference of drivers) algorithm19. As expected, despite marked profiling technology and sample preparation protocol differences between these datasets, HDAC6 activity predicted by the original and updated HDAC6 regulon were strongly overlapping (p = 1.7E-33, Extended Data Fig. 1b and Supplementary Table1). Additionally, they captured known functions of HDAC6 such as unfolded protein response (Extended Data Fig. 1c) and, consistent with previous findings, confirmed higher HDAC6 activity levels in IBCs9 (Extended Data Fig. 1d).
Using the updated regulon, we measured HDAC6 scores across all patient-derived samples and cell lines in the three data sets described above. Here, we observed that a significant fraction of all primary BCs ~30% had higher HDAC6 scores than the median IBC HDAC6 score, suggesting potential sensitivity to HDAC6 inhibitors (Extended Data Fig. 1d). Interestingly, high HDAC6 scores were not equally represented across BC subtypes but rather significantly enriched in the HR+ and HER2+ clinical subtypes (Fig. 1a) and the luminal-B and HER2-enriched molecular subtypes (Extended Data Fig. 1e). This association was also evident in cell lines (Fig. 1b). We also wondered if the association of high HDAC6 scores with HR+ and HER2+ BC subtypes will be maintained in metastatic BCs. There are no metastatic BC samples in METABRIC12 cohort and only 7 in TCGA11 cohort. Thus, we utilized the data available in the Metastatic Breast Cancer Project (MBC)13 to calculate the HDAC6 score for all ductal metastatic breast cancers containing both gene expression profiles and IHC-based subtyping information (n=45). Notably, we observed the same pattern from these metastatic samples as observed in primary BCs (Extended Data Fig. 1f).
Fig. 1. The HDAC6 score identifies breast cancers sensitive to the HDAC6 inhibitor ricolinostat.
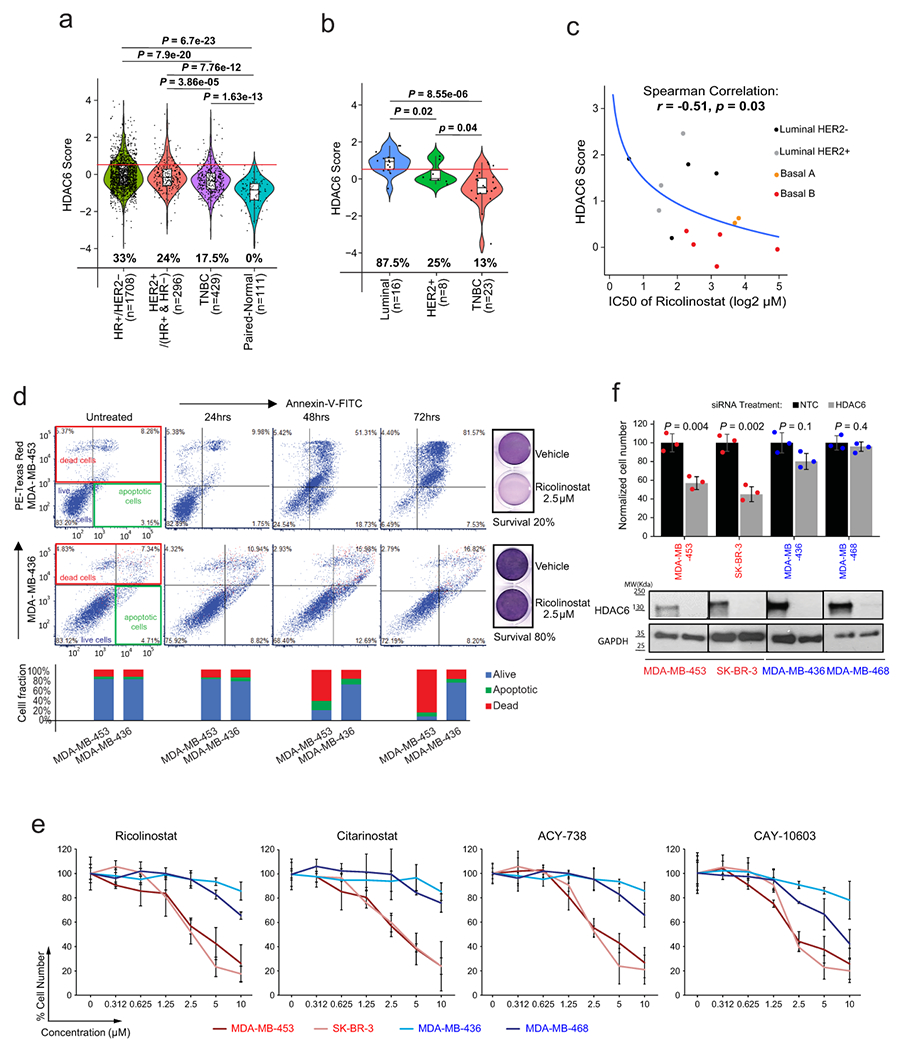
HDAC6 scores of BC patients from TCGA and METABRIC cohorts (a) and BC cell lines from CCLE database (b) are divided into subtypes. The red line represents the median of the HDAC6 scores in IBC samples and the numbers under each whisker plot indicate the percentage of samples over this value in each clinical subtype. Sample size of each group was indicated in the axis labels. P value was estimated using two-tailed t test. For (a) and (b) the center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5x interquartile range. (c) Graphical representation showing the correlation between HDAC6 score and response to ricolinostat among 14 breast cancer cell lines (n=8 independent experiments/per ricolinostat concentration). The curve was fitted by stat_smooth algorithm using lm smoothing method and y~log2(x) formula. The correlation coefficient (R) and P value were estimated using the Spearman correlation test. (d) Annexin-V/PE staining comparing the apoptotic response after Ricolinostat treatment of sensitive (MDA-MB-453) vs resistant (MDA-MB-436) BC cells. Quantification of alive, apoptotic, and dead cells is provided and visualized in stacked bar plot at the bottom of the panel. (e) Growth curves of sensitive (S) vs resistant (R) BC cells treated with four different HDAC6 inhibitors (n=3 independent experiments/per drug concentration). (f) Normalized cell number 6 days after transfection with siRNAs targeting HDAC6 or non-targeting control (NTC) (n=3 independent experiments/siRNA). The WT-blots show the silencing efficiency (The WT-blot results were reproduced n=3 times from independent experiments/siRNA). All error bars represent Mean±SD. P value was estimated by two-tailed t test.
Next, we evaluated the correlation between the HDAC6 score and ricolinostat sensitivity in 14 BC lines representative of the major molecular subtypes20, covering the full spectrum of HDAC6 scores (inferred from RNA-seq data from the CCLE). Specifically, we generated dose-response curves (8 doses ranging from 0 to 30μM) to measure cell line-specific IC50 values. The analysis revealed a strong inverse correlation between HDAC6 score and sensitivity (R = −0.51, by Spearman’s correlation, p = 0.03), thus showing that cancer cell lines with high HDAC6 score present higher ricolinostat sensitivity on average (Fig. 1c and Supplementary Table2).
We had previously shown that ricolinostat treatment induces apoptosis on sensitive IBC cancer cell lines9. Thus, we selected two lines with either low (MDA-MB-453) or high (MDA-MB-436) IC50 from the list to investigate if this alco occurs in non-IBC cells. For this we first identified the lowest concentration of ricolinostat that induces robust accumulation of acetylated α-Tubulin—a well-known HDAC6 substrate, without affecting accumulation of acetylated histones—established markers of pleiotropic class-I HDAC inhibition (i.e. off-target effects) (Extended Data Fig. 2a). Annexin-V staining following ricolinostat treatment with that concentration of ricolinostat (2.5μM) revealed progressive accumulation of apoptotic MDA-MB-453 cells (sensitive), with only very minor effects in MDA-MB-436 cells (resistant) (Fig. 1d).
To further demonstrate the specificity of the effect, we generated IC50 values for 3 additional HDAC6 specific inhibitors in BC cells presenting the highest (MDA-MB-453 and SK-BR-3) and lowest (MDA-MB-436 and MDA-MB-468) sensitivity to ricolinostat. These inhibitors include citarinostat—a more soluble analog of ricolinostat 4,9—and two next-generation inhibitors that are structurally unrelated to ricolinostat, ACY-738 21 and CAY10603 22 (Extended Data Fig. 2b). These analyses confirmed that all HDAC6 inhibitors reduced the growth of ricolinostat-sensitive cell lines with only minor effects in ricolinostat-resistant ones, at the same doses (Fig. 1 e). Finally, we also used RNAi to inhibit the expression of HDAC6. Consistently with the results obtained with the small molecule inhibitors, efficient RNAi induced silencing of HDAC6 reduced the viability of ricolinostat sensitive MDA-MB-453 and SK-BR-3 but not in MDA-MB-436 or MDA-MB-468 cells (Fig. 1f and Extended Data Fig. 2c).
Ricolinostat shows activity in vivo in selected cells
While the data described above show ricolinostat’s anticancer activity as a single agent, achieving a clinically relevant response in vivo generally requires combining targeted and standard chemotherapy. Primary chemotherapy for BCs typically includes the use of anthracyclines and taxanes23,24. Thus, we assessed the therapeutic value of combining paclitaxel and doxorubicin with ricolinostat. For this, we used two commonly used methods: Isobole curve25 and combination index (by Chou-Talalay equation)26. These two analyses showed that both chemo toxic agents synergize with ricolinostat, in ricolinostat-sensitive cells (Extended Data Fig. 2d), but not in ricolinostat-resistant cells (Extended Data Fig. 2d).
To transition our studies with MDA-MB-453 and MDA-MB-436 to an in vivo context these cells were injected as mouse xenografts in the flanks of γ-SCID mice. After the tumors grew to ~100-150mm3, they were randomly assigned to one of several therapeutic regimens, including ricolinostat and paclitaxel as single agents and ricolinostat plus paclitaxel in combination (combo). Paclitaxel was eventually selected over doxorubicin because it is widely used for BC standard of care. Confirming in vitro results, ricolinostat demonstrated significant antitumor growth activity as a single agent in MDA-MB-453 but not in MDA-MB-436 xenografts. WT-blot measuring Ac-α-Tubulin and Ac-His-K27 in tumor extracts from treated animals confirmed that the effect was associated with specific HDAC6 inhibition, with minimal effect on other class-I HDACs. Interestingly, while a small tumor mass was still detectable in the sensitive cells at the end of the treatment period with ricolinostat (1 month), combination treatment with paclitaxel-induced complete response (Fig. 2a). Intratumoral evaluation of the treated animals showed that the ricolinostat response in MDA-MB-453 tumors was associated with higher apoptosis levels (activated Caspase-3) while no such effect was seen in ricolinostat resistant MDA-MB-436 cells (Extended Data Fig. 2e).
Fig. 2. Anticancer activity of ricolinostat in vivo.
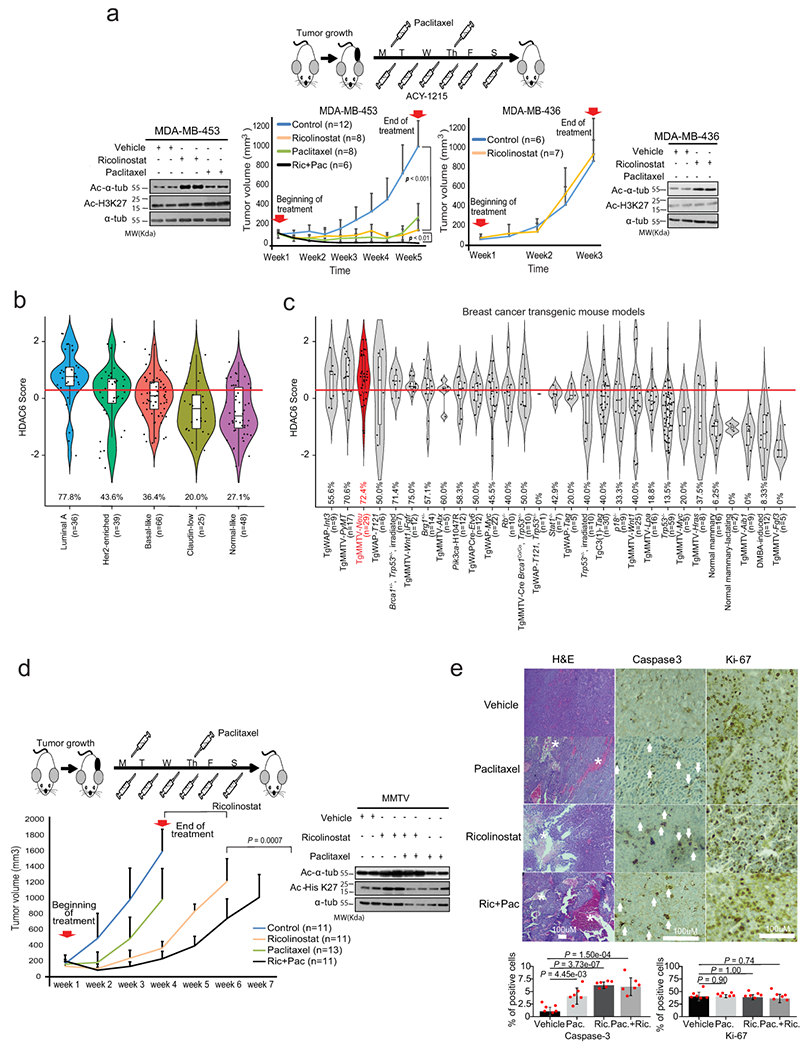
(a) Treatment of ricolinostat sensitive (MDA-MB-453) and resistant (MDA-MB-436) growing as xenografts in SCID mice. The cartoon illustrates the treatment regimen. The combinatorial effect with paclitaxel was also investigated in sensitive cells. On resistant cells, only ricolinostat was used because no effect was observed with the combo in vitro. The western blots show the accumulation of acetylated tubulin in tumors treated with ricolinostat. Additionally, the absence of off-target effect in class-I HDACs is shown by the minimum changes seen in the levels of acetylated Histone-3-K27 (two independent tumor samples are shown), (WT-blot results were reproduced n=3 times from independent experiments). For the growth curve, n=number of tumors for each of the treatment cohorts. (b) HDAC6 scores in tumors emerging in transgenic mouse models that recapitulate the molecular characteristics of human BCs (the number of samples is shown for each group in the figure). (c) HDAC6 scores in tumors emerging in 27 different transgenic mouse models of BC (with and without molecular characteristics of human BCs), (the number of samples is shown for each group in the figure). In C and D the red line represents the mean of the HDAC6 scores in IBC samples. In c and d, the red line represents the mean of the HDAC6 scores in IBC samples; The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5x interquartile range. (d) Treatment of BC tumors emerging in the MMTV_Neu transgenic mouse model. The beginning and end of treatment are indicated by the red arrows. The cartoon illustrates the treatment regimen. The combinatorial effect of ricolinostat plus paclitaxel was also investigated. The western blots indicate the same that in panel B (WT-blot results were reproduced n=3 times from independent experiment). For the growth curve, n=number of tumors for each of the treatment cohorts. (e) Histological intratumor evaluation of H&E, Caspase-3, and Ki-67 in tumor samples from panel E. Quantification is also shown in bar graphs. The white asterisks indicate necrotic areas and the white arrows indicate Caspase-3 positive stained cells. All error bars represent mean±SD and P value was estimated by two-tailed t test.
To complement our pre-clinical in vivo studies with more pathophysiologically-relevant models, we evaluated response to ricolinostat in spontaneous tumors using transgenic mouse models. Numerous murine models of BC have been created to mimic the genetic aberrations found in human tumors. In particular, gene expression profiles of 385 tumors representative of 27 different genetically-engineered mouse models (GEMMs) of BC have been described and compared with human counterparts27 (Extended Data Fig. 2f). Consistent with the human tumor studies, HDAC6 score analysis using these murine gene expression profiles confirmed that mouse models recapitulating the molecular characteristics of human luminal breast cancers presented with the highest HDAC6 scores (Fig. 2b). Since MMTV-Neu models tend to generate a homogeneous group of luminal tumors27 (Fig. 2c and Extended Data Fig. 2f), we investigated their response to the same ricolinostat regimens used to treat the MDA-MB-453 xenografts. Remarkably, when comparing single-agent treatments, we observed that ricolinostat alone was more effective than chemotherapy in MMTV-Neu tumors (Fig. 2d and Extended Data Fig. 2g). However, as also seen in MDA-MB-453 xenografts, the combination of ricolinostat and paclitaxel produced the strongest antitumor effect. Consistently, intratumoral evaluation of treated animals showed that ricolinostat anticancer activity was associated with higher cell death levels with multiple areas of necrosis and apoptosis and that this effect was stronger in the tumors treated with the drug combination (Fig. 2e).
