
Wu et al. 10.1073/pnas.0701817104. |
Fig. 5. Isolation and analysis of semisynthetic Rab7 NF. (A) Magnified picture of the Coomassie stained SDS/PAGE loaded with fraction eluted from gel filtration column loaded with Rab7-NF ligation mixture. Migration positions of Rab7 DC2 -thioester (Rab7-Th) and Rab7-NF are marked by arrows. Fractions 19-21 containing Rab7-NF at >80% purity were collected and used for further analysis. (B) HPLC elution profile of Rab7-NF alone (17.8 min, black line) or in vitro prenylated with GGPP (19.1 min, red line). The control reaction where REP was omitted is shown as a blue line (17.8 min.). The HPLC fluorescence detector was set to lex/em values of 490 and 535 nm. (C) MALDI-MS analysis of Rab7-NF (black, Mcalc = 23,770 Da) and its in vitro geranylgeranylated form (blue, Mcalc = 24,043 Da), i.e., Rab7C(geranylgeranyl)SC(NBD-farnesyl).
Fig. 6. Gel filtration chromatography of of Rab7-NF (black solid line) and Rab7-NF in the presence of GDI. The samples Superdex 200 gel filtration column and eluted proteins were detected either by absorbance at 280 nm (black dashed line) or by fluorescence (blue solid line) of NBD.
Fig. 7. RabGGTase in vitro prenylation assay using NBD-farnesyl pyrophosphate of Rab7-G in complex with RabGGTase (Rab7-G:GGT) or bGGT (Rab7-G: bGGT). The reaction mixtures were resolved on SDS/PAGE and analyzed by Coomassie blue staining (Top) and fluorescent scanning (Middle). Equal amount of Rab7C207S mutant were included as a positive control. (Bottom) The quantification of incorporation of NBD-farnesyl into Rab in the corresponding reactions.
Fig. 8. RabGGTase in vitro prenylation assay using NBD-farnesyl pyrophosphate of Rab7dans-G in complex with bGGT (Rab7d-G: bGGT). The reactions were performed and analyzed as above.
Fig. 9. Titration of bGGT to 40 nM Rab7-NF. (ex:479, em:542 nm). The Kd value was obtained by fitting the data to a quadratic equation.
Fig. 10. (A) Titration of REP to 5 nM Rab7-NF in the presence of the indicated concentrations of Rab7-G:bGGT. (B) Titration of GDI to 19 nM Rab7-NF in the presence of indicated concentrations of Rab7-G:bGGT.
Fig. 11. (A) Titration of REP to 7 nM Rab7-NF in the presence of the indicated concentrations of Rab7d-G:bGGT. (B) Titration of GDI to 19 nM Rab7-NF in the presence of indicated concentrations of Rab7d-G:bGGT.
Fig. 12. Titration of REP (A) and GDI (B) to 25 nM Rab7d-GG in the presence of indicated concentrations of delipidated BSA.
Fig. 13. (A and C) The emission spectra of Rab7NBD-G:bGGT in the absence (solid line) and in the presence (dashed line) of REP (A) or GDI (C), lex = 479 nm. (B and D) Titration of REP (B) and GDI (D) to the indicated concentrations of Rab7NBD-G:bGGT.
Fig. 14. (A) Elution profile of a Superdex 200 gel filtration column loaded with Rab7d-GG refolded in the presence of BSA (black line), Rab7d-GG:BSA in the presence of REP (red line), Rab7d-GG refolded by REP (blue line) and Rab7d-GG:REP low molecular mass fractions from gel filtration (lime line), detected by fluorescence with excitation at 340 nm and emission at 480 nm. (B) The same as A, elution profile was recorded by absorbance at 280 nm.
Fig. 15. Interaction analysis of diprenylated dansylated Rab7 with REP and GDI. The emission spectra of Rab7d-GG in the absence and the presence of REP (A) or GDI (B), lex = 340 nm. Titration of REP (C) or GDI (D) to 30 nM Rab7d-GG. (lex/em:340/479 nm). Kd values were obtained by fitting the data to a quadratic equation. Titration of REP (E) or GDI (F) to a mixture of 25 nM Rab7d-GG and 25 nM Rab7-G. (lex/em 340/479 nm). Kd values were obtained by fitting the data by numerical simulation where the Kd values of 0.06 nM and 1.5 nM of Rab7-G for REP and GDI respectively were fixed.
