Galkin et al. 10.1073/pnas.0609412103. |
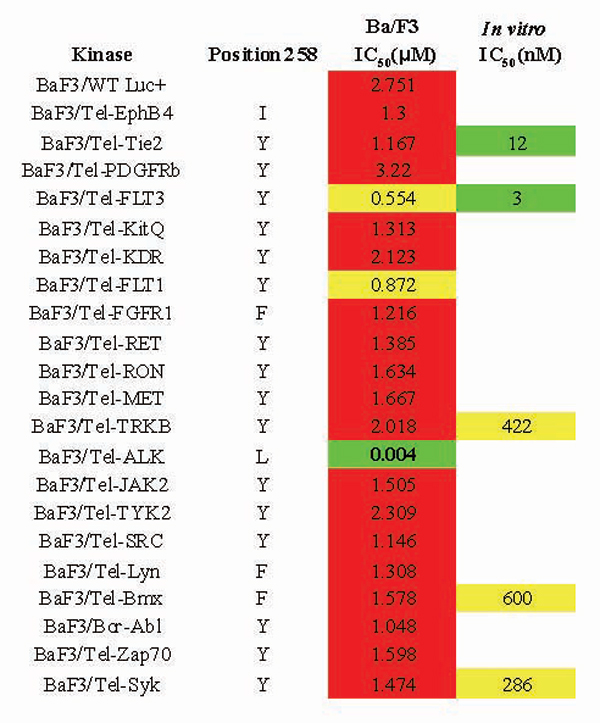
Fig. 7. Summary of inhibitory activity of TAE684 in cellular (Ba/F3 IC50s) and enzymatic assays (in vitro IC50s). Sequence alignments of kinases available in the Ba/F3 panel revealed that unlike ALK, most kinases have bulkier residues at the 258 position, which may play an important role in kinase selectivity determinants for TAE684.

Fig. 8. Effects of TAE684 treatment on Ba/F3 NPM-ALK or Ba/F3 BCR-ABL disease progression in vivo. (A) Representative bioluminescence images of mice after 3 weeks of treatment with either 10 mg/kg of TAE684 or vehicle solution. Dosing was initiated 3 days after mouse tail vein injection with one million Ba/F3 NPM-ALK cells. (B) Percent bioluminescence emission was determined with the Xenogen Imaging System after 3 weeks of TAE684 (10 mg/kg) or vehicle solution administration to mice injected with Ba/F3 cells expressing either NPM-ALK or BCR-ABL fusions (n = 10 mice per group). (C) Effect of TAE684 treatment on mean spleen weight of mice described above.

Fig. 9. TAE684 treatment reduces CD30 receptor expression in Karpas-299 cells in vitro. (A) Histogram representation of CD30 expression in Karpas-299 cells treated with either DMSO or 50 nM TAE684 for 48 h as determined by flow cytometry. DMSO-treated Karpas-299 cells incubated with FITC-conjugated anti-mouse secondary antibody alone were used as a negative control. (B) Percent of CD30 expression in Karpas-299 cells calculated with FlowJo Data Analysis Software after 24 and 48 h of treatment with 25-100 nM concentrations of TAE684. Representative graph of three experiments is shown.
Supporting Materials and Methods
Vectors and Cloning.
pMSCV IRES puro/Luc was generated by first introducing a luciferase gene between the NcoI and ClaI site downstream of the IRES sequence of pMSCV IRES GFP, replacing the GFP cassette (pMSCV IRES Luc). The puromycin gene was amplified from pcDNA3.1puro and introduced into the EcoRI/SalI sites of pEGFP N2 (Clontech, Mountain View, CA) to replace the GFP cassette (pLUC N2). The puromycin gene, including parts of the pLUC N2 multiple cloning site, was then PCR amplified and cloned into the NcoI site of pMSCV IRES Luc downstream of the IRES and upstream of the luciferase gene. Both BCR-ABL (a gift from Richard Van Etten,Tufts New England Medical Center, Boston, MA) and NPM-ALK (isolated from Karpas-299 cells) were cloned into the EcoRI site of pMSCV IRES puro/Luc upstream of the IRES sequence. The generation of various vectors encoding Tel-tyrosine kinase fusions are described in ref. 12.Cell Lines.