Phase 1b trial of ricolinostat combined with nab-paclitaxel
Taken together, these studies suggest a significant association of ricolinostat sensitivity in BC with a high HDAC6 score. To translate this hypothesis to a clinical context we designed an open-label phase Ib trial using ricolinostat in combination with nab-paclitaxel for patients with metastatic breast cancer (MBC) (Fig. 3a) that have progressed on multiple previous lines of therapy (Supplementary Table 3). The primary objective of this study was to establish the safety, tolerability, and identify the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) of ricolinostat when combined with nab-paclitaxel. Additionally, the secondary objectives were to assess progression-free survival (PFS), overall response rate (ORR), clinical benefit rate (CBR) and to evaluate the HDAC6 score as a predictive biomarker by investigating the correlation between the HDAC6 score and clinical endpoints.
Fig. 3. Phase 1b trial of ricolinostat combined with nab-paclitaxel in metastatic breast cancer.
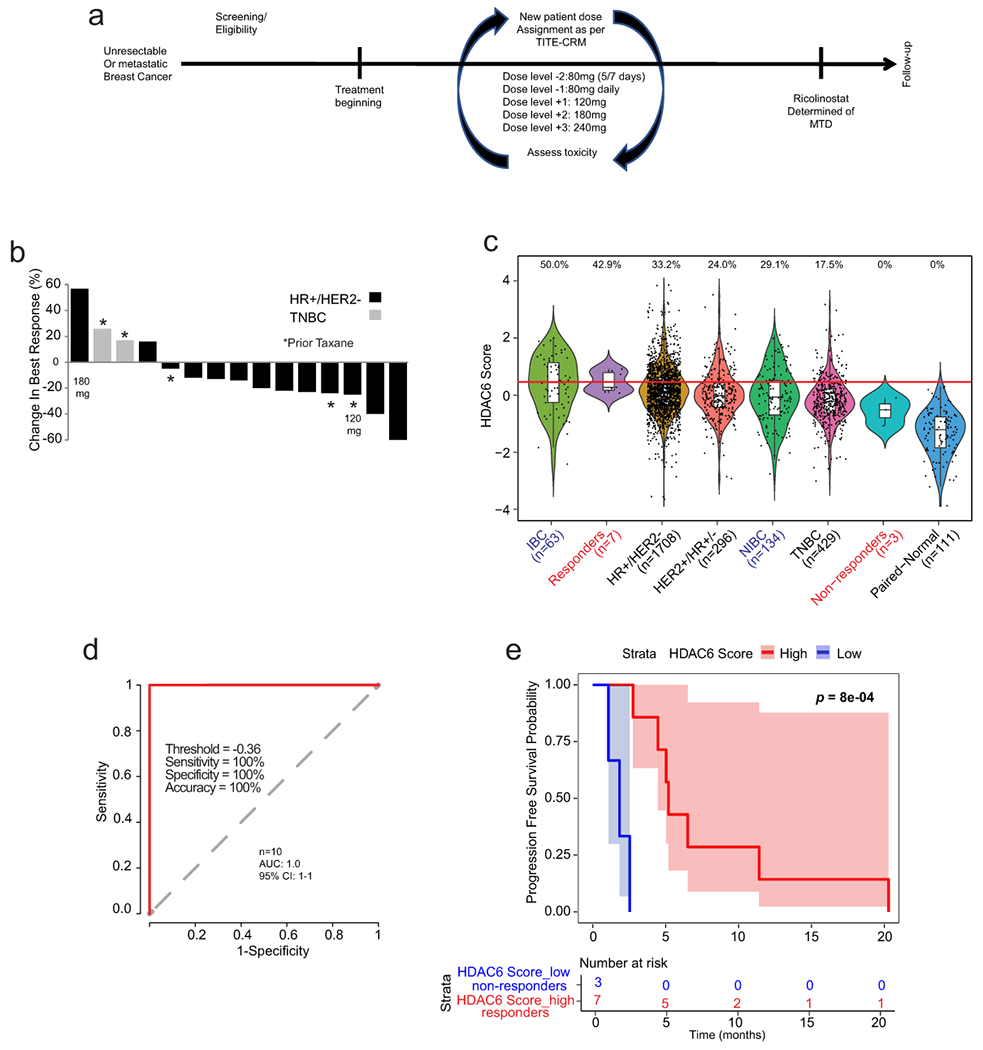
(a) Graphical description of the clinical study. (b) Waterfall plot showing the tumor best response for patients with measurable disease. Of the 16 patients, 3 were TNBCs (1 showed stable disease (SD) and 2 showed progressive disease (PD). The rest 13 patients were of the HR+/HER2− subtype (2 showed a partial response (PR), 9 showed SD and 2 showed PD. Note that one evaluable patient with stable disease did not have measurable disease and is not included in the waterfall plot (n=15). (c) The bar graph shows the HDAC6 scores in the patients in the trial (labeled in red) together with all the BC samples evaluated and separated by subtype. Labeled in blue are the IBC and the matched non-IBC series. The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5x interquartile range. The red line represents the median of the HDAC6 scores in IBC samples and the numbers above each whisker plot indicate the percentage of samples over this value in each clinical subtype. Sample size of each group was indicated in the axis labels. The full list of p values can be found summarized in the Source Data and was estimated using two-tailed t test. (d) Receiver operating characteristic (ROC) curve plot for evaluation of HDAC6 score to predict the response of patients with breast cancer to ricilinostat from the clinical trial. The recommended cutoff of the HDAC6 score and corresponding sensitivity, specificity, and accuracy were inside the box (n=2 independent HDAC6 score replicates per patient) (e) Kaplan – Meier graphic showing the survival of the patients in the study separated by HDAC6 score (high/low= higher and lower than −0.36, the cutoff ofHDAC6 score based on the ROC analysis in the study. In this study 10 out of 16 evaluable patients had tissue available for translational analyses. P value was estimated using two-tailed Log-Rank test.
In this trial, patients received ricolinostat orally (liquid) for 21 consecutive days of each 28-day cycle with nab-paclitaxel dosed at 100 mg/m2 on days 1, 8, and 15 until progression of the disease or unacceptable toxicity. Entry criteria included men or women with any metastatic BC subtypes. Measurable disease was not required. The MTD of ricolinostat with nab-paclitaxel was estimated via dose-escalation employing a time-to-event continual reassessment method (TITE-CRM)28. The MTD was defined as the dose combination associated with a target probability of dose-limiting toxicity (DLT) of 0.25. The TITE-CRM used an empirical dose-toxicity model see full description in material and methods). A total of five pre-defined doses of ricolinostat were selected for the dose-escalation process (Fig. 3a). Seventeen patients were accrued between March 2016 and February 2018. Of these, 16 patients had an evaluable disease, as one patient dropped out at cycle 2 due to no longer wishing to participate in the trial and in the absence of any related toxicity. In the 16 evaluable patients, the median age was 57.5 years (range: 41-78), 14 were female (87.5%), 3 had triple-negative MBC, and 13 were HR+/HER2− MBC. The median number of prior lines was 3 (range: 0-10) (Extended Data Fig. 3a). The first patient started at 120 mg/m2 qd, the second at 180 mg/m2 qd, and the remaining 14 patients were treated at 240 mg/m2 qd. No DLTs were seen in the DLT window of 8 weeks (first two cycles), and thus the MTD was not reached. Grade III events related to nab-paclitaxel included neutropenia (n=1), peripheral neuropathy (n=1), and 1 grade IV neutropenia. Grade III syncope related to ricolinostat was observed in 2 patients (Supplementary Tables 4–6). All of these events occurred after the DLT window. In the 16 evaluable patients, the following were best responses: 2 partial responses (PR), 10 stable diseases (SD), and 4 progressive diseases (PD: 2 TNBC, 2 HR+/HER2−) (Fig. 3b). All patients had measurable disease (Fig. 3b), except for one evaluable patient without target lesions who were reported to have SD for 9 months. Three patients who previously received a taxane in the metastatic disease achieved SD with ricolinostat plus nab-paclitaxel. One patient with SD remains on treatment since Feb 2018 (17 months). The clinical benefit rate was 31.25%: 5/16 patients (2 PR + 3 SD > 6 months). All of these patients were diagnosed with HR+/HER2− MBC, except 1 stable disease with TNBC. Median PFS 5.3 months [95% confidence interval (CI): 4.45-11.0] (Supplementary Table 7–9).
We were able to obtain tumor specimens in the form of paraffin sections (FFPE) with >50% in tumor content for 10 of the 16 evaluable patients (3 achieving PD and 7 showing SD or PR). RNA was obtained from these samples, subject to genome-wide RNA-seq, and the expression profiles obtained were used to calculate the HDAC6 scores. Interestingly, when we compared the HDCA6-scores between patients showing PD (non-responders) and those with either SD or PR (responders), a statistically significant higher HDAC6 score was seen in responder patients (p=2.1E-3 Extended Data Fig. 4a). HDAC6 score analysis integrating our clinical study with TCGA, METABRIC, and IBC cohorts confirmed our previous hypothesis that patients with HR+/HER2− breast cancer respond better than those with TNBC (Fig. 3c). Further, we used the receiver operating characteristic (ROC) curves 29 to characterize the sensitivity/specificity of the HDAC6 score using the trial data. Briefly, ROC curve analysis is a graphical plot that illustrates the diagnostic ability of a binary classifier system as its discrimination threshold is varied. The best cutoff value can be calculated by ROC analysis for continuous variables to predict dichotomous variables with the best sensitivity and specificity. In this analysis, the ROC curve analysis for the HDAC6 score achieved an area under the curve (AUC) of 1.0 (Fig. 3d). Although the perfect AUC was likely influenced by the small number of patients, at a cutoff value of −0.36, the HDAC6 score gave rise to a clear separation between responders and non-responders with 100% accuracy, and it outperformed the subtype (HR+/HER2 or TNBC) that had an accuracy of 80% (2 out of 10 patients were mispredicted, including 1 HR+/HER2− and 1 TNBC). Finally, we examined the predictive power of the HDAC6 score for patient prognosis, measured by PFS. We classified the patients into HDAC6 score high and low groups using the cutoff of −0.36 calculated by the ROC analysis. Patients with a high HDAC6 score had a median PFS of 6.51 months (95% CI: 5.19-NA), which was significantly better (p = 8.0E-4, Fig. 3e) than patients with low HDAC6 score who had a median PFS of 1.84 months (95% CI: 1.08-NA).
Transitioning any molecular biomarker to the clinic requires using a CLIA-certified test. The Darwin OncoTarget test, which is based on the VIPER (Virtual Inference of Protein-activity by Enriched Regulon analysis) algorithm30, was developed precisely to compute the activity of druggable proteins in cancer patients, based on the expression of their ARACNe-inferred transcriptional targets. The test has recently received CLIA certification from the NY State Dpt. of Health31. As a result, we assessed whether HDAC6 activity measured via this clinical-grade test was also predictive of patient sensitivity to the ricolinostat/nab-paclitaxel combination therapy in the Phase Ib study. Consistent with the results discussed in the previous sections, the HDAC6 score measured by Darwin OncoTarget was equally effective in stratifying patient sensitivity (p = 9.4E-3, Extended Data Fig. 4b), achieving a classification of the 7 responders and 3 non-responders with an AUC = 0.9 [95% CI: 0.68-1.0] based on ROC analysis (Extended Data Fig. 4c).
Because of the nature of the phase-Ib trials, our study did not include a paclitaxel-only control group. Thus, to investigate if the correlation between the HDAC6-score and the response to ricolinostat plus nab-paclitaxel in the trial was influenced by taxanes, we investigated a publically available series of breast cancers (n=106) treated only with paclitaxel32. As expected, the distribution of the HDAC6 scores in the series mimicked the results described for TCGA and METABRIC, and the HR+/HER2− and the luminal-B subtypes had the highest values while the triple-negative and basal subtypes showed the lowest ones (Extended Data Fig. 5a and 5b). Importantly the HDAC6 scores showed no correlation with pathologic response to paclitaxel (Extended Data Fig. 5c) or with the patient survival (Extended Data Fig. 5d). Thus, these results demonstrate that the correlation between the HDAC6 score and the response to treatment is linked to the use of ricolinostat.
High HDAC6 scores are found in a variety of human cancers
Since our studies confirmed the prognostic value of the HDAC6 score in human patients, we decided to systematically assess the HDAC6 scores across a large repertoire of human primary malignancies and cancer cell lines. Specifically, we analyzed >10,000 gene expression profiles, representing 32 molecularly distinct human malignancies represented in the TCGA database (https://www.cancer.gov/tcga). First, we generated tumor-specific HDAC6 regulons using the same approach successfully tested in breast cancer and used them to calculate the HDAC6 scores for all TCGA samples in a cancer type-specific manner. As expected, the number of genes that overlapped among the different tumor types was highly significant, although tumor type-specific differences were also noticeable (Extended Data Fig. 6a, and Supplementary Table 10–11). Next, we aimed to investigate whether a correlation exists between the HDAC-6 scores and the response to therapy in other tumor types. For this, we performed dose-response studies to assess ricolinostat IC50 in 58 additional cancer lines, representing 11 different tumor types. Notably, confirming the breast cancer-specific findings, a significant anticorrelation was detected between HDAC6 score and IC50 (R = −0.44, p = 5.2E-5) (Fig. 4a; Extended Data Fig. 6b and 6c, and Supplementary Table 12). Finally, we assessed the HDAC6 scores in 1156 different cancer cell lines available in the CCLE cohort, representing 20 tumor types17, as well as in 32 primary tumors (TCGA database) (Fig. 4b and 4c and Supplementary Table 13). We also compared the HDAC6 score in cancer cell lines and primary tumors. We used the 20 cancer types with data for both cohorts and reranked each cancer type by HDAC6 score in each cohort separately. Then we visualized the ranks of the 20 cancer types in a scatter plot and employed the Spearman method to calculate the correlation (Fig. 4d). Notably, we observed a strong correlation of the ranks between cell lines and primary samples, showing the consistency of HDAC6 score distribution in these two cohorts and supporting the use of cell lines to investigate the response to HDAC6i.
Fig. 4. The HDAC6 score correlates with the response to ricolinostat in other cancer types.