Fig. 16. (A and B) The emission spectra of Rab7C(dans)SCC in the absence and in the presence of GDI (A) and REP (B), lex = 340 nm. (C) Titration of REP to 75 nM Rab7C(dans)SCC. (ex/ em:340/486 nm). The Kd value was obtained by fitting the data to a quadratic equation.
Fig. 17. 280 nM Rab7d-GG:REP complex (A) or 240 nM Rab7d-G:REP complex (B) was incubated and at the moment of indicated by arrow 17.2 mM bGGT was added. (ex/ em: 340/503 nm). The red line shows the fit to a single exponential decay function, giving observed rate constant of 0.0019s-1.
Table 2. The Kd values of Rab7-G with REP and GDI
Concentration of Rab7-G:GGT, nM | K d(Rab7-G:REP), nM | K d(Rab7-G:GDI), nM |
5 | 1.1 | |
7.5 | 0.058 | |
10 | 0.88 | |
15 | 0.081 | |
19 | 0.079 |
The affinity of Rab7?CK(dans)SC(G)SC-OMe (Rab7d-G) to REP and GDI.
Table 3. The Kd values of Rab7dans-G with REP and GDI
Concentration of Rab7d-G:GGT, nM | K d(Rab7d-G:REP), nM | K d(Rab7d-G:GDI), nM |
6 | 0.41 | 3.5 |
10 | 2.8 | |
15 | 0.47 | 5.9 |
40 | 0.46 |
Table 4. The Kd values of Rab7NBD-G with REP and GDI
Concentration of Rab7NBD-G:GGT, nM | K d(Rab7NBD-G:REP), nM | K d(Rab7NBD-G:GDI), nM |
9 | 2.0 | |
13 | 2.5 | |
18 | 0.13 | 2.7 |
27 | 0.23 | |
36 | 0.22 |
Table 5. Observed Kd values of Rab7d-GG with REP and GDI in the presence of different concentrations of delipidated BSA
Concentration of BSA, M | Observed Kd(Rab7d-GG:REP), nM | Observed Kd(Rab7d-GG:GDI), nM |
0.24 | 1.2 | 2.2 |
1 | 1.4 | |
2 | 1.1 | |
5 | 1.0 | 7.3 |
10 | 2.6 | 8.5 |
SI Appendix
1.
Isolation of Rab7-NF and confirmation of its correct folding by its ability to undergo prenylation and form a complex with GDI.
2.
Analysis of the ability of semisynthetic Rab7-G, Rab7dans-G, Rab7NBD-G stabilized by bGGT to serve as substrates of RabGGTase.
3.
Analysis of Rab7-NF interaction with bGGT
4.
Analysis of the influence of bGGT on the interactions of Rab7G with REP and GDI
The affinity of Rab7-G to REP and GDI
The affinity of Rab7DCK(dans)SC(G)SC-OMe (Rab7d-G) to REP and GDI
The affinity of Rab7DCK(NBD)SCSC(G)-OMe (Rab7NBD-G) to REP and GDI
5.
Conformation of native folding of Rab7d-GG by complex formation with REP.
6.
The influence of BSA on the affinity of Rab7d-GG to REP and GDI.
7.
Interaction analysis of diprenylated dansylated Rab7 with REP and GDI.
As shown in SI Fig. 15 A and B the interaction between Rab7d-GG and REP or GDI results in quenching of dansyl fluorescence and a shift of emission maximum from 480 nm to 489 nm, probably reflecting reduction of hydrophobicity in the vicinity of the dansyl group. The presence of BSA at the concentration up to 10M did not have observe influence on the interaction of Rab7d-GG with REP and GDI (SI Fig. 13 and SI Table 5). The REP/GDI-induced decrease in dansyl fluorescence is specific for prenylated Rab7. No fluorescent change was observed when GDI was added to unprenylated Rab7C(dansyl)SCC, consistent with previous observations that GDI stably associates with Rab proteins only when they are modified with geranylgeranyl moiety(s) (SI Fig. 16A (1, 2)
9.
Analysis of interaction of mono- and diprenylated C terminus of Rab7 with REP in the prenylated complexes
10.