The murine pro-B cell line Ba/F3, the human t(2;5)-positive Karpas-299 and SU-DHL-1 ALCL cell lines were obtained from DSMZ (Berlin, Germany). Cells were maintained in RPMI medium1640 supplemented with 10% FBS (Sigma-Aldrich, St. Louis, MO). Ba/F3 cells were grown in the presence of IL-3 (10 ng/ml) (R&D Systems, Minneapolis, MN). The rat hepatoma cell line H-4-II-E and 293-T cells were obtained from ATCC (Manassas, VA) and maintained in MEME or DMEM, respectively, supplemented with 10% FBS (Sigma-Aldrich).Cell lines expressing luciferase or NPM-ALK, BCR-ABL and TEL fusion constructs (Ba/F3-luc, Ba/F3 BCR-ABL, Ba/F3 NPM-ALK Karpas-299-luc, SU-DHL-1-luc) were generated by retroviral transduction of cells with pMSCV IRES puro/Luc vector. Stocks of retrovirus were generated from 293-T cells transiently transfected with pMSCV retroviral and packaging vector constructs (EcoPac or pAmpho) as previously described. Briefly, 293-T cells were co-transfected with 60 mg of retroviral plasmid and 30 mg of ecotropic (kindly provided by Richard van Etten) or amphotropic (Clontech) packaging plasmid using CaCl2 precipitation. Two days after transfection, the viral supernatant was harvested and filtered. 2 ml of viral supernatant was then added to 4 ´ 106 cells contained in 2 ml of infection medium [RPMI medium 1640, 20% FBS, 2% 1 M Hepes buffer and 4 mg/ml polybrene (Chemicon International, Temecula, CA.)]. IL-3 (10 ng/ml) was included in the infection media for Ba/F3 cells. Cells were centrifuged at 200 × g for 90 min at room temperature. The following day, cells were expanded into a T75 flask and were selected for stable transfectants in the presence of 1 mg/ml of puromycin (Sigma-Aldrich). Puromycin-selected Ba/F3 cells expressing kinase fusion constructs were subsequently transferred to IL-3-free growth medium and monitored for growth-factor independent outgrowth by light microscopy. The generation of Ba/F3 cell expressing TEL fused tyrosine kinases will be described elsewhere. NPM-ALK, BCR-ABL and TEL-kinase fusion expression was confirmed by Western blotting (12).
Immunoblotting.
A standard immunoblotting protocol was used to assay protein expression in cell and lymph node lysates. Briefly, DMSO and TAE684 treated cells were lysed with a protein extraction buffer (150 mM Tris 1 M, pH 8.0/150 mM NaCl/10% Glycerol/1% Nonidet P-40/10 mM EDTA) supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich), incubated on ice for 15 min and then centrifuged at 20,000 × g for 25 min at 4C.To assess the effect of TAE684 on the phosphorylation of InsR, H4IIE cells were plated into a six-well plate at 1 ´ 106 cells per well in 4 ml of DMEM/10%FBS and incubated overnight at 37°C in a tissue culture incubator. After a 24-h serum-starvation period, H4IIE cells were preincubated with compound for 30 min at 37°C and stimulated with 2,000 nM bovine insulin (I-0516; Sigma, St. Louis, MO). After 10 min of insulin stimulation, cells were washed twice with ice-cold PBS (GIBCO, Carlsbad, CA) and lysed with 250 ml of lysis buffer [1% Nonidet P-40/10% Glycerol/20 mM Tris (pH 8.0)/150 mM NaCl/10 mM EDTA; 1× protease inhibitor mixture, 1× phosphatase inhibitor mixture I, 1× phosphatase inhibitor mixture II) (all reagents obtained from Sigma). Lysates were removed from wells, transferred to microcentrifuge tubes, and cleared by spinning at 20,000 × g at 4°C for 15 min.
For the pharmacodynamic study of NPM-ALK and STAT3 inhibition in tumor infiltrated lymph nodes, tissue was homogenized in the lysis buffer described above with stainless steel beads (5 mm) (Qiagen, Valencia, CA) in a TissueLyser (Qiagen). Lysates derived from cells or lymph nodes were centrifuged for 10 min at 12,000 × g at 4°C, and the resultant supernatants were used for immunoblotting. BCA assay (Pierce, Rockford, IL) was used to determine protein concentration. Equivalent amounts of proteins were loaded onto a NuPage 10% Bis-Tris gel (Invitrogen), followed by transfer to nitrocellulose membrane (Amersham Pharmacia Biosciences, Piscataway, NJ). After blocking in 5% milk, membranes were incubated with primary antibodies against specific proteins in 5% BSA Tris-buffered-saline/Tween 20 overnight at 4°C, followed by incubation with secondary antibodies, ECL Rabbit and Mouse IgG, HRP Linked Whole Antibodies (Amersham Pharmacia Biosciences). Western blots were developed with enhanced chemiluminescence reagents (Pierce). Primary antibodies (ALK, phospho-ALK (Tyr-1640), Stat3, phospho-Stat3 (Tyr-705), Stat5, phospho-Stat5 (Tyr-694), phospho-Akt (Ser-473), phospho-ERK(Thr-202/Tyr-204), phospho-FKHR(Thr-24)/FKHRL1(Thr-32), and pTyr1146 InsR) were obtained from Cell Signaling Technology (Beverly, MA) and used at 1:1,000 dilution.