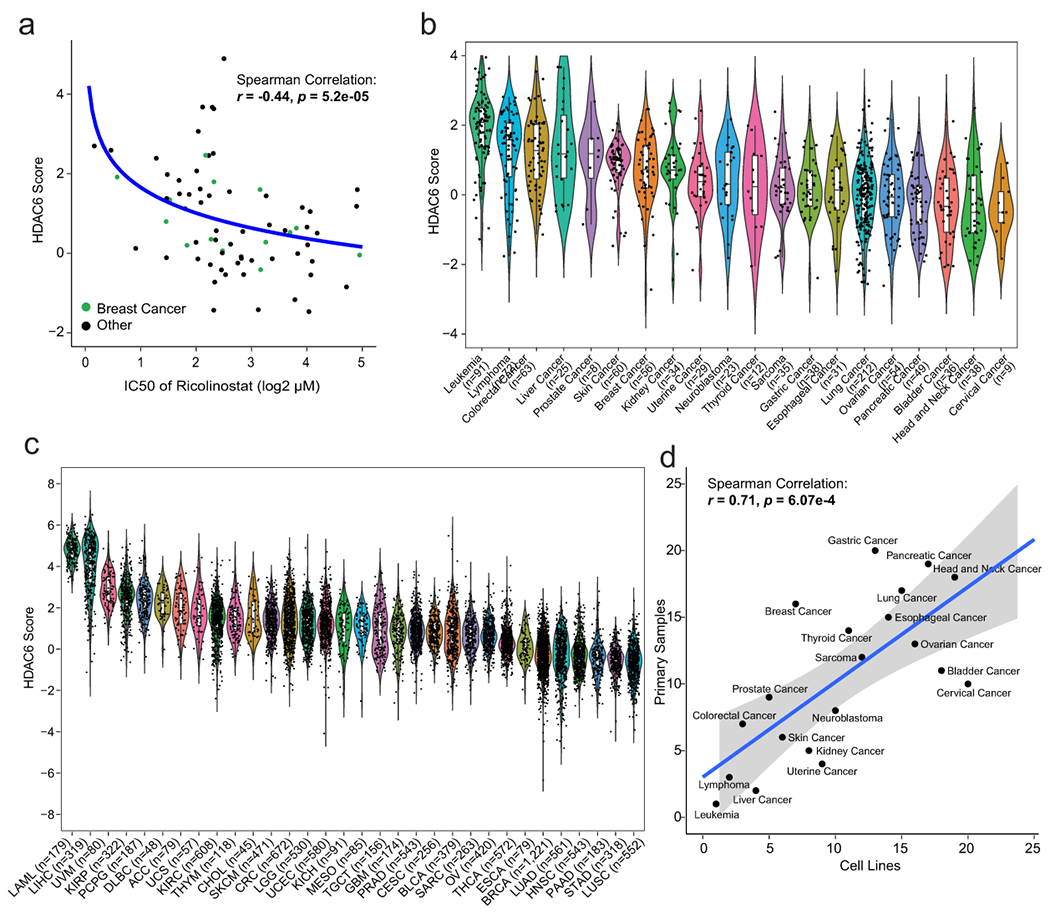
(a) Graphic showing the correlation between HDAC6 score and response to ricolinostat in 72 human cancer cell lines. The curve was fitted by stat_smooth algorithm using lm smoothing method and y~log2(x) formula. The correlation coefficient (R) and P value were estimated using two-tailed Spearman correlation test. N=6 independent experiments per cell line and ricolinostat dose. (b) HDAC6 scores were calculated for 1,156 different cancer cell lines available in the CCLE database representing 20 different human cancers and (c) for over 10,000 molecular profiles representing 32 different types of human cancer that have been collected in the TCGA database. For b and c the number of cell line/patient samples is shown for each group on the figure. The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5x interquartile range. As N we used all independent replicates available in the CCLE and TCGA data sets, precise information for each sample is available in CCLE and TCGA. LAML: Acute Myeloid Leukemia; LIHC: Liver Hepatocellular Carcinoma; UVM: Uveal Melanoma; KIRP: Kidney Renal Papillary Cell Carcinoma; PCPG: Pheochromocytoma and Paraganglioma; DLBC: Diffuse Large B-Cell Lymphoma; ACC: Adrenocortical Carcinoma; UCS: Uterine Carcinosarcoma; KIRC: Kidney Renal Clear Cell Carcinoma; THYM: Thymoma; CHOL: Cholangiocarcinoma; SKCM: Skin Cutaneous Melanoma; CRC: Colorectal Carcinoma; LGG: Brain Lower Grade Glioma; UCEC: Uterine Corpus Endometrial Carcinoma; KICH: Kidney Chromophobe; MESO: Mesothelioma; TGCT: Testicular Germ Cell Tumors; GBM: Glioblastoma; PRAD: Prostate Adenocarcinoma; CESC: Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma; BLCA: Bladder Urothelial Carcinoma; SARC: Sarcoma; OV: Ovarian Serous Cystadenocarcinoma; THCA: Thyroid Carcinoma; ESCA: Esophageal Carcinoma; BRCA: BC; LUAD: Lung Adenocarcinoma; HNSC: Head and Neck Squamous Cell Carcinoma; PAAD: Pancreatic Adenocarcinoma; STAD: Stomach Adenocarcinoma; LUSC: Lung Squamous Cell Carcinoma. (d) Correlation of ranks of HDAC6 score among 22 cancer types between cancer cell lines from CCLE and patient cancer samples from TCGA. The cell lines used for this comparison were the same used in panels b and c (n=patients and cancer lines used in those panels). The curve was fitted by stat_smooth algorithsm using lm smoothing method and y~x formula, and the 95% confidence interval of the regression line is displayed as shaded. The correlation coefficient (R) and P value were estimated using two-tailed Spearman correlation test.
Additionally, to the effect that HDAC6i can cause directly on cancer cells, it may be additional effects on the tumor microenvironment. Thus, we evaluated the expression profiles of the TCGA-BRCA samples using the algorithm ESTIMATE33 to predict infiltration of immune cells. Here, we found a negative correlation between immune infiltration and the HDAC6 score (Extended Data Fig. 7a). Although this is not surprising due to the association of higher HDAC6 scores and lower immune scores with HR+ and HER2+ BC tumors compared with TNBCs (Extended Data Fig. 7b), it warrants additional studies on how HDAC6i/s may impact immune response.
Overall, our analyses showed that most human cancer types present a wide distribution of HDCA6-scores. Because of the correlation between the HDAC6 score and the response to ricolinosat these results suggested that a significant subset of patients, across multiple tumor types, may benefit from treatment with this HDAC6i.
The response to ricolinostat is linked to reduction of c-MYC
To investigate the molecular mechanism involved in the response to ricolinostat we compared the transcriptional profiling of three ricolinostat-sensitive cell lines as well as MMTV-Neu tumors treated with ricolinostat for 12 hours with control counterparts. Interestingly, gene set enrichment analysis (GSEA) revealed that hallmark signatures associated with MYC activity were the topmost downregulated signatures in cells treated with ricolinostat (Extended Data Fig. 8a). Based on this result, we expanded our studies by profiling ricolinostat-sensitive cell lines treated with ricolinostat at earlier (6 hours) and later (24 hours) time points and looking in greater detail in all the signatures associated with MYC networks. Here, we found a strong and robust loss of MYC activity across the vast majority of MYC signatures that was evident in all the sensitive cells even at the earliest time point (Fig. 5a). To complete these studies, we also used the ARACNe algorithm described above to infer MYC activity during treatment. In agreement with the GSEA studies, we found a strong reduction of MYC activity in ricolinostat-sensitive cells (Extended Data Fig. 8b). Prompted by these results we compared the MYC expression at a protein level between a series of ricolinostat resistant and sensitive cell lines (Fig. 5b). This study revealed a strong reduction in MYC protein and MYC activity in ricolinostat-sensitive cell lines while these were basically unchanged in resistant cells (Fig. 5b and Extended Data Fig. 8c). Loss of Myc activity and protein expression was associated with a mild reduction in MYC mRNA in some cell lines but not in others (Extended Data Fig. 8d and 8e). As expected, the same specific loss of protein was observed when the cells were treated with other HDAC6 inhibitors or when HDAC6 was silenced by RNAi (Fig. 5c).
Fig. 5. Treatment with ricolinostat induces a robust reduction of MYC expression and activity.
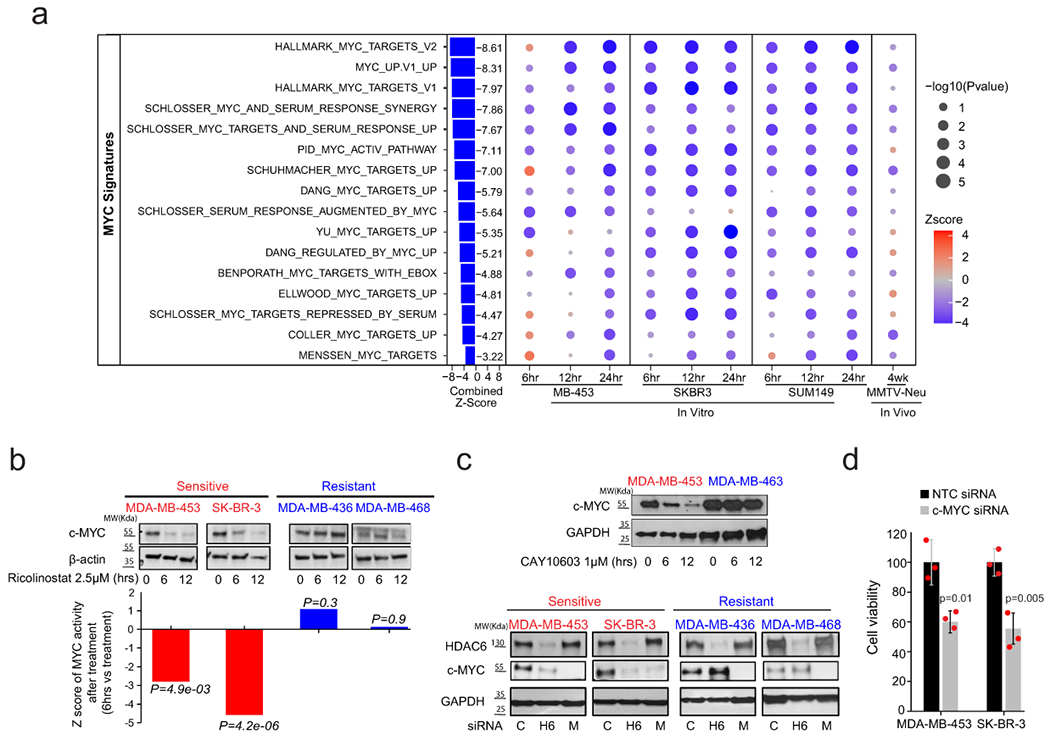
(a) Bubble plot representing GSEA analysis of MYC signatures during ricolinostat exposure in sensitive breast cancer cells. The size and intensity of the bubble indicate the statistical significance. N = 2 independent experiments for each time point of each BC cell line or mouse model. P value was estimated by two-tailed t test. The Z-scores were transformed from these P values and further combined using Stouffer’s method. (b) The upper panel (WT-blots) shows the reduction of MYC protein expression after ricolinostat treatment (2.5 μM) in sensitive but not resistant cancer cells. WT-blot results were reproduced n=3 times from independent experiments. The lower panel shows the summarized z-scores, comparing Ricolinostat-treated (6hrs) vs. untreated in the same cell lines shown in the upper panel. The Z-score was transformed from the P value estimated by two-tailed t test. (c) WT-blots showing the reduction of MYC in sensitive cell lines when HDAC6 was inhibited by the small molecule inhibitor CAY10603 (1μM, upper panel) or by RNAi (100nM, lower panel) (c=non-targeting siRNA control, H6=siRNA targeting HDAC6, M= siRNA targeting MYC). WT-blot results were reproduced n=3 times from independent experiments. (d) Cell viability (cell number) after MYC is silenced by siRNA in ricolinostat sensitive cell lines (notice that the western blot showing efficient silencing of MYC is shown in Fig. 5c). N=3 independent experiments per siRNA and cell line. Data are presented as mean values +/−SD. P values was estimated by two-tailed t test.
Genome-wide CRISPR screens have indicated that a large majority of breast cancer cells depend on MYC expression34 and we validated this dependency on ricolinostat-sensitive cells using RNAi (Fig. 5d). Based on these data, we conclude that the loss of viability seen in ricolinostat-sensitive cells is linked to the loss of MYC expression induced by ricolinostat treatment (Fig. 5d).
Finally, we also wondered whether the different responses to ricolinostat between sensitive and resistant cells could be due to a lack of HDAC6 inhibition in resistance cells. To check this, we calculated the HDAC6 score before and after treatment with ricolinostat to estimate the HDAC6 activity. We found that this was not the case and in both, sensitive and resistant cells, ricolinostat clearly reduced the HDAC6 score (Extended Data Fig. 8f).
c-MYC is a substrate of HDAC6
Only a handful of HDAC6 substrates have been identified and validated14–16. Acetylation levels of these substrates can influence protein stability, activity, or both15,35. Thus, we decided to further investigate whether c-Myc could be a substrate of HDAC6.
First, we performed co-immunoprecipitation (co-IP) to study if there is a physical interaction between c-Myc and HDAC6. Briefly, given the short half-life of c-Myc (~30min.)36, we transduced HEK-293T cells with a construct expressing a more stable form of c-Myc (with a mutated degron domain37) under doxycycline (dox) inducible conditions. Upon induction with dox, a clear direct interaction between HDAC6 and c-Myc was detected, and we observed both co-IP of c-Myc when using HDAC6 antibodies and co-IP of HDAC6 using c-Myc antibodies (Fig, 6a). To complement our studies and obtain information regarding potential amino acids (aa/s) residues influenced by HDAC6 we performed tandem mass tag (TMT) proteomics to compare the spectrum of acetylated proteins before and after inactivating HDAC6. For this, we first generated both knock-out (KO) and HDAC6 mutant (H651A) variants of HAP1 cells using CRISPR technology (see material and methods). This cell line was selected as it is a haploid model that facilitates the generation of point mutants by homolog recombination (Fig. 6b). Mutation of histidine 651 was selected as it has been shown to present a 150-fold diminished affinity for ligand binding 38. As expected for HDAC6 deficient cells, HDAC6 mutants presented elevated levels of acetylated α-Tubulin than control WT counterparts (Fig. 6b). For TMT studies, HAP1 variants were processed to obtain protein extracts. Then acetylated proteins were extracted by IP before being finally quantified (Fig. 6b). This study was able to quantify 3,359 acetylation sites in 1,618 proteins (Supplementary Table 14). Of those, 57 sites presented a statistically significant increase in acetylation (FDR<0.05; fold change >1.5) in HDAC6-KO cells; 49 of them (86%) were also found significantly increased in H651A mutant (FDR>0.05). Interestingly, acetylation of c-Myc in the Lys148 (K148) was also one of the top putative sites on this list showing a significant increase ((FDR<0.05; fold change >1.5) in both HDAC6-KO and -mutant (Fig. 6b and Extended Data Fig. 9a). Remarkably, acetylation of this site by TTC539 and EP300 40 acetyltransferases have been linked to c-Myc degradation mediated by the proteasome36,41.
Fig. 6. HDAC6 modulates the acetylation level of MYC.
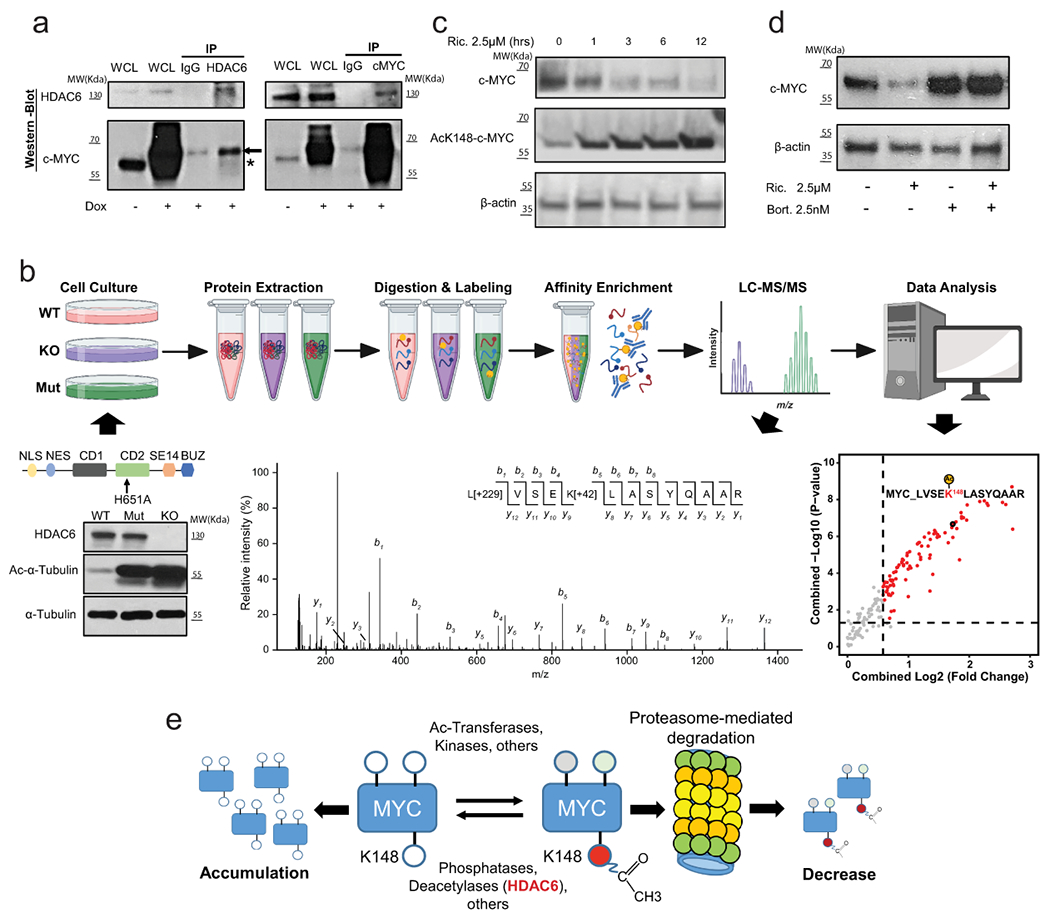
(a) Co-IP of MYC and HDAC6 in HEK-293T cells. The asterisk and arrow indicate the endogenous and the transduced c-MYC respectively (transduced construct expresses a slightly larger form). IP results were reproduced n=3 times from independent experiments. (b) Schematic description of the proteomic study described in the text. The WT-blot shows the accumulation of acetylated α-Tubulin in HDAC6 deficient HAP1 cells. The MS/MS spectrum shows an example assigned to the peptide containing MYC K148 acetylation site with b- and y- ions corresponding to the N- and C-terminal fragments, respectively. Peaks that match to theoretically calculated fragmented ions of the lysine-acetylated peptide are indicated. Modifications on specific residuals are indicated for TMT (+229 Da) and acetylation (+42 Da), respectively. WT-blot results were reproduced n=3 times from independent experiments. The dot plot shows the top differentially acetylated proteins from the proteomic study. The vertical and horizontal dash lines indicated the cutoffs of fold change > 1.5 and P < 0.05, respectively. N = 2 for each group. P value was estimated using two-tailed t test. (c) Degradation of MYC correlates with the accumulation of the ac-K148 form during treatment of MDA-MB-453 with ricolinostat (2.5μM). WT-blot results were reproduced n=3 times per time point from independent experiments. (d) Proteasome inhibition by Bortezomib (2.5nM) blocks the reduction of c-MYC protein induced by ricolinostat (2.5μM). WT-blot results were reproduced n=3 times per drug combination from experimental replicates. (e) Mechanistic model of the effect of HDAC6 inhibition on MYC expression. C-MYC stability is influenced by posttranslational modification. Acetylation of K148 promotes degradation by the proteasome and it is prevented by HDAC6. Thus, HDAC6 inhibition leads to hyperacetylation of MYC leading to its degradation.