Synthesis of NH2-Cys(StBu)-Ser-Cys(NBD-farnesyl)-OH
Strategy for the Synthesis of Lipidated Fluorescent Cysteine Building Block 8
Reagents and conditions: (a) DHP, PPTS, CH2Cl2, r.t. quantitative; (b) t-BuOOH, SeO2, salycilic acid, CH2Cl2, r.t. 27%; (c) phthalimide, PPh3, DIAD, THF, r.t. 66%; (d) NH2NH2, ethanol, r.t. 53%; (e) NBD-Cl, CH3CN/25 mM NaHCO3, r.t. 60%; (f) PPTS, ethanol, 60°C, 83%; (g) NCS, DMS, CH2Cl2, -30°C to 0°C, quantitative; (h)cysteine, 7N NH3/methanol, r.t.; (i) Et3N, Fmoc-OSu, CH2Cl2, 27% for two steps
Strategy for the Synthesis of Lipidated Fluorescent Peptide 9
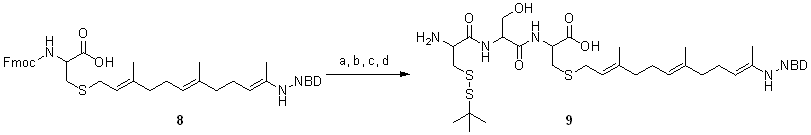
Reagents and conditions: (a) 2-Chlorotrityl chloride resin, DIPEA, CH2Cl2; (b) 20% piperidine/DMF (c) Fmoc-Ser(Trt)-OH, DIC, HOBT, DMF; b and c were repeated for Fmoc-Cys(St-Bu)-OH; (d) 1% TFA, 1% TES, CH2Cl2; overall yield 11%
Synthesis of Compound 1
![]()
To transtrans-farnesol (4.4g, 20 mmol) in 20 ml CH2Cl2 was added 2,3-dihydropyran (DHP) (2.6g, 30 mmol) and pyridinium-para-toluenesulfonate (PPTS) (0.5g, 2mmol) at room temperature and stirred overnight. The mixture was washed with saturated solution of NaHCO3, brine, and dried over MgSO4. The solution was filtered and concentrated under reduced pressure to obtain 5.7g (100%) of 1 as colourless oil. Yield: Quantitative. Rf = 0.7 (cyclohexane : ethyl acetate = 4:1). 1H NMR (400 MHz, CDCl3): d = 5.37 (dq, J = 7.5, 1.3 Hz, 1H), 5.16-5.04 (m, 2H), 4.63 (dd, J = 4.2, 2.8 Hz, 1H, O-CH-O), 4.24 (ddd, J = 11.9, 6.4, 0.7 Hz, 1H, = CH-CH2-O), 4.03 (ddd, J = 11.9, 7.4, 0.5 Hz, 1H, = CH-CH2-O), 3.90 (ddd, J = 11.3, 7.6, 3.7 Hz, 1H, O-CH2), 3.55-3.47 (m, 1H, O-CH2), 2.18-2.01 (m, 6H), 2.01-1.93 (m, 2H), 1.90-1.78 (m, 1H), 1.78-1.70 (m, 1H), 1.68 (s, 6H), 1.60 (s, 6H), 1.58-1.46 (m, 4H) ppm. 13C NMR (100 MHz, CDCl3): d = 140.2, 135.2, 131.2, 124.3, 123.8, 120.6, 97.7, 63.6, 62.2, 39.7, 39.6, 30.7, 26.7, 26.3, 25.6, 25.5, 19.6, 17.6, 16.4, 16.0 ppm.
Synthesis of Compound
2![]()
To THP protected transtrans-farnesol 1 (5.7g, 20mmol) was dissolved in 40 ml CH2Cl2 at room temperature, t-BuOOH (7.2g, 80mmol, 70% aqueous solution), SeO2 (221 mg, 2mmol) and salycilic acid (276 mg, 2mmol) were added respectively. The reaction mixture was stirred vigorously at room temperature for 2.5 h. The excess t-BuOOH was co-evaporated with toluene. The residue was re-dissolved in Et2O and the organic phase was washed with saturated solution of NaHCO3, brine and dried over MgSO4. The solution was filtered and concentrated in vacuo. The crude product was purified by silica gel chromatography to obtain 1.7g of 2 as colourless oil. Yield: 27%. Rf = 0.2 (cyclohexane : Ethyl acetate = 4:1). 1H NMR (400 MHz, CDCl3): d = 5.46-5.28 (m, 2H), 5.11 (dt, J = 6.7, 6.5, 1.1 Hz, 1H), 4.62 (t, J = 4.2 Hz, 1H, O-CH-O), 4.23 (dd, J = 11.6, 6.6 Hz, 1H, = CH-CH2-O), 4.03 (dd, J = 12.5, 6.7 Hz, 1H, = CH-CH2-O), 3.99 (s, 2H, HO-CH2), 3.89 (ddd, J = 10.9, 7.5, 3.2 Hz, 1H, O-CH2), 3.56-3.45 (m, 1H, O-CH2), 2.17-1.97 (m, 8H), 1.91-1.77 (m, 2H), 1.68 (s, 3H), 1.66 (s, 3H), 1.60 (s, 3H), 1.59-1.48 (m, 4H) ppm.
