
Zuber et al. 10.1073/pnas.0700154104. |
Fig. 6. Immunoblot of postnuclear cellular membrane fractions from HepG2 cells were prepared and loaded onto a 2.5-30% discontinuous Optiprep gradient as described in Materials and Methods. Fractions collected in the order of increasing density and resolved on a reducing SDS/PAGE were probed for EDEM1 (a-EDEM1).

Fig. 7. Spatial relationship of EDEM1 to Sec61b. Confocal double immunofluorescence with HepG2 cells was performed. Colocalization, as indicated by shades of yellow, is detectable in some punctate and elongated structures (arrowheads). Sec61b unreactive EDEM1 structures are labeled with arrows. (Scale bar, 10 mm.)

Fig. 8. Spatial relationship of EDEM1 to Derlin-2. Confocal double immunofluorescence with HepG2 cells was performed. Colocalization, as indicated by shades of yellow, is detectable in some punctate (arrows) and elongated structures (arrowhead). (Scale bar, 10 mm.)

Fig. 9. Spatial relationship of EDEM1 to PDI and ERGIC-53. Double confocal immunofluorescence images of HepG2 hepatoma cells are displayed. Staining for EDEM1 (A and D), PDI (B), and ERGIC-53 (E). In the overlay from single confocal optical sections, restricted punctate co-distribution of EDEM1 and PDI along cisternal elements is obvious (arrowheads in C Insets). No evidence for codistribution EDEM1 with ERGIC-53 was obtained (F). (Scale bars, 4 mm in A-C and 10 mm in D-F.).
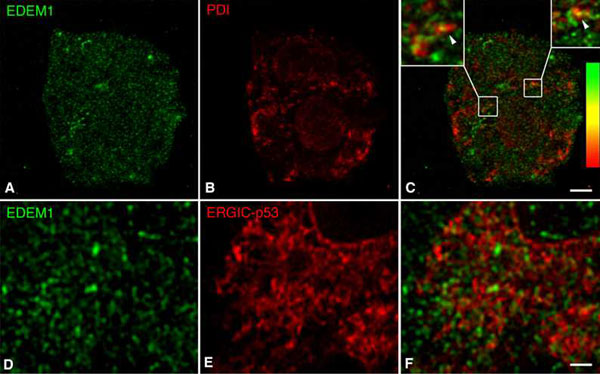
Fig. 10. EDEM1 structures represent vesicles. A series of four consecutive 80 nm thin sections of HepG2 cells with preembedding immunoperoxidase labeling for EDEM1 are displayed. Arrows in A2 and A3 as well as in B2 and B3 mark an EDEM1-positive vesicle which is absent in the first and fourth section of the respective series of the sections. ER: this ER cisternal element can be followed through all four thin sections. (Scale bars, 500 nm.)

Fig. 11. EDEM1 is undetectable in coated buds of transitional ER and the associated vesiculotubular cluster. Eight serial sections from a HepG2 cell showing immunolabeling for EDEM1 in restricted parts of a cisterna of the rough endoplasmic reticulum (ER in # 1) which forms an exit site with coated buds (arrowheads in #5 - #7) and a vesiculotubular cluster (VTC in #7 and #8). Arrows denote cytoplasmic immunolabeling for EDEM1. PO: peroxisomes. Sections #5-#8 are part of Fig. 4. Preembedding immunoperoxidase labeling was performed. (Scale bar, 425 nm.)