Thus, next, we used c-Myc ac-K148 specific antibodies and confirmed that ricolinostat treatment in MDA-MB-453 and SK-BR-3 BC cells induces the accumulation of acetylated c-Myc (Fig. 6c and Extended Data Fig. 9b). Finally, we reasoned that if the accumulation of Ac-K148-cMyc and degradation of c-Myc were linked to proteasomal-mediated degradation then proteasomal inhibition will restore its protein levels. In fact, this was the case, and treating cells with bortezomib completely block c-Myc reduction induced by ricolinostat (Fig. 6d).
DISCUSSION
There are several key points related to the study presented in this manuscript that warrant further discussion. First, thanks to the development of specific small molecule inhibitors, such as ricolinostat, HDAC6 is emerging as a promising therapeutic target. Anticancer activity of ricolinostat alone or in combination with additional drugs has been recently reported in pre-clinical models of multiple myeloma (MM)3, pancreatic and ovarian cancer4, esophageal cancer 5, melanoma6 and lymphoma7. Our studies presented here showed that ricolinostat is also active in a variety of BCs enriched in HR+ and HER2+ characteristics. Our clinical study is an early-phase trial and the enrolled patients had shown disease progression through multiple lines of therapy before starting the treatment. Despite being a heavily pretreated cohort, the clinical benefit rate in our patients was 31.25%. Thus, our study provides initial evidence supporting the use of ricolinostat plus nab-paclitaxel for patients that have already exhausted the standard of care options. Remarkably, the activity of the combo was also observed in patients that had previously progressed after taxane-base therapy (Fig. 3b).
Second, several early-stage clinical trials using ricolinostat in MM, lymphoid malignancies, leukemia, gynecological cancers, and our own in BC are currently being evaluated. Because these are early-stage studies they have been mainly focused on describing the MTD of this HDAC6i alone or in combination with other drugs. Overall, and in agreement with our results, the clinical data from these studies confirmed that, at the range of doses used in our clinical trial, ricolinostat is safe, well-tolerated, and active42–44.
Third, targeted therapies like ricolinostat represent exciting novel strategies to treat human cancers as they do not have the general undesired toxicity that is commonly seen with standard chemotherapy. However, these therapies do not have the wide spectrum of anti-cancer activity of the former therapies and generally need to be delivered based on the assessment of predictive molecular markers45. In fact, multiple specific inhibitors that have demonstrated efficient anticancer activity in some contexts have shown disappointing clinical results due to the lack of predictive biomarkers that can identify the correct patient population. Critically, the HDAC6 score developed in this manuscript provides an effective predictive biomarker to identify patients most likely to benefit from HDAC6i therapy. In terms of immediate clinical translation, we have shown that the NY CLIA certified Darwin OncoTarget test produces HDAC6 scores that are highly predictive of patient sensitivity to ricolinostat plus nab-paclitaxel combination therapy.
Fourth, we have used the HDAC6 score to study a large variety of human cancer and found that cancer types such as AML46,47, lymphoma7, and melanoma6, where ricolinostat and other HDAC6 inhibitors have shown anticancer activity in pre-clinical models present the highest HDAC6 scores. Additionally, a fraction of the most common tumor types such as prostate, colorectal, or B-cell lymphoma and some of the deadliest ones such as melanoma or glioblastoma were also at the top of the list. This opens the exciting possibility to use HDAC6i such as ricolinostat beyond BC in a variety of other tumors.
Fifth, our mechanistic studies have linked the anticancer activity of ricolinostat with the reduction of c-Myc levels. MYC is one of the most potent oncogenes and is activated in more than half of human cancers. Importantly, MYC activation often results in the well-known phenomenon of oncogene addiction, and attenuation of MYC reduces tumor cell viability48. Although MYC inhibition has been proposed as a powerful therapeutic strategy for the treatment of many types of cancers, direct targeting of MYC remains a challenge49. Hence, the reduction of MYC expression through ricolinostat treatment represents an exciting alternative that can potentially impact a large variety of tumor types. How does HDAC6 inhibition modulate MYC expression? Our mechanistic studies support a model where MYC is a substrate of HDAC6 controlling the balance between the acetylated and deacetylated forms. HDAC6 inhibition shifts this balance and promotes the accumulation of acetylated MYC which is rapidly degraded by the proteasome (Fig. 6e).
Our study has some limitations that are worth discussing. While we have demonstrated that inhibition of HDAC6 induces accumulation of acetylated c-Myc leading to its degradation, we still do not know how HDAC6i resistant cells prevent degradation of MYC. Multiple possibilities exist. Resistance cells may somehow prevent accumulation of acetylated MYC. Alternatively, protein stability and function are often regulated by a compendium of posttranslational modifications (PTMs). In fact, several PTMs (Thr-58 and Ser-62 and Lys-323), have been experimentally linked with c-Myc stability50,51. Thus, other PTMs in c-Myc may modify the response to HDAC6i/s creating a “PTM MYC code” that defines the response to HDAC6i. We have observed that although accumulation of c-Myc ac-K148 acetylation is reduced in some HDAC6i resistance cells, it is not fully abrogated (Extended Data Fig. 10). Additional studies are necessary to fully understand the regulation of MYC in HDAC6i resistant cells.
Although there is no doubt that MYC levels influence the viability of cancer cells it is unlikely that this is the only effect induced by HDAC6 inhibition. For instance, HDAC6 is necessary for the formation of the aggresome and establishing an efficient protective response to the accumulation of misfolded intracellular protein14–16. Cells deficient in HDAC6 have a limited capacity for clearing toxic protein aggregates14,15,52. Additionally, misfolded polypeptides can be corrected by chaperones35. Notably, the Hsp90 chaperone is a known substrate of HDAC6 whose activity is affected when HDAC6 is inhibited53. Dysfunctional mitochondria aggregate into aggresome-like structures also dependent on HDAC6, called the mito-aggresome35,54. Accumulation of defective mitochondria also generates toxicity that compromises cell viability55–60.
Only a few bona fide HDAC6 substrates such as α-Tubulin16 and Hsp9053 have been well-validated, and over another half dozen have been reported in the literature only a few times. Although it is out of the scope of this report, our proteomic and genomic studies provide additional evidence of multiple putative substrates, other than c-Myc, that are worth investigating. The acetyltransferase EP300, one of the main regulators of gene transcription with an essential role in cell growth and proliferation was also found hyperacetylated in deficient HDAC6 cells (Supplementary Table 14). Remarkably, upregulation of the EP300 activity through acetylation is well-know61. Because acetylation of c-MYC by EP300 is associated with increase turnover40, we could speculate that the acetylation status of c-MYC is balanced by HDAC6 through a multiple layered regulation. Thus, HDAC6 could not only deacetylate c-MYC directly but also by deacetylating and reducing the activity of EP300. While this remains highly speculative it illustrates the potential relevance that other putative targets may have in the anticancer activity of HDAC6 inhibition and that their relevance may differs among cancer cells. Finally, in our studies, we have used multiple specific HDAC6i/s as well as genetics (RNAi). While these complementary approaches provide supporting evidence of the anticancer activity of HDAC6 blockage we should consider that each of them may have its unknown off-target effects.
A recent report has described a noncanonical role of HDAC6 in the regulation of the immune response showing that HDAC6i can improve immune checkpoint blockade (ICB)62. Thus, the addition of ICB could be considered in future trials containing HDAC6i like ricolinostat. In this regard, it is worth mentioning that although it is not surprising that our study found a negative correlation between immune infiltration and the HDAC6 score due to the association of higher HDAC6 scores and lower immune scores with HR+ and HER2+ BC tumors compared with TNBCs, it warrants additional studies on how HDAC6i/s may impact immune response.
The ability to calculate the HDAC6 score for some of the patients recruited to our clinical trial study provides the first evidence that the HDAC6 score has predictive potential in the clinical setting. Because our study is a phase Ib the number of patients is still limited. Additionally, our trial cannot meaningfully distinguish which effects are simply from abraxane and which from ricolinostat. New studies with larger cohorts are needed to further confirm these results and help to precisely define a threshold of HDAC6 score for patient stratification.
METHODS
All research complies with all relevant ethical regulations.
Cell culture
All breast cancer cell lines were obtained from the American Type Culture Collection (ATCC).
Flow cytometry and cell viability
Cells were analyzed for phosphatidylserine exposure by annexin-V FITC / propidium iodide double staining using BD FITC Annexin V Apoptosis Detection Kit (Cat# 556547) according to the manufacturer’s instructions.
Cell viability 5,000 cells/well were seeded in a 96 well white with clear bottom plates and treated with respective drugs for 72hrs and quantified for viability using CellTiter-Glo® Luminescent Cell Viability Assay (Cat# G7571). After treatment with the drug was complete, media containing the drug was removed and washed once with 1x PBS and replaced with an equal volume of media and Cell Titer-Glo reagent and incubated for 5minutes on a platform rocker. Plates were then read using a SpectraMax M5 microplate reader at the endpoint and luminescent setting.
Drug synergy Using the same conditions and reagents as stated above, cells were treated with ricolinostat (0-20uM), paclitaxel (0-437.5nM), and doxorubicin (0-200nM) for 72 hrs to determine the dose curves. After validating 2.5uM of ricolinostat as the lowest concentration of the drug that induces robust accumulation of acetylated α-tubulin, the cells were treated with a combination of ricolinostat at 2.5uM and the same range of concentrations of paclitaxel and doxorubicin as indicated above for 72hrs and quantified for viability. The synergistic activity between ricolinostat and paclitaxel or doxorubicin was evaluated by two approaches: isobole curves and combination index using the Chou-Talalay equation.
Cell viability by MTS Cells were plated in 96-well plates with 150 μl culture media at an optimized cell density. Twenty-four hours later, testing compounds were added, and the time zero plates were measured by MTS assay as G0 reference. Cell proliferation was measured by MTS assay after compound treatment for 3 days. Compounds dilution: 20mM stock solution of ACY-1215 in DMSO. On the day of treatment, compounds were freshly diluted from the stock solution to a working solution with 8 data points with final concentrations ranging from 0-30uM in culture medium.
Crystal violet staining 50,000cells/well were seeded in a 24 well plate and treated with respective drugs for 72hrs. All wells were then washed once with 1xPBS and stained with 0.5% crystal violet solution for 20min while placed on a rocker. Plates were then washed three times with distilled water and dried overnight and resuspended with methanol and quantified with a plate reader at 540nm wavelength.
RNAi All siRNAs were purchased from Dharmacon as a pool of 4 siRNAs targeting HDAC6, MYC, and Non-targeting control (NTC). Cultured cells were plated at 70% confluence 24hrs prior to transfection. The following day, the cells were supplied with fresh media and transfected with siRNA using the Lipofectamine RNAiMAX transfection reagent (Cat # 13778075) according to the manufacturer’s protocol. Knock-down efficiency was evaluated 72hrs post-transfection using western blot.
CRISPR All sgRNAs were cloned into the lentiCRISPR v2 vector backbone (Addgene plasmid #52961) and were prepared using Qiagen Maxiprep Kit (Cat# 12162). 3ug of plasmids were then transfected into the haploid cell line (HAP1) using JetPei DNA transfection reagent (Polyplus Cat #101-10N) overnight. SgRNA sequence H651A (5’ GCTGAGTTCCATTACCGTGGTGG). For the generation of HDAC6 point mutants, the sgRNA was cotransfected with 100nM of an ssDNA oligo to promote homologous recombination (CGCCATGCCCAGACTATCAGTGGGCATGCCCTACGGATCCTGATTGTGGATTGGGATGTCCACCACGGTAATGGAACTCAGCACATGTTTGAGGATGACCCCAGTGTGCTATATGTGTCCCTGCACCGCTATGATCATGG). Individual cells were then plated into 96 well plates to grow single clones. After expansion, genomic DNA was extracted using Qiagen kit (Cat # 69504) and sequenced for verification of the presence of point mutations.
Clinical Trial (NCT02632071)
This study was approved by the Columbia University Institutional Review Board (IRB-Q3709). This is an open-label phase Ib trial using ricolinostat in combination with nab-paclitaxel for patients with metastatic breast cancer (MBC) (Fig. 3a) that have progressed on multiple previous lines of therapy (Supplementary Table 3). The primary objective of this study was to establish the safety, tolerability, and identify the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) of ricolinostat when combined with nab-paclitaxel. Additionally, the secondary objectives were to assess progression-free survival (PFS), overall response rate (ORR), clinical benefit rate (CBR) and to evaluate the HDAC6 score as a predictive biomarker by investigating the correlation between the HDAC6 score and clinical endpoints.
Definition of DLT: a) Any grade 4 hematologic toxicity, b) Grade 3 thrombocytopenia associated with clinically significant bleeding; c)≥ Grade 3 non-hematologic toxicity and not related to underlying malignancy except the following: 1. nausea, vomiting, or diarrhea lasting < 72 hr and controlled by optimal antiemetic/antidiarrheal therapy; 2. Clinical laboratory abnormalities that are reversible to ≤ Grade 1 or baseline status within 72 hr with outpatient care and/or monitoring, or that are considered not clinically significant by the treating investigator; 3. Grade 3 amylase and/or lipase elevation that is not associated with either clinical or radiographic evidence suggestive of pancreatitis.
Definition of patient evaluability for toxicity: Patients were evaluable for toxicity from the time of their first treatment with ACY-1215 and nab-paclitaxel. To qualify as DLT evaluable, a minimum of 80% dose should be taken.
-
Definition of patient evaluability for the efficacy evaluation: Evaluable for objective response: Only those patients who have measurable disease present at baseline, have received at least one cycle of therapy, and have had their disease re-evaluated will be considered evaluable for response.
Evaluable Non-Target Disease Response: Patients who have lesions present at baseline that are evaluable but do not meet the definitions of measurable disease, have received at least one cycle of therapy, and have had their disease re-evaluated will be considered evaluable for non-target disease.
TITE-CRM description: The TITE-CRM used an empirical dose-toxicity model with a prior distribution of the model parameter assumed to be normal with a mean of 0 and a variance of 1.34. The dose-toxicity model was calibrated such that the method will eventually select a dose that yields between 17% and 33% DLT 63 and had good operating characteristics across a wide range of potential scenarios of toxicity profiles. The proposed toxicity evaluation period for this study was set at 8 weeks with a minimum of 1 week of observation between two consecutive patients, which resulted in a maximum of four patients in the DLT window at any given time. A linear function was used to assign weights to patients according to their follow-up time and toxicity status. In this trial, we assumed that the weight given to a patient’s outcome increases at a constant rate while being monitored for DLT, with full weight being achieved at the end of the observation period. If a death unrelated to the study drug occurred within the observation window, the model would into consideration the time to death for that patient, and death (event) will be censored. The design did not allow for dose skipping during dose escalation or escalation immediately after a dose-limiting toxicity.
-
Trial information: Seventeen patients were accrued between March 2016 and February 2018. Of these, 16 patients had an evaluable disease, as one patient dropped out at cycle 2 due to no longer wishing to participate in the trial and in the absence of any related toxicity. In the 16 evaluable patients, the median age was 57.5 years (range: 41-78), 14 were female (87.5%), 3 had triple-negative MBC, and 13 were HR+/HER2− MBC. The median number of prior lines was 3 (range: 0-10). Detailed information of each patient is given in extended data tables and source data.
Ages Eligible for Study: 18 Years and older.
Sexes Eligible for Study: All
Accepts Healthy Volunteers: No
-
Inclusion Criteria:
Patients were recruited by personal interview with the oncologist. Subjects have histologically confirmed adenocarcinoma of the breast -- all breast cancer subtypes are allowed. Unresectable or metastatic breast cancer. Locally recurrent disease must not be amenable to any local treatment with curative intent. Metastatic disease must be demonstrated either radiographically or histologically. Patients may have measurable disease only, non-measurable disease only, or both (RECIST 1.1). ECOG performance status of 0-1. Must have recovered from the acute toxic effects of all prior therapy prior to registration for this study to grade 1 or less.