Synthesis of Compound 3

To a 20 ml solution of 2 (1.7g, 5.3mmol), phthalimide (853 mg, 5.8mmol) and PPh3 (1.5g, 5.8mmol) in THF were added DIAD (1.17g, 5.8mmol) dropwise for 0.5 h at room temperature and the reaction mixture was stirred overnight. The mixture was evaporated to dryness and Et2O was added to precipitate Ph3PO. The white suspension was filtered and the filtrate was concentrated in vacuo. The crude product was purified by silica gel chromatography to obtain 1.6 g of 3 as colourless oil. Yield: 66%. Rf = 0.3 (cyclohexane : ethyl acetate = 6:1). 1H NMR (400 MHz, CDCl3): d = 7.85 (dd, J = 5.4, 3.0 Hz, 2H), 7.71 (dd, J = 5.4, 3.0 Hz, 2H), 5.40-5.27 (m, 2H), 5.10-5.03 (m, 1H), 4.62 (dd, J = 4.3, 2.8 Hz, 1H, O-CH-O), 4.22 (dd, J = 12.4, 5.8 Hz, 1H, = CH-CH2-O), 4.19 (s, 2H, N-CH2), 4.02 (dd, J = 11.7, 7.2 Hz, 1H, = CH-CH2-O), 3.89 (ddd, J = 11.2, 7.4, 3.5 Hz, 1H, O-CH2), 3.59-3.43 (m, 1H, O-CH2), 2.25-1.91 (m, 8H), 1.90-1.78 (m, 1H), 1.77-1.68 (m, 1H), 1.66 (s, 3H), 1.64 (s, 3H), 1.56 (s, 3H), 1.56-1.51 (m, 4H) ppm. 13C NMR (100 MHz, CDCl3): d = 168.2, 140.1, 134.7, 133.8, 132.1, 129.1, 127.4, 124.2, 123.2, 120.6, 97.7, 63.6, 62.2, 44.9, 39.5, 39.1, 30.7, 26.9, 26.4, 25.5, 19.6, 15.9, 14.6 ppm. HRMS (FAB, m-NBA) m/z calc. for C28H37NO4 451.2723, found 452.2786 [M+H]+
Synthesis of Compound 4
![]()
To a 40 ml ethanol solution of 3 (1.6g, 3.5mmol) was added hydrazine and the reaction mixture was stirred at room temperature overnight. The solution was evaporated to dryness.100 ml CH2Cl2 was added and the white solid was filtered. The filtrate was concentrated in vacuo and purified by silica gel chromatography to obtain 600 mg of 4 as colourless oil. Yield: 53%. Rf = 0.25 (ethyl acetate : Et3n = 99:1). 1H NMR (400 MHz, CDCl3): d = 5.36 (ddq J = 8.7, 5.0, 1.2, 1.2, 1.2 Hz, 1H), 5.32-5.24 (m, 1H), 5.11 (dt, J = 6.8, 6.6, 1.1 Hz, 1H), 4.62 (dd, J = 4.2, 2.8 Hz, 1H, O-CH-O), 4.23 (ddd, J = 11.7, 6.3, 0.6 Hz, 1H, = CH-CH2-O), 4.03 (dd, J = 11.9, 7.4 Hz, 1H, = CH-CH2-O), 3.89 (ddd, J = 11.1, 7.6, 3.5 Hz, 1H, O-CH2), 3.55-3.46 (m, 1H, O-CH2), 3.18 (s, 2H, NH2-CH2), 2.22-1.95 (m, 8H), 1.93-1.70 (m, 2H), 1.68 (s, 3H), 1.64 (s, 3H), 1.60 (s, 3H), 1.58-1.47 (m, 4H) ppm. 13C NMR (100 MHz, CDCl3): d = 140.1, 139.8, 134.9, 124.4, 124.0, 120.6, 97.8, 63.6, 62.2, 49.7, 39.6, 39.4, 30.7, 26.3, 26.2, 25.5, 19.6, 16.4, 16.0, 14.5 ppm. HRMS (FAB, m-NBA) m/z calc. for C20H35NO2 321.2668, found 322.2756 [M+H]+
Synthesis of Compound 5

