
Fig. 2. In silico evaluation of ProteinMPNN.
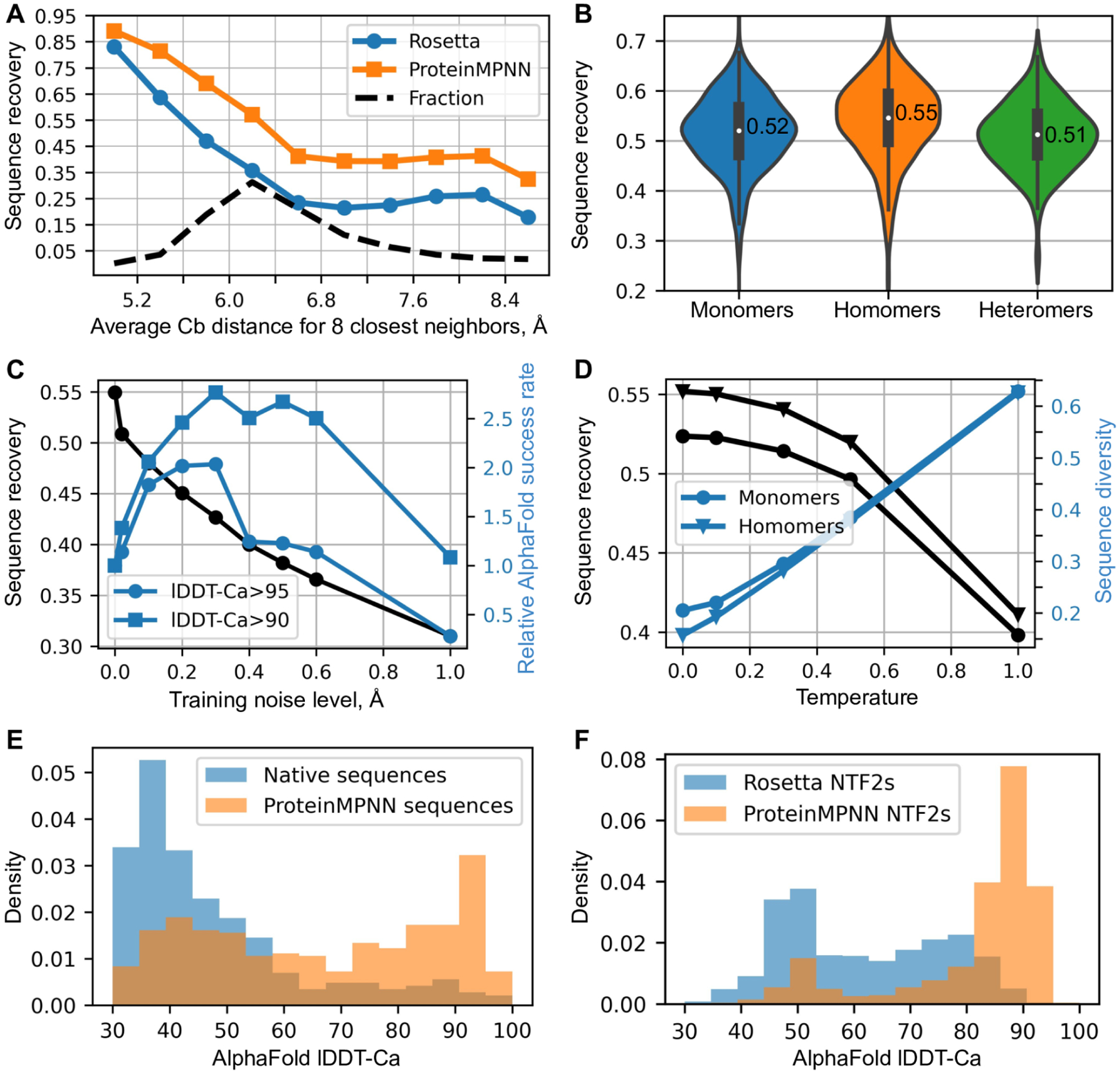
(A) ProteinMPNN has higher native sequence recovery than Rosetta. The average Cb distance of the 8 closest neighbors (x axis) reports on burial, with most buried positions on the left and more exposed on the right; ProteinMPNN outperforms Rosetta at all levels of burial. Average sequence recovery for ProteinMPNN was 52.4%, compared to 32.9% for Rosetta. (B) ProteinMPNN has similarly high sequence recovery for monomers, homo-oligomers, and hetero-oligomers; violin plots are for 690 monomers, 732 homomers, 98 heteromers. (C) Sequence recovery (black) and relative AlphaFold success rates (blue) as a function of training noise level. For higher accuracy predictions (circles) smaller amounts of noise are optimal (1.0 corresponds to 1.8% success rate), while to maximize prediction success at a lower accuracy cutoff (squares), models trained with more noise are better (1.0 corresponds to 6.7% success rate). (D) Sequence recovery and diversity as a function of sampling temperature. Redesign of native protein backbones with ProteinMPNN considerably increases AphaFold prediction accuracy compared to the original native sequence using no multiple sequence information. Single sequences (designed or native) were input in both cases. (F) ProteinMPNN redesign of previous Rosetta designed NTF2 fold proteins (3,000 backbones in total) results in considerably improved AlphaFold single sequence prediction accuracy.