Women and men of all races and ethnic groups are eligible for this trial. Minimum number of prior treatments required given standard nab-paclitaxel dosing:
If HER2 negative: none
If HER2 positive: two prior regimens containing HER2 targeted therapies in the inoperable locally advanced and/or metastatic setting. Prior therapy for inoperable locally advanced/metastatic disease should include trastuzumab plus pertuzumab as well as ado-trastuzumab. Pertuzumab and ado-trastuzumab must have been previously used, unless for reasons that include, but are not limited, to the following: intolerance to pertuzumab and/or ado-trastuzumab, medical contraindication, regimen declined by patient, treating investigator discretion, or medical insurance coverage issues which prevented administration of pertuzumab or ado-trastuzumab. These reasons must be reviewed with the study chairs and documented in the medical record and care report form. Patients who relapse within 12 months of completing neoadjuvant/adjuvant pertuzumab or ado-trastuzumab would be considered as having progressed on that regimen.
There is no maximum number of prior treatments allowed in the metastatic setting.
Patients must have normal organ and marrow function as defined below:
leukocytes ≥3,000/mcL
absolute neutrophil count ≥1,500/mcL
platelets ≥100,000/mcL
hemoglobin ≥9 g/dL
total bilirubin ≤ 1.5 × the upper limit of normal
AST(SGOT)/ALT(SGPT) ≤2.5 × institutional upper limit of normal
Serum creatinine ≤ 1.5 × the upper limit of normal or calculated creatinine clearance ≥ 60 mL/min
-
Subject is capable of understanding the informed consent process.
The effects of ACY-1215 on the developing human fetus are unknown. For this reason and because the effects of chemotherapy are known to be teratogenic, women of child-bearing potential and men must agree to use adequate contraception (hormonal or barrier method of birth control; abstinence) prior to study entry and for the duration of study participation. Should a woman become pregnant or suspect she is pregnant while she or her partner is participating in this study, she should inform her treating physician immediately. Men treated or enrolled on this protocol must also agree to use adequate contraception prior to the study, for the duration of study participation, and 2 weeks after completion of ACY-1215 administration.
-
Exclusion Criteria:
Patients who have had chemotherapy, hormonal therapy, or radiotherapy within 2 weeks prior to entering the study or those who have not recovered from adverse events due to agents administered more than 2 weeks earlier. Concomitant treatment with bone-targeted therapies such as RANKL inhibitors or bisphosphonates is allowed.
Patients who are receiving any other investigational agents concurrently or have received investigational agents within 2 weeks or 5 half-lives of the compound or active metabolites, whichever is longer before the first dose of the study treatment.
Patients who have received HDAC inhibitors (including valproic acid, entinostat, vorinostat) are excluded
Subject is pregnant or nursing. Pregnant women are excluded from this study because ACY-1215 is an investigational therapy with unknown potential for teratogenic or abortifacient effects. Because there is an unknown but potential risk for adverse events in nursing infants secondary to treatment of the mother with ACY-1215, breastfeeding should be discontinued if the mother is treated with ACY-1215.
Symptomatic or unstable brain metastases. (Note: Asymptomatic patients with metastatic brain disease who have been on a stable dose of corticosteroids for treatment of brain metastases for at least 14 days prior to registration are eligible to participate in the study).
HIV+ with a CD4 count <200 are ineligible because these patients are at increased risk of lethal infections when treated with marrow-suppressive therapy. Appropriate studies will be undertaken in patients receiving combination antiretroviral therapy when indicated.
Patients receiving any medications or substances that are strong inhibitors of CYP450 3A4 isoenzyme.
History of allergic reactions attributed to compounds of similar chemical or biologic composition to nab-paclitaxel.
Uncontrolled intercurrent illness including but not limited to, ongoing or active infection, symptomatic congestive heart failure, unstable angina pectoris, cardiac arrhythmia, or psychiatric illness/social situations that would limit compliance with study requirements.
Corrected QT interval (QTc) value > 480 msec at screening; family or personal history of long QTc syndrome or ventricular arrhythmias including ventricular bigeminy at screening; previous history of drug-induced QTc prolongation or the need for treatment with medications known or suspected of producing prolonged QTc intervals on electrocardiogram (EKG). If QTc prolongation on screening ECG is felt to be related to electrolyte imbalance, an EKG can be repeated after correction of electrolytes.
-
Primary Outcome:
Maximum tolerated dose (MTD) of ACY-1215 (Ricolinostat) [ Time Frame: 28 days ]
The maximum tolerated dose (MTD) combination is defined as the dose combination associated with a target probability of dose limiting toxicity (DLT) of 0.25. A dose-limiting toxicity is defined as the MTD with DLTs defined as any grade 3 non-hematologic toxicities despite maximal supportive care or any grade 4 hematologic toxicity. The MTD will be estimated using the time to event continual reassessment method (TITE-CRM). The TITE-CRM will use an empirical dose-toxicity model, with a sample size of 24. The dose-toxicity model is calibrated such that the method will eventually select a dose that yields between 17% and 33% DLT, which will be the recommended phase II dose (RP2D).
-
Secondary Outcome Measures :
Number of adverse events related to ACY-1215 (Ricolinostat) [ Time Frame: up to 14 days following the last administration of study treatment ]
All patients will be evaluable for toxicity from the time of their first treatment with the study drug. Toxicities will be graded based upon CTCAE v4.0.2.
Animal studies
All mouse experiments were conducted using protocols (IACUC-2014-0104) approved by the Institutional Animal Care and Use Committee (IUCAC) at the Icahn School of Medicine at Mount Sinai. The maximum tumor size allowed by the ethic committee was 1.5 cc. This limit was not exceeded in our studies.
Transgenic and xenograft mouse models FVB/N-Tg(MMTVneu)202Mul/J transgenic mice were purchased from Jackson laboratory and bred. Six to eight-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ female mice were also obtained from Jackson laboratory. Mice were subcutaneously injected under each flank at 3 month old with 10x10^6 MDA-MB 453 and MDA-MB-436 cells resuspended in an equal volume of respective media and matrigel matrix (Corning Ref #354234). Treatment began when tumors reached a volume of 100-150mm3 for both transgenic and xenograft mice. A minimum of 6 mice per cohort was used in the study.
Drug administration Ricolinostat, obtained from Acetylon Pharmaceuticals, and was administered for 6 days a week at 50mg/kg. Paclitaxel was obtained from the Mount Sinai Hospital and administered at 6mg/kg twice a week. Both treatments were given intraperitoneally at a volume of 200ul. Tumors were measured twice a week for the duration of the experiment using the ellipsoid volume formula 1/2 × L × W × H. At the experimental endpoint, tumors were collected, formalin-fixed, and flash frozen. Statistical differences were evaluated with one-tailed t-test (n >= 6 per cohort).
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed paraffin-embedded (FFPE) tumor tissue sections by the Neuropathology Brain Bank Core at Mount Sinai. All Slides were sectioned, mounted, and stained for hematoxylin and eosin, Ki-67(Abcam #ab15580 1:200) and c-caspase 3(Cell signaling #9664s at 1:50). All slides were processed using Ventana Benchmark XT machine by the core facility.
Statistical analysis of experimental data
Data on experimental graphs related to cell lines and animal studies are represented as mean ± standard deviation unless specified otherwise. Results were analyzed by Student’s t-test and a p-value below 0.05 was considered statistically significant.
Statistic & Reproducibility
All studies in the report were performed a minimum of 3 independent times if not otherwise described in the legends. No sample was excluded except when otherwise described in the text (e.g. tumor samples with low tumor content). No statistical method was used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Transcriptomics data collection and processing
Cancer cell lines. The RNA-seq transcripts per million (TPM) data of 1,165 cell lines representing 29 cancer types from the Cancer Cell Line Encyclopedia (CCLE) project, together with cell line annotations and gene dependency scores were downloaded from the portal of the Dependency Map (DepMap) project (https://depmap.org/portal, version: Public 19Q1).
Transgenic mouse models of breast cancer. The gene expression profiles of 385 samples from 27 genetically engineered mouse models (GEMMs) of breast cancer and 2 normal mammary tissues were collected in a study by Dr. Charles Perou and downloaded from the Gene Expression Omnibus (GEO) (GSE42640, Agilent Technology gene expression microarray platforms, 22K, 4x44K or 4x180K
The Cancer Genome Atlas (TCGA). The TCGA RNA-seq data at both isoform and gene levels for 32 human primary cancer types including breast cancer were extracted from the QIAGEN OncoLand.
The Molecular Taxonomy of Breast Cancer International Consortium (METABRIC). We downloaded the normalized METABRIC gene expression profiles (Illumina HT 12 arrays, N=1,981) from Synapse (https://www.synapse.org/#!Synapse:syn1757063).
Inflammatory breast cancer (IBC). The pre-processed gene expression profiles of 63 IBC samples and 134 non-IBC patient samples were collected by an IBC study and downloaded from the GEO (GSE23720, Affymetrix Human Genome U133 Plus 2.0). We normalized the data by quantile method and then removed probe sets with no EntrezGene IDs, resulting in 51,997 probe sets representing 20,517 genes. The gene-level expression values were estimated by taking average the expression of all probe sets for the same gene. The data QC was assessed by the “draw.eset.QC” function in the NetBID software. Two outlier samples (T60 and T61) were identified and removed from subsequent analysis.
Breast cancer patient samples treated by Paclitaxel only. The pre-processed gene expression profiles of 106 breast cancer patient samples treated by Paclitaxel only were collected from the GEO (GSE25066, Affymetrix Human Genome U133A Array). We removed probe sets with no gene symbols and estimated the gene-level expression values by taking average of the expression of all probe sets for the same gene, resulting in 14,208 genes for subsequent analysis.
RNA-seq analysis of the clinical trial
FFPE sample collection. Out of 16 evaluable patients in our clinical trial we were able to obtain 11 FFPE biopsy samples before entering the clinical trial.
Data generation. RNA samples were isolated using the RNeasy Mini Kit (QIAGEN) and subjected to RNA-seq sequencing. The sequencing was performed by Illumina HiSeq 2500 with ~25 million SE-100 reads at the Genomics Core Facility at Mount Sinai.
Data QC and processing. Raw fastq data was assessed by FastQC (v-0.11.5) 64. Salmon (v-0.9.1) 65 was used to quantify the expression of transcripts and genes based on the reference genome hg38 (GRCh38) with gene annotation from GENCODE (release 28). Data has been deposited in the GEO portal (GSE148623).
Batch effect removal. The RNA-seq experiments were performed in three batches based on sample received time. We then used the “removeBatchEffect” function in the limma R package (v-3.42.2) to remove the batch effects.
RNA-seq analysis of two sensitive and two resistant cell lines
RNA samples of two sensitive cell lines (MDA-MB-453 and SK-BR-3) and two resistant cell lines (MDA-MB-436 and MDA-MB-468) were isolated using the RNeasy Mini Kit (QIAGEN) and subjected to RNA-seq sequencing, which was performed by Illumina NovaSeq 6000 with ~100 million PE-150 reads in the CMPB Genomics Laboratory at St Jude Children’s Research Hospital. Raw fastq data was assessed by FastQC (v-0.11.5) (Andrews, 2010) and trimmed by cutadapt (v-2.10) under default parameters. Salmon (v-0.9.1) 65 was used to quantify the expression of transcripts and genes based on the reference genome hg38 (GRCh38) with gene annotation from GENCODE (release 28). Data has been deposited in the GEO portal (GSE148623).
Microarray analysis of breast cancer cell lines and mouse models
RNA samples of three sensitive cell lines (MDA-MB-453, SK-BR-3, and SUM-149) and a mouse model (MMTV-Neu) were extracted using the RNAeasy Kit (Qiagen). The amount and quality of RNA were assessed with DU-640 UV Spectrophotometer. The samples with the optical density 260/280 ratio greater than 1.8 and the total RNA yield greater than 1 μg were considered qualified and further profiled on the Agilent sure printG3 Human gene expression V3 and Mouse gene expression V2 chip following the standard protocol. Agilent Feature Extraction software was used to generate probe-level intensities and quality measures including median intensity, percent of probe set outliers, and percent of single probe outliers for each chip. The data normalization was performed by limma R package (v-3.42.2) with “loess” method within array and “quantile” between arrays. The probe sets mapping to protein-coding genes were selected for subsequent analysis, resulting in 27726 probes/18498 genes for cell lines and 38143 probes/20051 genes for the mouse model. The data QC was assessed by the “draw.eset.QC” function in the NetBID software.
Integration of human breast cancer transcriptomics data from different sources
Gene expression profiles from four datasets of human breast cancer, including TCGA, METABRIC, IBC, and RNA-seq data from our clinical trial, were merged by using the genes shared across all four datasets. Batch effects were detected by NetBID QC and removed by limma as described in the batch removal of clinical trial RNA-seq data.
Protein Extraction and Protein Quantification
A total of 12 million cells from each sample were lysed in 8M urea as previously described66. Briefly, the frozen cell pellets were homogenized in the lysis buffer (50 mM HEPES, pH 8.5, 8 M urea, and 0.5% sodium deoxycholate) with 1x PhosSTOP phosphatase inhibitor cocktail (Sigma-Aldrich). Protein concentration was measured by the BCA assay (Thermo Fisher) and then confirmed by Coomassie-stained short SDS gels.
Protein Digestion and Tandem-Mass-Tag (TMT) Labeling
The analysis was performed with a previously optimized protocol 67. For acetylome profiling, quantified protein samples (~1 mg in the lysis buffer with 8 M urea) for each TMT channel were proteolyzed with Lys-C (Wako, 1:100 w/w) at 21 °C for 2 h, diluted with 50 mM HEPES (pH 8.5) by 4-fold to reduce urea to 2 M before the addition of trypsin (Promega, 1:50 w/w) to continue the digestion at room temperature overnight. The digestion was terminated by the addition of 1% trifluoroacetic acid. After centrifugation, the supernatant was desalted with the Sep-Pak C18 cartridge (Waters), and then dried by Speedvac. Each sample was resuspended in 50 mM HEPES (pH 8.5) for TMT labeling, and then mixed equally, followed by desalting for the subsequent fractionation. Samples were labeled with 11-plex TMTs from Thermo Fisher (Waltham, MA, USA) according to the manufacturer’s recommendations.
Two-Dimensional Liquid Chromatography-Tandem Mass Spectrometry (LC/LC-MS/MS) and Acetylated Peptide Enrichment
The TMT labeled samples were fractionated by offline basic pH reverse phase LC during a 50 min gradient on an XBridge C18 column (3.5 μm particle size, 4.6 mm x 25 cm, Waters; buffer A: 10 mM ammonium formate, pH 8.0; buffer B: 95% acetonitrile, 10 mM ammonium formate, pH 8.0)67. Eighty fractions were collected and concatenated into 10 fractions. Each of the 10 fractions was subjected to enrichment by PTMScan Acetyl-Lysine Motif (Cell Signaling Technology) antibody. Briefly, tryptic peptides dissolved in 1X IPA buffer (50mM MOPS/NaOH pH7.2; 10mM Na2HPO4) were incubated with pre-washed antibody beads at 4°C for 2 h with gentle shaking. The beads were then washed two times with cold 1X PBS. The bound peptides were eluted with 0.15% trifluoroacetic acid (TFA) and desalted on C18 StageTip and died. Each concatenated fraction was depleted 3 times with antibody. The resulting peptides were resuspended in 2uL of 5% formic acid (FA). In the acidic pH LC-MS/MS analysis, each fraction was run sequentially on a column (75 μm x 25 cm for the acetylome proteome, 1.9 μm C18 resin from Dr. Maisch GmbH, 65 °C to reduce backpressure) interfaced with an Q Exactive HF Orbitrap or Fusion MS (Thermo Fisher). Peptides were eluted by an 80 min gradient (buffer A: 0.2% formic acid, 5% DMSO; buffer B: buffer A plus 65% acetonitrile). MS settings included the MS1 scan (410-1600 m/z, 60,000 resolution, 1 x 106 AGC and 50 ms maximal ion time) and 20 data-dependent MS2 scans (fixed first mass of 120 m/z, 60,000 resolution, 1 x 105 AGC, 100-150 ms maximal ion time, HCD, 35-38% normalized collision energy, ~1.5 m/z isolation window with 0.3 m/z offset, and ~15 s dynamic exclusion).
Data Analysis (Proteomics)
Raw mass spectra were pre-processed by the JUMP program68 and searched using SEQUEST engine69 against UniProt human database and concatenated with a reversed protein sequence decoy database. Searches were performed with a mass tolerance of 25 ppm for precursor ions and 15 ppm mass tolerance for fragment ions, fully tryptic restriction with two maximum missed cleavages, three maximum modification sites, and assignment of the a, b, and y ions. TMT tags on lysine residues and N-termini (+229.162932 Da) and the carbamidomethylation of cysteine residues (+57.021 Da) were used for determining static modifications, and methionine oxidation (+15.99492 Da) and lysine acetylation (+42.01056) was considered a dynamic modification. Mass spectra were filtered by mass accuracy and matching scores to reduce the false discovery rate (FDR) to approximately 1%. The lysine-acetylated peptides were quantified by summarizing reporter ion counts across all matched peptide spectrums with the JUMP software suite70, followed by differential expression analysis using the limma R package (v-3.42.2) [https://link.springer.com/chapter/10.1007/0-387-29362-0_23].