To a solution of 4 (600 mg, 1.8mmol) in 30 ml acetonitrile/25 mM NaHCO3 (1:1) was added NBD-Cl (335 mg, 1.8mmol) at room temperature. The reaction mixture was stirred for one hour, another equivalent of NBD-Cl (335 mg, 1.8mmol) was added and the mixture was stirred overnight. The mixture was poured into 200 ml CH2Cl2 and extracted twice with brine. The organic solution was dried over MgSO4 and concentrated in vacuo. The crude product was purified by silica gel chromatography to obtain 543 mg of 5 as red brown oil. Yield: 60%. Rf = 0.1 (CH2Cl2). 1H NMR (400 MHz, CDCl3): d = 8.47 (d, J = 9.1 Hz, 1H), 6.19 (d, J = 8.6 Hz, 1H), 5.53-5.45 (m, 1H), 5.38-5.30 (m, 1H,), 5.14-5.06 (m, 1H), 4.62 (dd, J = 4.2, 2.8 Hz, 1H, O-CH-O), 4.24 (ddd, J = 11.9, 6.2, 0.7 Hz, 1H, = CH-CH2-O), 4.09-4.02 (m, 1H, = CH-CH2-O), 4.02-3.99 (m, 2H, NBD-NH-CH2), 3.94-3.85 (m, 1H, O-CH2), 3.59-3.44 (m, 1H, O-CH2), 2.45-1.92 (m, 8H), 1.92-1.78 (m, 2H), 1.73 (s, 3H), 1.67 (d, J = 0.6 Hz, 3H), 1.60 (d, J = 0.8 Hz, 3H), 1.58-1.47 (m, 4H) ppm. 13C NMR (100 MHz, CDCl3): d = 143.8, 140.8, 139.8, 136.2, 134.3, 132.9, 129.3, 128.8, 128.5, 124.6, 120.7, 99.1, 97.9, 63.7, 62.4, 51.5, 39.5, 39.0, 30.7, 26.2, 26.2, 25.4, 19.6, 16.4, 15.9, 14.7 ppm.
Synthesis of Compound 6

To a solution of 5 (543 mg, 1.1mmol) in 20 ml ethanol was added pyridinium-para-toluenesulfonate (PPTS) (562 mg, 2.24mmol). The reaction mixture was stirred at 60°C for 2 h and poured into 200 ml Et2O. The mixture was washed twice with brine and dried over MgSO4. The organic solution was filtered and evaporated to dryness. The crude product was purified by silica gel chromatography to obtain 370 mg of 6 as red brown oil. Yield: 83%. Rf = 0.3 (cyclohexane : ethyl acetate = 2:1). 1H NMR (400 MHz, CDCl3): d = 8.46 (d, J = 8.6 Hz, 1H), 6.72 (t, J = 4.9, 4.9 Hz, 1H, NH), 6.16 (d, J = 8.6 Hz, 1H), 5.45 (dt, J = 7.1, 7.1, 1.0 Hz, 1H), 5.37 (dt, J = 7.0, 7.0, 1.3 Hz, 1H), 5.08 (dt, J = 6.8, 6.6, 1.0 Hz, 1H), 4.17 (d, J = 6.8 Hz, 2H, NBD-NH-CH2), 4.00 (d, J = 5.5 Hz, 2H, = CH-CH2-OH), 2.28-2.05 (m, 6H), 2.03-1.96 (m, 2H), 1.73 (s, 3H), 1.66 (s, 3H), 1.59 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3): d = 144.2, 144.0, 139.2, 136.3, 134.3, 128.8, 128.5, 124.5, 124.2, 123.4, 123.1, 99.1, 59.4, 51.3, 39.3, 38.9, 26.0, 26.0, 16.2, 15.9, 14.7 ppm. HRMS (FAB, m-NBA) m/z calc. for C21H28N4O4 400.2111, found 400.2126 [M]+
Synthesis of Compound 7

To NCS (40 mg, 0.30mmol) in dry CH2Cl2 under argon atmosphere was added Dimethylsulfide (DMS) (42 mg, 0.67mmol) dropwise at -30°C. The mixture was stirred for 5 min at 0°C and cooled again to -30°C. A solution of the alcohol 6 (120 mg, 0.30mmol) in 1 ml dry CH2Cl2 was added and the mixture was stirred for 2 h at 0°C. The mixture was poured into CH2Cl2 and the organic phase was washed twice with brine, dried with MgSO4 and concentrated in vacuo. The crude product 7 was used for the next step without further purification. Yield: quantitative. Rf = 0.4 (cyclohexane : ethyl acetate = 3:1).