Fig. 12. EDEM1 is undetectable in Golgi-associated ER exit sites. Coated buds of transitional ER (TE), the Golgi-associated vesiculotubular cluster and the Golgi cisternal stack (GA) of a human MRC5 fibroblast are not labeled for EDEM1. Note the ER-associated (arrowhead) and cytoplasmic (arrows) EDEM1 immunolabeling. Silver-enhanced nanogold immunolabeling was performed. (Scale bar, 400 nm.)
SI Materials and Methods
Generation and affinity-purification of anti-EDEM1 antibody.
Bacterial soluble EDEM1 was generated by PCR amplifying base pairs of the human EDEM1 gene corresponding to aa31-657 (lacking the N-terminal signal sequence). The DNA product was ligated into a pET 21d vector with a C-terminal hexahistidine tag. The vector was transformed into Rosetta-gami B (Novagen, San Diego, CA) bacterial cells to facilitate bacterial protein production and purification. The bacteria were grown to log phase (OD600 nm = 0.4) followed by induction of protein expression using 0.2 mM isopropyl-b-D-thioglucoside overnight at 25°C. The protein was purified using Ni-NTA Sepharose (Novagen, San Diego, CA) according to the manufacturer's instructions. In brief, the bacteria were lysed using 100 mg/ml of lysozyme in binding buffer (1 x PBS, pH 7.3, 20 mM imidazole) for 3 h at 4°C, followed by three rounds of sonication on ice for 1.5 min each. Bacterial debris was pelleted by centrifugation. Ni-NTA resin was added to the clarified supernatant and the slurry was rotated for 1 h at 4°C. The slurry was loaded into an empty column allowing the supernatant drain leaving just the resin. The Ni-NTA column was washed with 10 volumes of binding buffer. The protein was eluted using elution buffer (1 x PBS, pH 7.3, 150 mM imidazole). The purified bacterial soluble EDEM1 protein was dialyzed into coupling buffer (0.1 M NaHCO3, pH 8.3, 0.5 M NaCl) and covalently linked to activated cyanogen-bromide Sepharose (GE Healthcare, Pittsburgh, PA) according to the manufacturer's instructions. The anti-EDEDM1 antiserum was incubated with the EDEM1-cyanogen-bromide Sepharose at 4°C for overnight. The column was washed with 20 column volumes of 1 x PBS pH 7.3, 0.02% sodium azide. The bound antibody was eluted using 0.1 M glycine (pH 2.5) and was immediately neutralized to pH 7.0 with 50 mM Tris (pH 8.0). Purified EDEM1 antibody was dialyzed against PBS (pH 7.3) containing 5% glycerol and 0.02% sodium azide and was stored in PBS (pH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide at 4°CReagents.
Rabbit polyclonal anti-Sec61b antibody was kindly provided by Dr. B. Dobberstein (Heidelberg, Germany), mouse monoclonal anti-p53 (anti-ERGIC-53) antibody by Dr. H.-P. Hauri (Basel, Switzerland), rabbit anti-Derlin antibodies by Dr. H. Ploegh (Boston, MA, USA) and purified rabbit anti-glucosidase II antibodies were available from previous studies (1). In addition to our own anti-peptide EDEM1 antibodies, affinity-purified antibodies against a C-terminal peptide of human EDEM1 from Santa Cruz Biotechnology (Santa Cruz, CA, USA) were used. Antibodies against calnexin and PDI were purchased from Stressgen (Victoria, BC, Canada), GM130 from BD Biosciences (San Jose, CA), Sec23p from Abcam (Cambridge, UK), alpha-1-antitrypsin from Dako (Glostrup, Denmark) and FLAG from Sigma (St. Louis, MO). Unlabeled or Cy 2 and Cy 5-conjugated Fab fragments of goat anti-rabbit IgG, Cy 2-conjugated affinity-purified goat anti-mouse IgG, peroxidase-conjugated Fab2 fragments of goat anti-rabbit IgG were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA), and Alexa 488/nanogold-conjugated Fab2 fragments of goat anti-rabbit IgG and nanogold silver enhancer from Molecular Probes (Eugene, OR). Protein A-gold was prepared according to standard protocol.EasyTag [35S] methionine/cysteine was purchased from PerkinElmer Life Sciences (Wellesley, MA), flexi rabbit reticulocyte lysate (RRL) from Promega (Madison, WI), restriction enzymes, PNGase F, and RNA cap structure analog from New England Biolabs (Ipswich, MA), RosettaGami, pET21d-His vector, and Ni-NTA Sepharose from Novagen (Madison, WI), and protein A-Sepharose and cyanogen-bromide activated-Sepharose from GE Healthcare Life Sciences (Piscataway, NJ). MEM, FBS, penicillin, streptomycin and L-glutamine were obtained from Invitrogen (Carlsbad, CA), and OptiPrep along with all other remaining reagents were from Sigma (St. Louis, MO).
Confocal immunofluorescence.
Cells were grown on glass coverslips and fed with fresh medium 16 h before fixation in freshly prepared 3% formaldehyde (Fluka, Buchs, Switzerland) and were permeabilized with 0.2% saponin. Single (for EDEM1) and double (EDEM1 and calnexin, Sec23p, p53, PDI, Sec61b, Derlin-1 and Derlin-2, respectively) immunolabeling incubations were performed. Immunofluorescence was recorded with Leica Confocal Laser Scanning Microscopes SP2 and SL2 using the 100 x objective (1.4). In double immunofluorescence overlays, effects of z axis pixel shifts were corrected.Immunoelectron microscopy.