Next-generation HDAC6 breast cancer regulon inference
To reconstruct HDAC6 regulon with high accuracy and applicability across all breast cancer subtypes, gene expression profiles was extracted from each of the 9 breast cancer subtypes, including 3 (HR+/HER2−, HER2+ (HR+ and HR-), triple-negative breast cancer or TNBC) defined by IHC assays of HR and HER2, and 6 (Luminal A, Luminal B, HER2overexpressed, Claudinlow, Basal-like and Normal-like) defined by PAM50, from over 3,000 primary breast cancer samples from TCGA and METABRIC. We then used SJARACNe, a scalable software tool for gene network reverse-engineering from big data, to reconstruct signaling networks for each of the 9 breast cancer subtypes.
HDAC6 cancer type–specific regulon inference
TCGA dataset included RNA-seq profiles representing 32 human primary cancer types. In addition to the updated HDAC6 regulon for breast cancer as described above, we used the TCGA RNA-seq data as detailed in the previous section and reconstructed HDAC6 regulons for each of the other 31 cancer types by using the SJARACNe algorithm. The parameters were the same as those used to generate HDAC6 breast cancer regulons.
HDAC6 score inference by NetBID
Given a gene expression profile in a cohort of study, we calculated the raw HDAC6 score that summarized the activity of the HDAC6 regulon by using the “cal.Activity” function with the method of “mean” in NetBID software. For the HDAC6 score of human breast cancer samples, we used the updated HDAC6 regulon for breast cancer. For HDAC6 score across the 31 TCGA cancer types, we used the cancer type matched HDAC6 regulon. To calculate the HDAC6 score in GEMMs of mouse breast cancer, we used the human gene ids transferred based on the human/mouse homology map that was done in the breast cancer GEMM study. For graphical representation, all HDAC6 scores in a study were normalized considering all the samples in that specific series of samples.
HDAC6 score inference by VIPER (Virtual Inference of Protein-activity by Enriched Regulon analysis)
HDAC6 relative protein activity based on our next-generation HDAC6 breast cancer regulon was inferred by the VIPER algorithm implemented in the DarwinOncoTarget test, which has been approved by the NYS Department of Health CLIA/CLEP Validation Unit as an offering in the category of “Molecular and Cellular Tumor Markers for Oncology” (Neal, Michael, Assay Validation Review, Wadsworth Center, NY State Department of Health, PFI: 7313, Project ID: 63859, March 8, 2019).
Overlaps of cancer type-specific HDAC6 regulons
The HDAC6 regulons of 32 TCGA cancer types inferred by SJARACNe, as described above, were summarized in Supplementary Table 10. The regulon overlapping statistics, including the size of overlapped regulon genes and the p-value of Fisher’s Exact test was summarized in Supplementary Table 11. The scatter plot was made by R package ggplot2 (v-3.3.0).
Progression-free survival (PFS) analysis against HDAC6 score in the clinical trial
The PFS in month was calculated from the dates of the trial assignment and disease progression or last follow-up by the “difftime” function in lubridate (v-1.7.8). The PFS analysis of the clinical trial against HDAC6 score, including the COX model fitting and Kaplan–Meier plot, was performed by using the R package survminer (v-0.4.6) 717374. The HDAC6 score high and low patients were defined at the cutoff of the mean of HDAC6 scores.
Receiver operating characteristic (ROC) curve analysis of HDAC6 score in the clinical trial
The ROC curve analysis to evaluate the performance of HDAC6 score in predicting clinical response of patients, including statistics and plot, was performed by using the R package pROC (v-1.6.2) 72. We used −0.36 as the cutoff of HDAC6 score from the ROC analysis to define HDAC6 high and low patients.
Data Analysis (Statistics)
Multiple testing correction was not applied otherwise unless specified in the figure legends.
Extended Data
Extended Data Fig. 1. HDAC6 score in BC.
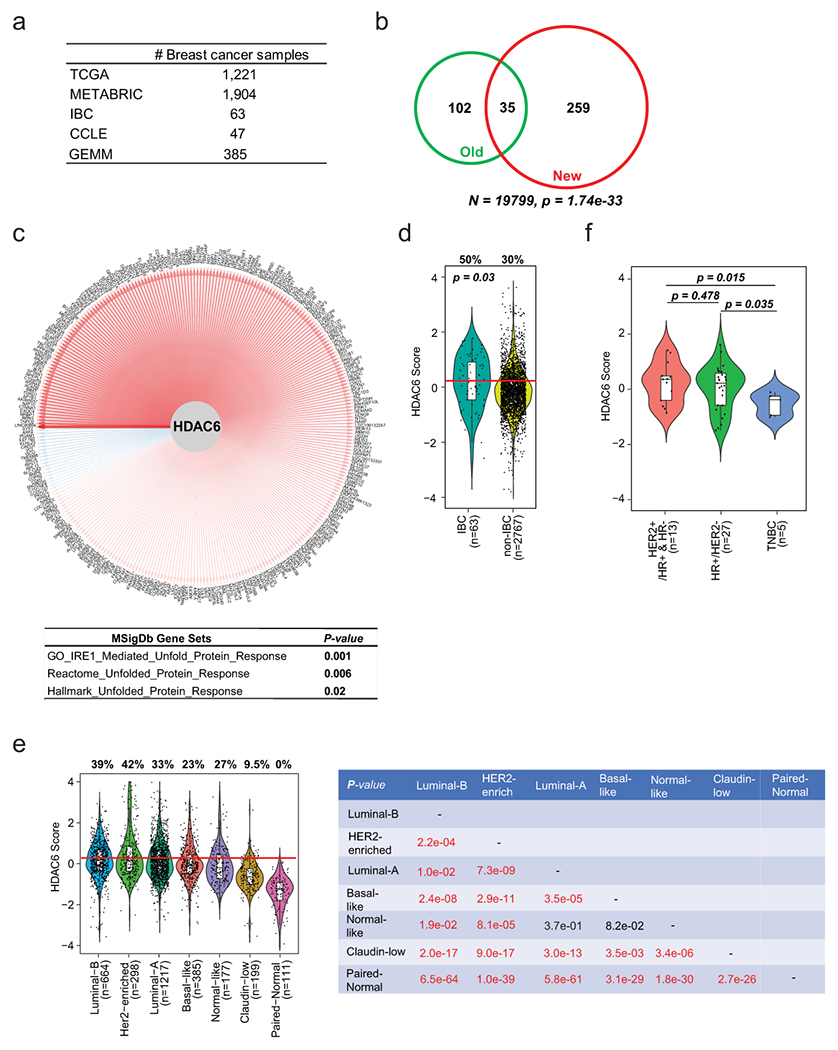
(a) The number of samples and data set of origin used to evaluate the HDAC6 regulon. (b) Overlap of the original and updated HDAC6 regulons. P value was estimated using two-tailed Fisher’s exact test. N was determined by the total number of genes for network inference. (c) The network plot of HDAC6 and updated HDAC6 regulon. Edge width is corresponding to the correlation strength measured by mutual information. Red and blue edges indicate positive and negative correlations between HDAC6 and each of the regulon genes. The table below shows the pathway enrichment of the genes in the regulon, showing its association with unfolded protein response. P value was estimated using two-tailed Fisher’s exact test. (d) New HDAC6 score comparing IBCs vs non-IBCs. (e) HDAC6 scores of all BCs from TCGA and METABRIC are divided into molecular subtypes. (f) HDAC6 scores in 45 ductal metastatic breast cancer samples from the MBC Project divided into histological molecular subtypes. In d, e and f, the center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5x interquartile range; the red line represents the median of the HDAC6 scores in IBC samples and the numbers over each whisker plot indicate the percentage of samples over this value in each clinical subtype. Sample size (n=number of samples) of each group was indicated in the axis labels. P value was estimated using two-tailed t test.
Extended Data Fig. 2. Anticancer activity of HDAC6 inhibitors.
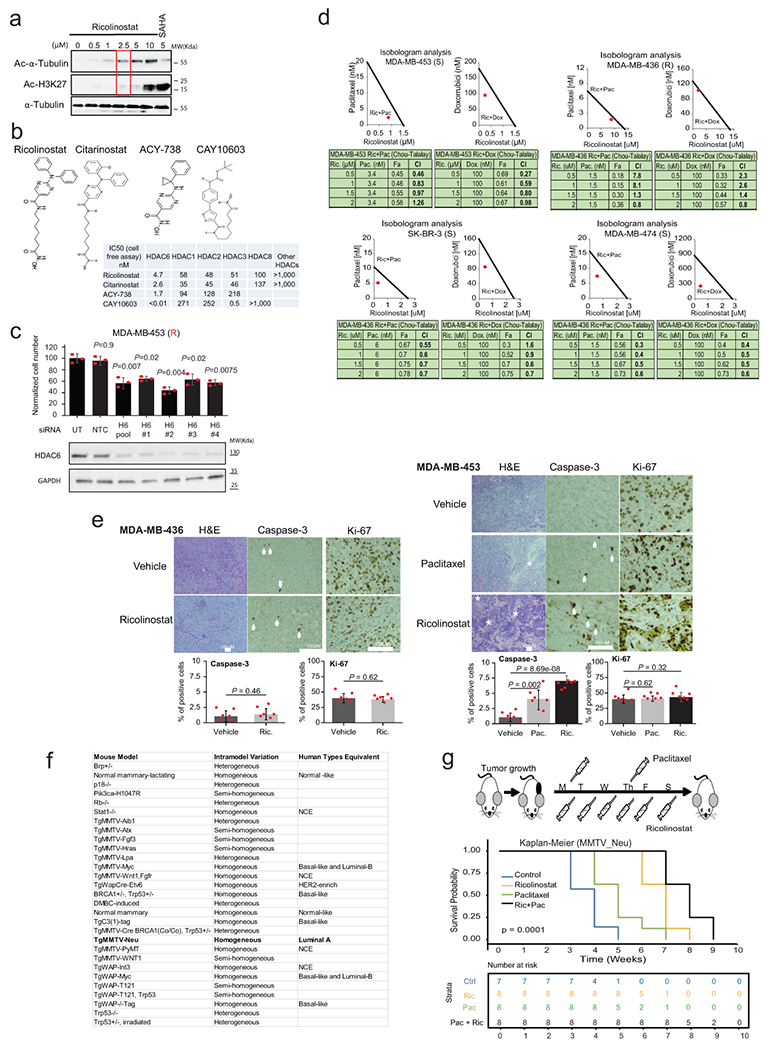
(a) The western blot shows the titration of ricolinostat to identify an effective dose (accumulation of Ac-α-Tubulin) without off-target effects in class-I HDACs (accumulation of Ac-H3K27). SAHA is used as a control Pan-HDAC inhibitor. WT-blot results were reproduced n=3 times from independent experiments. (b) Chemical structure of the different HDAC6 inhibitors used in Fig. 1E. (c) Normalized cell number 6 days after transfection with individualized siRNAs targeting HDAC6 or non-targeting control (NTC) (n=3 independent independent experiments per siRNA). The WT-blots show the silencing efficiency. WT-blot results were reproduced n=3 times from independent experiments. All error bars represent Mean±SD. P value was estimated by two-tailed t test. (d) The graphic shows the lack of synergistic activity between ricolinostat and commonly used chemotherapy (paclitaxel and doxorubicin) in MDA-MB-436 cells. In contrast, cells sensitive to ricolinostat MDA-MB-453, SK-BR-3 and MDA-MB-474 show synergistic activity between ricolinostat and chemotherapy. R and S indicate ricolinostat resistance and sensitivity respectively. N=3 independent replicate experiments per drug combination and concentration. (e) Histological intratumoral evaluation of H&E, Caspase-3, and Ki-67 in tumor samples from Fig. 1B. Quantification is also shown in bar graphs. Notice that the combo treatment (Pac+Ric) is not shown because all tumors regressed with this treatment. The white asterisks indicate necrotic areas and the white arrows indicate Caspase-3 positive stained cells. All error bars represent Mean±SD. P value was estimated by two-tailed t test. N= 6 samples from individual tumors. (f) The list shows all the transgenic mouse models evaluated by the HDAC6 score in Fig. 2c and d and indicates their correlation with human BCs. (g) Kaplan – Meier graphic showing the survival of the MMTV tumors in Fig. 2e. Control n=7; ricolinostat (Ric) n=8; paclitaxel (Pac) n=8; Ric+Pac n=8. P value was estimated using two tailed Log-Rank test.
Extended Data Fig. 3.
Characteristics of the evaluable patients enrolled in the clinical trial
Extended Data Fig. 4. Biomarker evaluation of HDAC6 score calculated by using the NetBID and VIPER algorithms in the phase Ib trial.
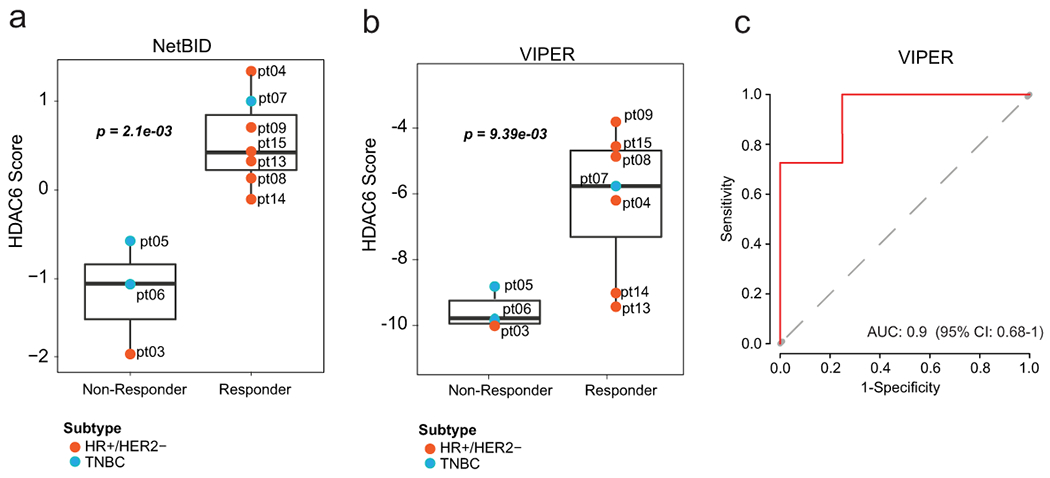
The Supplementary Extended Data Fig. shows the similarities between the HDAC6 scores inferred by NetBID (a) and VIPER (b) in responders and non-responders, and (c) ROC curve plot of HDAC6 score inferred by VIPER (similar plot for NetBID is in Fig. 3d). In a and b, one dot represents one patient sample and the center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5x interquartile range. P value was estimated by two-tailed t test. For all panels n=3 non-responders and n=7 responders.
Extended Data Fig. 5. The HDAC6 score does not correlate with the response in paclitaxel-only treated patients.