In vitro
Enzyme Assays. All in vitro enzyme assays were done at Upstate Biotechnology (Dundee, U.K.) with the exception of InsR and IGF1R. Detail protocols for the respective assays are available from www.upstate.com. To determine the IC50 of TAE684 against InsR and IGF1R a homogeneous time-resolved fluorescence assay was performed. ATP (10 mM) and 20 mg/ml biotinylated PolyEY (Glu, Tyr 4:1) (CIS Bio International, Bedford, MA) were combined with 50 nl of serial dilutions of compound and 4 ng of InsR enzyme (Upstate Biotechnology, Charlottesville, VA) in the presence of the kinase reaction buffer (20 mM Tris×HCl, pH 7.5/10 mM MgCl2/3 mM MnCl2/1 mM DTT/10 mM NaVO4/0.1 mg/ml of BSA). Assays were incubated for 1 h at ambient temperature. Reactions were terminated by adding 10 ml of the detection solution containing 50 mM EDTA, 500 mM KF, 0.5 mg/ml of BSA, 5 mg/ml Eu3+ cryptate-labeled anti-phosphotyrosine antibody Mab PT66-K, and 5 mg/ml Streptavidin-XLent (CIS bio International). The reaction was incubated for half an hour, and fluorescence signals were read on Analyst GT (Molecular Devices, Sunnyvale, CA).Flow Cytometry.
Ba/F3-NPM-ALK, Karpas-299 and SU-DHL-1 cells were treated with DMSO or various concentrations of NVP-TAE684 for 24, 48, and 72 h before analysis of cell cycle distribution and apoptosis by flow cytometry. For cell cycle analysis, cells were washed with PBS, fixed in 70% ethanol, incubated with RNaseA (0.1 mg/ul) for 30 min, followed by incubation with propidium iodide (10 mg/ml). Cells were analyzed on a LSRII Flow Cytometer (BD Biosciences, San Jose, CA). Cell viability and apoptosis were determined by using the Annexin V-PE Apoptosis Detection Kit (BD Biosciences). Annexin V staining was carried out according to manufacturer's instructions. Briefly, cells were washed with PBS, resuspended in provided 1× binding buffer and incubated with Annexin V-PE and 7-AAD for 15 min before analysis. Cell cycle distribution and percent of preand apoptotic cells was determined with FlowJo Data Analysis Software (Tree Star, Ashland, OR). For CD30 expression, Karpas-299 cells were washed with PBS, incubated with anti-CD30 primary antibody (Dako, Carpinteria, CA) for 1 h, followed by incubation with fluorescein-anti-mouse IgG secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h in the dark at 4C. Cells were then immediately analyzed by flow cytometry.In vivo
experiments. Four- to 6-week old female Fox Chase SCIDBeige mice were obtained from Charles River Laboratories (San Diego, CA) and were maintained in pressurized, ventilated caging. Institutional guidelines for the proper and humane use of animals in research were followed. For in vivo compound efficacy studies, treatment was initiated 72 h after tail vein injection of 1 ´ 106 Karpas-299, Ba/F3 NPM-ALK or BCR-ABL-expressing cells. Mice (n = 10 per group) were administered either TAE684 resuspended in 10% 1-methyl-2-pyrrolidinone/90% polyethylene glycol 300 (Sigma) solution at 1, 3, and 10 mg/kg of body weight once daily for 3 weeks or the vehicle solution at the same dosing schedule. Disease progression and compound efficacy was monitored weekly with bioluminescence imaging. Mice were injected with D-luciferin (Xenogen, Alameda, CA) and imaged using the Xenogen Imaging System. To determine the efficacy of TAE684 on established disease, dosing was initiated on day 12, at which time the disease confirmed to be wide-spread by bioluminescence imaging. For analysis of downstream molecular effects in vivo, mice were injected with Karpas-299 cells and monitored for disease progression. Three weeks after injection, mice were administered vehicle solution or TAE684 (10 mg/kg of body weight) for 3 days. At the end of treatment mice, were killed, and lymph nodes were extracted for immunoblotting and histological analysis.Histology.
Lymph node tissue infiltrated with Karpas-299-luc cells 3 weeks after tail vein injection of cells was fixed in 10% formalin and subsequently embedded into paraffin. Five-micron-thick sections were stained with hematoxylin and eosin or incubated with primary antibodies after antigen retrieval (anti-CD30, anti-CD246 (Dako). The DAKO ARK Peroxidase Kit (Dako) and the Vector Rabbit ABC Kit (Vector Labs, Burlingame, CA) were used according to the manufacturer's instructions.