Cells were grown to near confluency and fed with fresh medium 16 h before fixation in freshly prepared 3% formaldehyde (Fluka, Buchs, Switzerland) or in a mixture of 2% formaldehyde-0.1% glutaraldehyde (vacuum distilled, Fluka) as described previously (2). Frozen ultrathin sections were prepared at -110°C according to Tokuyasu (3). For immunolabeling, grids were incubated with anti-EDEM1 antibodies diluted in PBS containing 1% BSA and 0.01% Tween 20 followed by several rinses in PBS and 8 nm protein A-gold (4, 5) diluted to an absorbance of 0.1 at 525 nm in PBS containing 0.2% defatted milk and 0.01% Tween 20 (6) for 1 h.Preembedding immunoperoxidase labeling was performed as described by Brown and Farquhar (7) and modified by Pavelka et al. (8). Following formaldehyde-fixation and permeabilization with 0.05% saponin, coverslips were incubated with anti-EDEM1 antibodies (IgG fraction or affinity-purified) or anti-A1AT antibodies followed by horseradish peroxidase-conjugated Fab2 goat-anti rabbit IgG (4 mg/ml in BSA-PBS) and rinses with buffer. Before the DAB reaction, cells on coverslips were postfixed with 2% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) containing 5% sucrose for 16 h at 4°C. Following several rinses with buffer, aldehyde groups were amidinated with 50 mM NH4Cl. All incubations using DAB (diaminobenzidine tetrahydrochloride) to reveal horseradish peroxidase activity were performed in the darkness. DAB was dissolved under stirring in 50 mM Tris-HCl (pH 7.4) containing 7.5% sucrose to a final concentration of 0.2% and the pH readjusted to pH 7.4. Coverslips were transferred to 1 ml of DAB solution to which 20 ml of 0.1% H2O2 were gradually added and incubated for 5 min. Three additional 20 ml of 0.1% H2O2 were added at 4-min intervals. For postfixation, 1% freshly reduced OsO4 in 0.1 M cacodylate buffer, pH 7.4, was used for 15 min followed by 1% OsO4 in 0.1 M cacodylate buffer for 4 h at 4°C. Dehydration was started with 70% ethanol over night at 4°C followed by several changes of 100% ethanol. Cells on coverslips were embedded in Epon-Araldite according standard protocol. Serial ultrathin sections in the plane of the cell monolayer were prepared using a Leica ultramicrotome Ultracut S and contrasted with 1% uranyl acetate and freshly prepared lead citrate. Optiprep fractions were fixed in 2% (para) formaldehyde and 0.1% glutaraldehyde in PBS, enclosed in Agar, permeabilized with 0.02% saponin and subjected to immunoperoxidase labeling for EDEM1 as described above.
Preembedding nanogold immunolabeling with silver enhancement was performed according to published protocols (9, 10). Following formaldehyde-fixation and saponin-permeabilization as described for immunofluorescence, coverslips were incubated in anti-EDEM1 antibodies (IgG fraction or affinity-purified) diluted in BSA-PBS for 2 h followed by 2 rinses in BSA-PBS for 2 min. The nanogold incubation and silver enhancement was performed according to manufacturer's instructions. Finally, cells were fixed with 1% OsO4 and embedded in Epon-Araldite. Serial ultrathin sections were prepared using a Leica ultramicrotome Ultracut S and contrasted with 1% uranyl acetate and freshly prepared lead citrate.
Immunocytochemical controls for EDEM1 labeling
. Different types of controls were performed which included the use of preimmune rabbit serum, the flow-through from the EDEM1 antibody affinity-purification and affinity-purified anti-EDEM1 antibody preabsorbed with Sepharose-immobilized EDEM1 expressed in E. coli. Furthermore, the EDEM1 antibody (either IgG fraction or affinity-purified) was replaced by an unrelated antibody (rabbit anti-chicken IgG) or was omitted from the incubation procedure.Quantification of immunogold labeling.
Micrographs from ultrathin frozen sections of CHO cells and Clone9 hepatocytes were taken at the original magnification of x 25,000. The percentage of gold particle labeling for EDEM 1 was estimated over the ER and smooth vesicles, clathrin-coated vesicles and Golgi apparatus as well as nucleus and mitochondria. For this, 80 micrographs from CHO cells and Clone9 hepatocytes were evaluated.1. Brada, D., Kerjaschki, D. & Roth, J. (1990) J. Cell Biol. 110, 309-318.
2. Zuber, C., Spiro, M. J., Guhl, B., Spiro, R. G. & Roth, J. (2000) Mol Biol Cell 11, 4227-4240.
3. Tokuyasu, K. (1978) J Ultrastruct Res 63, 287-307.
4. Roth, J., Bendayan, M. & Orci, L. (1978) J Histochem Cytochem 26, 1074-1081.
5. Roth, J. (1989) in Meth Cell Biol, ed. Tartakoff, A. M. (Academic, San Diego), Vol. 31, pp. 513-551.
6. Roth, J., Taatjes, D. J. & Warhol, M. J. (1989) Histochemistry 92, 47-56.
7. Brown, W. J. & Farquhar, M. G. (1989) in Methods in Cell Biology, ed. Tartakoff, A. M. (Academic, San Diego), Vol. 31, pp. 553-569.
8. Pavelka, M., Ellinger, A., Debbage, P., Loewe, C., Vetterlein, M. & Roth, J. (1998) Histochem Cell Biol 109, 555-570.
9. Hainfeld, J., Powell, R. & Furuya, F. (2002) in Gold and silver staining: Techniques in molecular morpholgy, eds. Hacker, G. & Gu, J. (CRC Press, Boca Raton), pp. 85-106.
10. Sawada, H. & Esaki, M. (2002) in Gold and silver staining: Techniques in molecular morpholgy, eds. Hacker, G. & Gu, J. (CRC Press, Boca Raton), pp. 169-176.