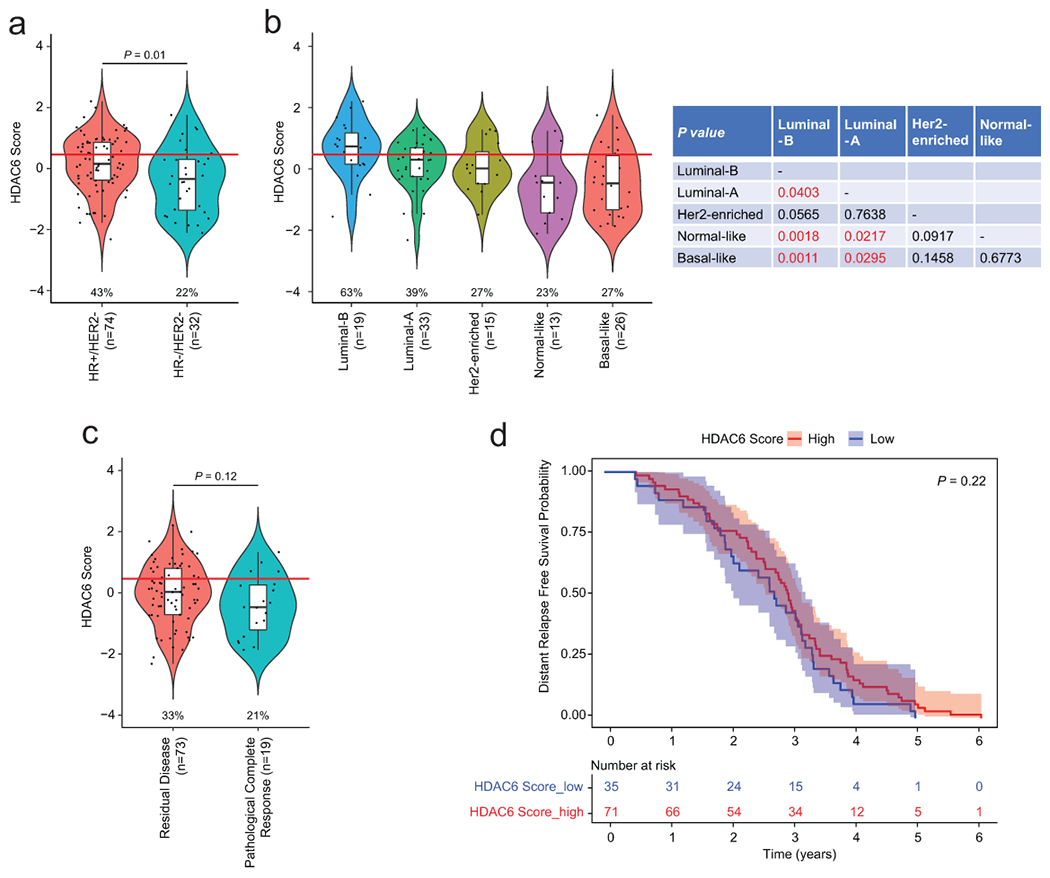
The Supplementary Extended Data Fig. shows the HDAC6 scores of all BCs from Hatzis et al. (JAMA, 2011) divided into clinical (a) and molecular (b) subtypes. (c) HDAC6 scores of patients divided by response to paclitaxel, Residual Disease (CD) and Pathological Complete Response (pCR), showing the lack of correlation between the HDAC6 score and the response to paclitaxel. In a, b and c the number of patient samples is indicate as (n). d, Kaplan – Meier graphic showing the survival of breast cancer patients treated exclusively with paclitaxel in the neoadjuvant setting separated by HDAC6 score (high/low=higher and lower −0.36, based on the ROC analysis in our clinical trial). N = 35 patient samples for HDAC6 score-low and N = 71 patient samples for HDAC6 score-high group. P value was estimated using two-tailed Log-Rank test. The 95% confidence interval of the regression lines were displayed. In a, b and c, the center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5x interquartile range; the red line represents the median of the HDAC6 scores in IBC samples and the numbers under each whisker plot indicate the percentage of samples over this value in each clinical subtype. Sample size of each group was indicated in the axis labels. P value was estimated using two-tailed t test.
Extended Data Fig. 6. HDAC6 scores in other human cancers.
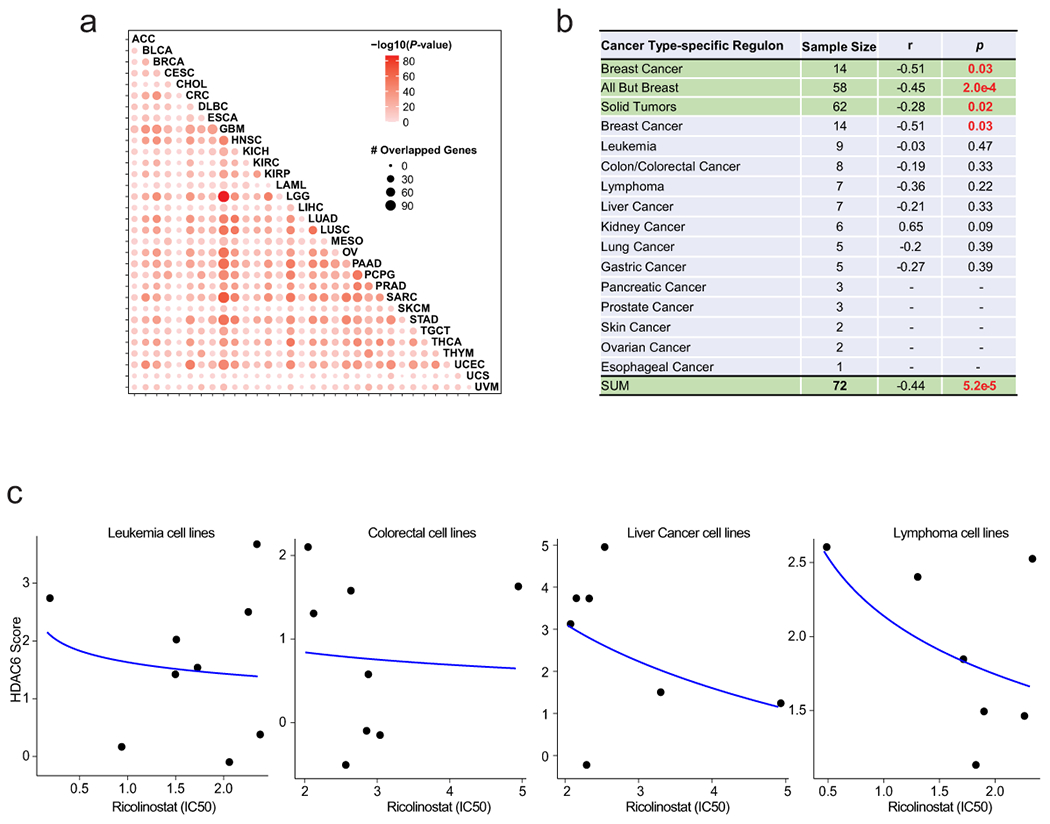
(a) Correlation between HDAC6 regulon in different tumor types. The tumors sets are the same as in figure 4c. The number of patient sample for each set is indicated there. LAML: Acute Myeloid Leukemia; LIHC: Liver Hepatocellular Carcinoma; UVM: Uveal Melanoma; KIRP: Kidney Renal Papillary Cell Carcinoma; PCPG: Pheochromocytoma and Paraganglioma; DLBC: Diffuse Large B-Cell Lymphoma; ACC: Adrenocortical Carcinoma; UCS: Uterine Carcinosarcoma; KIRC: Kidney Renal Clear Cell Carcinoma; THYM: Thymoma; CHOL: Cholangiocarcinoma; SKCM: Skin Cutaneous Melanoma; CRC: Colorectal Carcinoma; LGG: Brain Lower Grade Glioma; UCEC: Uterine Corpus Endometrial Carcinoma; KICH: Kidney Chromophobe; MESO: Mesothelioma; TGCT: Testicular Germ Cell Tumors; GBM: Glioblastoma; PRAD: Prostate Adenocarcinoma; CESC: Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma; BLCA: Bladder Urothelial Carcinoma; SARC: Sarcoma; OV: Ovarian Serous Cystadenocarcinoma; THCA: Thyroid Carcinoma; ESCA: Esophageal Carcinoma; BRCA: BC; LUAD: Lung Adenocarcinoma; HNSC: Head and Neck Squamous Cell Carcinoma; PAAD: Pancreatic Adenocarcinoma; STAD: Stomach Adenocarcinoma; LUSC: Lung Squamous Cell Carcinoma. P value was estimated by two-tailed Fisher’s exact test. (b) List with all the cell lines evaluated by dose-response to ricolinostat and HDAC6 scores. The correlation (R) and P value between the response to ricolinostat and HDAC6 scores were estimated by two-tailed Spearman correlation test. (c) Graphic showing the correlation between the HDAC6 score and the response to ricolinostat in individual cell types (only tumor types with more than 6 cell lines are shown). N=8 individual independent experiments for each ricolinostat dose. The curve was fitted by stat_smooth algorithsm using lm smoothing method and y~log2(x) formula.
Extended Data Fig. 7. The correlation of HDAC6 score with immune infiltrates.
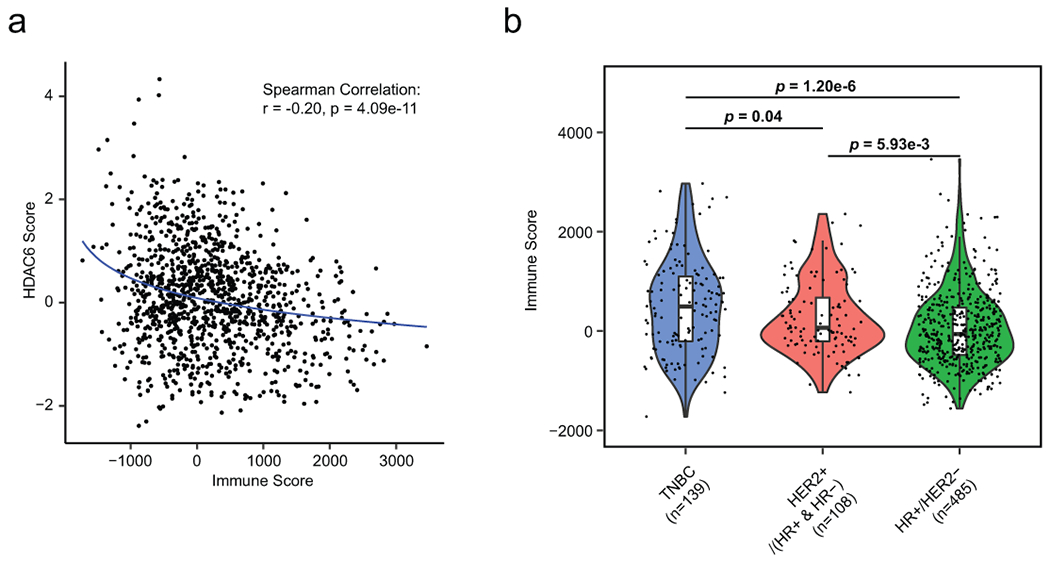
(a) Scatter plot showing the correlation between HDAC6 score and immune score by ESTIMATE in TCGA-BRCA primary patient samples (n=1109). The correlation coefficient (R) and P value were estimated using Spearman correlation test. (b), Violin plot showing the distribution of immune score across IHC-based breast cancer subtypes. Sample size of each group was indicated in the axis labels. P value was estimated using two-tailed t test. The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5x interquartile range.
Extended Data Fig. 8. Ricolinostat treatment reduces the expression of c-MYC in sensitive cell lines.
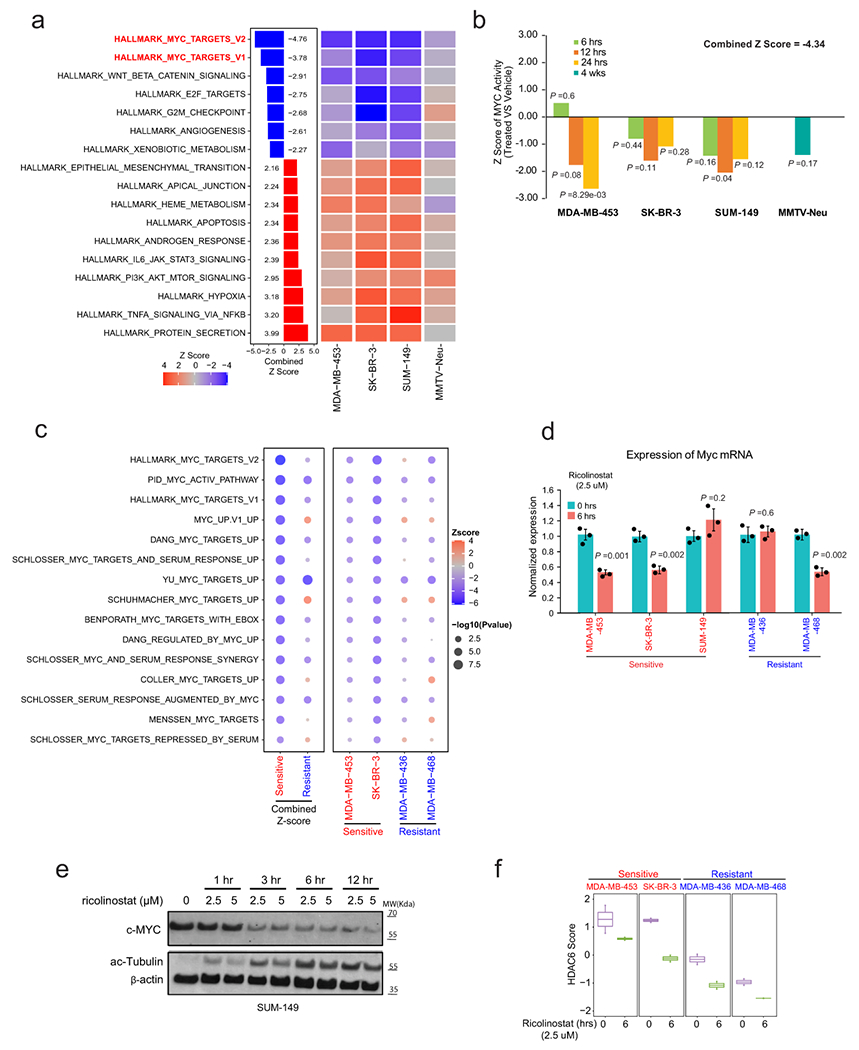
(a) Heatmap representing GSEA analysis of hallmark signatures during ricolinostat exposure in sensitive breast cancer cell lines and TgMMTV-Neu model. P value was estimated by two-tailed t test. The Z-scores were transformed from these P values and further combined using Stouffer’s method. Only significant (combined Z > 1.96 or < −1.96) sets are shown. (b) The graphic shows summarized z-scores in cell lines and TgMMTV-Neu sensitive to ricolinostat during a time curse treatment (6, 12, 24 hours and 4 weeks). For a and b N = 2 individual independent experiments for cell lines and N=3 individual tumors for TgMMTV-Neu. P value was estimated using two-tailed t test. (c) Bubble plot representing GSEA analysis of multiple MYC-associated signatures after ricolinostat in sensitive and resistant cells. N = 3 independent experiments per cell line. P value was estimated by two-tailed t test. The Z-score was transformed from the P values and further combined by Stouffer’s method. (d) QRT-PCR of c-Myc mRNA expression after 6 hours of exposure to ricolinostat. N = 3 independent experiments for each time point All effort bars represent Mean±SD. P value was estimated by two-tailed t test. (e) The WT-blot shows the changes in the protein expression of c-Myc and ac-Tubulin in SUM-149 cells after ricolinostat is added to the culture media. WT-blot results were reproduced n=3 times from independent experiments. (f) Graphic showing an efficient reduction in the HDAC6 score after treatment with ricolinostat in multiple cell lines, n≥2 independent experiments per time point. The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5x interquartile range.
Extended Data Fig. 9. Acetylation of c-Myc in Lys148 after inhibition of HDAC6.
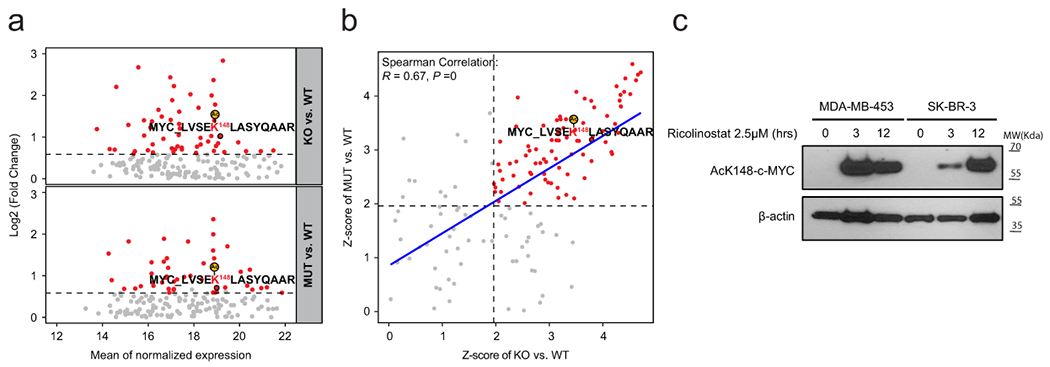
a) MA plots showing the peptides upregulated upon HDAC6 knockout (above) and HDAC6 catalytic domain 2 mutants (below). In the MA-plots, each dot represents a peptide. The significantly upregulated peptides were identified by fold change > 1.5 and p-value < 0.05 and highlighted in red. P value was estimated by two-tailed t test. (b) Scatter plot showing the correlation of the Z-score of the comparison between HDAC6 KO and wild type with that of the comparison between HDAC6 mutant and wild type. The curve was fitted by stat_smooth algorithm using lm smoothing method and y~x formula. The correlation coefficient (R) and P value were estimated using two-tailed Spearman correlation test. Each dot represents a peptide. For a and b the N = 2 independent proteomic replica studies per cell line. c) The western blot shows the accumulation of ac-K148-c-Myc after HDAC6 is inhibited by ricolinostat in MDA-MB-453 and SK-BR-3 BC lines. WT-blot results were reproduced n=3 times from independent experiments.
Extended Data Fig. 10. Acetylation of c-Myc in Lys148 after inhibition of HDAC6 in sensitive and resistant BC cancer cells.
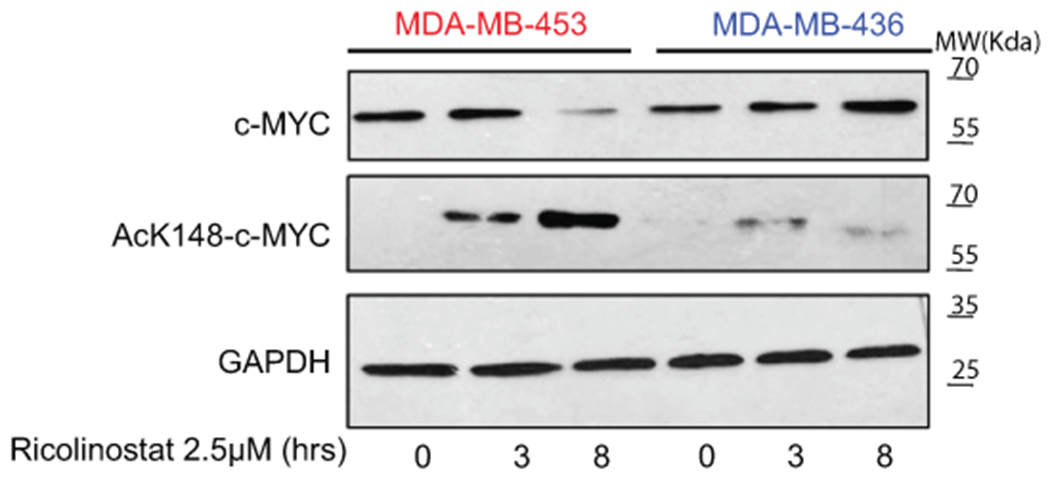
The western blot shows the accumulation of ac-K148-c-Myc after HDAC6 is inhibited by ricolinostat in MDA-MB-453 (sensitive) and MDA-MB-436 (resistant) lines. WT-blot results were reproduced n=3 times from independent experiments.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank members of the Silva and Yu laboratories for advice generating this manuscript and for technical assistance. Silva lab: Dayanira Alsina-Beauchamp and Rachel Werner; Yu lab: Xinran Dong, Koon-Kiu Yan, and Liang Ding. We also would like to thank Sui-Wan Lee from Mount Sinai’s CCMS core for technical assistance with mouse xenograft experiments. The authors also thank Keith A. Laycock, Ph.D., ELS, for scientific editing of the manuscript.
This research was partially funded through the DOD Breakthrough award 151500 (JS), ALSAC (JY), NIH R01 CA153233 (JS), NIH R01 CA153233–supplement (TZ), R01 GM134382 (JY), U01 CA217858 (AC), S10 OD012351 (AC), and S10 OD021764 (AC), and the Irving Scholar program (KK).
DECLARATION OF INTERESTS
MY and SJ were Acetylon employees when this project was initiated.
This research has been partially supported by a sponsor research agreement with Acetylon.
JY was a consultant of the computational analysis for the Phase Ib trial 2015-2017.
MJA is Chief Scientific Officer and equity holder at DarwinHealth, Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. AC is the founder, equity holder, consultant, and director of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth Inc.
The remaining authors declare no competing interest.
Footnotes
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
CODE AVAILABILITY
The codes for the HDAC6 score calculation and other analyses are freely available at https://github.com/jyyulab/HDAC6-score
DATA AVAILABILITY
The gene expression profile data is available at GEO under GSE180607. It includes two subseries, one for the RNA-seq data of clinical trial samples and 4 breast cancer cell lines (GSE128623), and another one for the microarray data of three cell lines and one mouse model (GSE180606). The acetylomics data, includiing Raw files and pepXML files for each sample, can be accessed at PRIDE under the accession number of PXD026010. Source data have been provided as Source Data files. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Previously published transcriptomic data that were re-analyzed here are available:
Cancer cell lines. The RNA-seq transcripts per million (TPM) data of 1,165 cell lines representing 29 cancer types from the Cancer Cell Line Encyclopedia (CCLE) project, together with cell line annotations and gene dependency scores were downloaded from the portal of the Dependency Map (DepMap) project (https://depmap.org/portal, release: Public 19Q1).
The Cancer Genome Atlas (TCGA). The TCGA RNA-seq data at both isoform and gene levels for 32 human primary cancer types including breast cancer were extracted from the QIAGEN OncoLand release TCGA_B38 2020v1.
The Molecular Taxonomy of Breast Cancer International Consortium (METABRIC). The human breast cancer microarray data (Illumina HT 12 arrays, N=1,981) were downloaded from Synapse (https://www.synapse.org/#!Synapse:syn1757063, version syn1757063).
Breast cancers treated only with paclitaxel GSE25066.
REFERENCES
- 1.Luo J, Solimini NL & Elledge SJ Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837, doi:S0092-8674(09)00200-1 [pii] 10.1016/j.cell.2009.02.024 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McClure JJ, Li X & Chou CJ Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv Cancer Res 138, 183–211, doi: 10.1016/bs.acr.2018.02.006 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Santo L et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 119, 2579–2589, doi: 10.1182/blood-2011-10-387365 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang P et al. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 8, 2694–2707, doi: 10.18632/oncotarget.13738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao J et al. Ricolinostat (ACY-1215) suppresses proliferation and promotes apoptosis in esophageal squamous cell carcinoma via miR-30d/PI3K/AKT/mTOR and ERK pathways. Cell Death Dis 9, 817, doi: 10.1038/s41419-018-0788-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang F, Zhong BW & Zhao ZR ACY 1215, a histone deacetylase 6 inhibitor, inhibits cancer cell growth in melanoma. J Biol Regul Homeost Agents 32, 851–858 (2018). [PubMed] [Google Scholar]
- 7.Cosenza M, Civallero M, Marcheselli L, Sacchi S & Pozzi S Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis 22, 827–840, doi: 10.1007/s10495-017-1364-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Uden DJ, van Laarhoven HW, Westenberg AH, de Wilt JH & Blanken-Peeters CF Inflammatory breast cancer: an overview. Critical reviews in oncology/hematology 93, 116–126, doi: 10.1016/j.critrevonc.2014.09.003 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Putcha P et al. HDAC6 activity is a non-oncogene addiction hub for inflammatory breast cancers. Breast Cancer Res 17, 149, doi: 10.1186/s13058-015-0658-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Margolin AA et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7 Suppl 1, S7, doi:1471-2105-7-S1-S7 [pii] 10.1186/1471-2105-7-S1-S7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70, doi: 10.1038/nature11412 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curtis C et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352, doi: 10.1038/nature10983 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parry M Introducing the Metastatic Breast Cancer Project: a novel patient-partnered initiative to accelerate understanding of MBC. ESMO Open 3, e000452, doi: 10.1136/esmoopen-2018-000452 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawaguchi Y et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Matthias P, Yoshida M & Khochbin S HDAC6 a new cellular stress surveillance factor. Cell cycle 7, 7–10 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Hubbert C et al. HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458, doi: 10.1038/417455a (2002). [DOI] [PubMed] [Google Scholar]
- 17.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607, doi: 10.1038/nature11003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khatamian A, Paull EO, Califano A & Yu J SJARACNe: a scalable software tool for gene network reverse engineering from big data. Bioinformatics 35, 2165–2166, doi: 10.1093/bioinformatics/bty907 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du X et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α+ dendritic cells. Nature 558, 141–145, doi: 10.1038/s41586-018-0177-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neve RM et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527, doi:S1535-6108(06)00314-X [pii] 10.1016/j.ccr.2006.10.008 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majid T, Griffin D, Criss Z 2nd, Jarpe M & Pautler RG Pharmocologic treatment with histone deacetylase 6 inhibitor (ACY-738) recovers Alzheimer’s disease phenotype in amyloid precursor protein/presenilin 1 (APP/PS1) mice. Alzheimers Dement (N Y) 1, 170–181, doi: 10.1016/j.trci.2015.08.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma XJ et al. HDAC-selective Inhibitor Cay10603 Has Single Anti-tumour Effect in Burkitt’s Lymphoma Cells by Impeding the Cell Cycle. Curr Med Sci 39, 228–236, doi: 10.1007/s11596-019-2024-4 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Dawood S, Ueno NT & Cristofanilli M The medical treatment of inflammatory breast cancer. Semin Oncol 35, 64–71, doi:S0093-7754(07)00237-0 [pii] 10.1053/j.seminoncol.2007.11.012 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Matro JM et al. Inflammatory Breast Cancer Management in the National Comprehensive Cancer Network: The Disease, Recurrence Pattern, and Outcome. Clinical breast cancer, doi: 10.1016/j.clbc.2014.05.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tallarida RJ Quantitative methods for assessing drug synergism. Genes Cancer 2, 1003–1008, doi: 10.1177/1947601912440575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou TC Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research 70, 440–446, doi: 10.1158/0008-5472.can-09-1947 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Pfefferle AD et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol 14, R125, doi: 10.1186/gb-2013-14-11-r125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung YK & Chappell R Sequential designs for phase I clinical trials with late-onset toxicities. Biometrics 56, 1177–1182, doi: 10.1111/j.0006-341x.2000.01177.x (2000). [DOI] [PubMed] [Google Scholar]
- 29.Obuchowski NA & Bullen JA Receiver operating characteristic (ROC) curves: review of methods with applications in diagnostic medicine. Phys Med Biol 63, 07TR01, doi: 10.1088/1361-6560/aab4b1 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Alvarez MJ et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet 48, 838–847, doi: 10.1038/ng.3593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez MJ & Califano A Darwin OncoTarget/OncoTreat: NY CLIA certified tests to identify effective drugs on an individual cancer patient basis from RNASeq profiles <https://www.pathology.columbia.edu/diagnostic-specialties/personalized-genomic-medicine/oncology-testing/darwin-oncotarget-tm-oncotreat> (2018).
- 32.Hatzis C et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 305, 1873–1881, doi: 10.1001/jama.2011.593 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshihara K et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4, 2612, doi: 10.1038/ncomms3612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyers RM et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49, 1779–1784, doi: 10.1038/ng.3984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan J Interplay between HDAC6 and its interacting partners: essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA and cell biology 33, 567–580, doi: 10.1089/dna.2013.2300 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Gregory MA & Hann SR c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol 20, 2423–2435, doi: 10.1128/MCB.20.7.2423-2435.2000 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chakraborty AA et al. A common functional consequence of tumor-derived mutations within c-MYC. Oncogene 34, 2406–2409, doi: 10.1038/onc.2014.186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hai Y & Christianson DW Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol 12, 741–747, doi: 10.1038/nchembio.2134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lynch JT, Somerville TD, Spencer GJ, Huang X & Somervaille TC TTC5 is required to prevent apoptosis of acute myeloid leukemia stem cells. Cell Death Dis 4, e573, doi: 10.1038/cddis.2013.107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faiola F et al. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol 25, 10220–10234, doi: 10.1128/MCB.25.23.10220-10234.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farrell AS & Sears RC MYC degradation. Cold Spring Harb Perspect Med 4, doi: 10.1101/cshperspect.a014365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee EK et al. Results of an abbreviated Phase Ib study of the HDAC6 inhibitor ricolinostat and paclitaxel in recurrent ovarian, fallopian tube, or primary peritoneal cancer. Gynecol Oncol Rep 29, 118–122, doi: 10.1016/j.gore.2019.07.010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogl DT et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin Cancer Res 23, 3307–3315, doi: 10.1158/1078-0432.CCR-16-2526 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yee AJ et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol 17, 1569–1578, doi: 10.1016/S1470-2045(16)30375-8 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Twomey JD, Brahme NN & Zhang B Drug-biomarker co-development in oncology - 20 years and counting. Drug Resist Updat 30, 48–62, doi: 10.1016/j.drup.2017.02.002 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Hackanson B et al. HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk Res 36, 1055–1062, doi: 10.1016/j.leukres.2012.02.026 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Shouksmith AE et al. Class I/IIb-Selective HDAC Inhibitor Exhibits Oral Bioavailability and Therapeutic Efficacy in Acute Myeloid Leukemia. ACS Med Chem Lett 11, 56–64, doi: 10.1021/acsmedchemlett.9b00471 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabay M, Li Y & Felsher DW MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med 4, doi: 10.1101/cshperspect.a014241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen H, Liu H & Qing G Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther 3, 5, doi: 10.1038/s41392-018-0008-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vervoorts J, Luscher-Firzlaff J & Luscher B The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem 281, 34725–34729, doi: 10.1074/jbc.R600017200 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Sears RC The life cycle of C-myc: from synthesis to degradation. Cell cycle 3, 1133–1137 (2004). [PubMed] [Google Scholar]
- 52.Boyault C et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev 21, 2172–2181, doi: 10.1101/gad.436407 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyault C, Sadoul K, Pabion M & Khochbin S HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 26, 5468–5476, doi: 10.1038/sj.onc.1210614 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Lee JY, Nagano Y, Taylor JP, Lim KL & Yao TP Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol 189, 671–679, doi: 10.1083/jcb.201001039 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinus RD et al. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. European journal of biochemistry / FEBS 240, 98–103 (1996). [DOI] [PubMed] [Google Scholar]
- 56.Hu F & Liu F Mitochondrial stress: a bridge between mitochondrial dysfunction and metabolic diseases? Cellular signalling 23, 1528–1533, doi: 10.1016/j.cellsig.2011.05.008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vives-Bauza C, de Vries RL, Tocilescu M & Przedborski S PINK1/Parkin direct mitochondria to autophagy. Autophagy 6, 315–316 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Narendra D, Tanaka A, Suen DF & Youle RJ Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183, 795–803, doi: 10.1083/jcb.200809125 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haynes CM & Ron D The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci 123, 3849–3855, doi: 10.1242/jcs.075119 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Carroll RG, Hollville E & Martin SJ Parkin Sensitizes toward Apoptosis Induced by Mitochondrial Depolarization through Promoting Degradation of Mcl-1. Cell reports 9, 1538–1553, doi: 10.1016/j.celrep.2014.10.046 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Thompson PR et al. Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol 11, 308–315, doi: 10.1038/nsmb740 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Banik D et al. HDAC6 Plays a Noncanonical Role in the Regulation of Antitumor Immune Responses, Dissemination, and Invasiveness of Breast Cancer. Cancer research 80, 3649–3662, doi: 10.1158/0008-5472.CAN-19-3738 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 63.Lee SM & Ying Kuen C Model calibration in the continual reassessment method. Clin Trials 6, 227–238, doi: 10.1177/1740774509105076 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andrews S (2010). [Google Scholar]
- 65.Patro R, Duggal G, Love MI, Irizarry RA & Kingsford C Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419, doi: 10.1038/nmeth.4197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bai B et al. Deep Profiling of Proteome and Phosphoproteome by Isobaric Labeling, Extensive Liquid Chromatography, and Mass Spectrometry. Methods Enzymol 585, 377–395, doi: 10.1016/bs.mie.2016.10.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pagala VR et al. Quantitative protein analysis by mass spectrometry. Methods Mol Biol 1278, 281–305, doi: 10.1007/978-1-4939-2425-7_17 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Wang X et al. JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol Cell Proteomics 13, 3663–3673, doi: 10.1074/mcp.O114.039586 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eng JK, McCormack AL & Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989, doi: 10.1016/1044-0305(94)80016-2 (1994). [DOI] [PubMed] [Google Scholar]
- 70.Niu M et al. Extensive Peptide Fractionation and y1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal Chem 89, 2956–2963, doi: 10.1021/acs.analchem.6b04415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.survminer: https://rpkgs.datanovia.com/survminer/index.html.
- 72.Robin X et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77, doi: 10.1186/1471-2105-12-77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The gene expression profile data is available at GEO under GSE180607. It includes two subseries, one for the RNA-seq data of clinical trial samples and 4 breast cancer cell lines (GSE128623), and another one for the microarray data of three cell lines and one mouse model (GSE180606). The acetylomics data, includiing Raw files and pepXML files for each sample, can be accessed at PRIDE under the accession number of PXD026010. Source data have been provided as Source Data files. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Previously published transcriptomic data that were re-analyzed here are available:
Cancer cell lines. The RNA-seq transcripts per million (TPM) data of 1,165 cell lines representing 29 cancer types from the Cancer Cell Line Encyclopedia (CCLE) project, together with cell line annotations and gene dependency scores were downloaded from the portal of the Dependency Map (DepMap) project (https://depmap.org/portal, release: Public 19Q1).
The Cancer Genome Atlas (TCGA). The TCGA RNA-seq data at both isoform and gene levels for 32 human primary cancer types including breast cancer were extracted from the QIAGEN OncoLand release TCGA_B38 2020v1.
The Molecular Taxonomy of Breast Cancer International Consortium (METABRIC). The human breast cancer microarray data (Illumina HT 12 arrays, N=1,981) were downloaded from Synapse (https://www.synapse.org/#!Synapse:syn1757063, version syn1757063).
Breast cancers treated only with paclitaxel GSE25066.