Synthesis of Building Block 8

To 7 (125 mg, 0.30mmol) in 4 ml methanol was added cysteine. HCl.H2O (52 mg, 0.3mmol) and 4 ml of 7N NH3 in methanol at room temperature. The reaction mixture was stirred overnight. The mixture was evaporated to dryness to obtain a red-brown solid. To the crude red-brown solid in 5 ml CH2Cl2 was added Et3N (61 mg, 0.6mmol) and Fmoc-OSu (101 mg, 0.3mmol) at 0°C. The mixture was stirred at room temperature for 2 h. The reaction mixture was poured into 50 ml Et2O and extracted with 1M HCl. The organic layer was washed with brine and dried over MgSO4. The organic solution was filtered and concentrated in vacuo. The crude product was purified by silica gel chromatography to obtain 60 mg 8 as red brown oil. Yield: 27%. Rf = 0.5 (CH2Cl2 : meOH = 9:1). LC-MS (C4) 726.0 [M+H]+, 748.2 [M+Na]+; Rt = 10.40 min. 1H NMR (400 MHz, CD3OD): d = 8.45 (d, J = 8.7 Hz, 1H), 7.84 (d, J = 7.2 Hz, 2H), 7.67 (t, J = 6.9, 6.9 Hz, 2H), 7.38 (t, J = 7.9, 7.9 Hz, 2H), 7.29 (t, J = 7.3, 7.3 Hz, 2H), 6.25 (d, J = 8.7 Hz, 1H), 5.80-5.62 (m, 1H), 5.45-5.25 (m, 1H), 5.23-4.94 (m, 2H), 4.59 (dd, J = 12.5, 5.5 Hz, 1H), 4.46-4.33 (m, 3H), 4.22 (t, J = 7.1, 7.1 Hz, 1H), 4.17 (d, J = 6.8 Hz, 2H), 3.40-3.07 (m, 2H), 3.05-2.85 (m, 2H), 2.07-1.89 (m, 8H), 1.65 (s, 3H), 1.63 (s, 3H), 1.57 (s, 3H) ppm. HRMS (ESI) m/z calc. for C39H43N5O7S 725.2883, found 726.2956 [M+H]+
Synthesis of Lipidated Fluorescent Peptide 9

Attachment of 8 to 2-Chlorotrityl Chloride Resin.
To 8 (25 mg, 3.4mmol) and DIPEA (17 mg, 1.4mmol) in dry CH2Cl2, the mixture was added to 2-Chlorotrityl chloride resin (30 mg, 4.1mmol) and stirred for 2 h at room temperature. The solvent was filtered off and the resin was washed three times with CH2Cl2/MeOH/DIPEA (17:2:1), three times with DMF, three times with CH2Cl2, and three times with DMF.
Removal of Fmoc Protecting Group.
The resin was washed 3 times with 3 ml 20% piperidine/DMF (5 min each time) to remove the Fmoc group from the amino acid. Subsequently, the resin was washed again three times with DMF, three times with CH2Cl2, three times with DMF (each time 2 to 5 ml).
Attachment of Fmoc-Ser(Trt)-OH.
To Fmoc-Ser(Trt)-OH (58 mg, 1mmol) and HOBT ( 16 mg, 1mmol) in 2 ml DMF was added DIC (13 mg, 1mmol), the mixture was stirred for 1 min and was added to the resin at room temperature. The solvent was filtered off and the resin was washed three times with DMF, three times with CH2Cl2, three times with DMF (each time 2 to 5 ml).
Release of Peptide 9 from 2-Chlorotrityl Chloride Resin.
To resin bound 9 in 2 ml CH2Cl2 was added 1% TFA and 1% TES. The mixture was shaken for 1 h. The solvent was filtered, collected, and washed three times with CH2Cl2, three times with methanol and three times with CH2Cl2. The collected solvent was co-evaporated with toluene till dryness. The crude product was purified by semipreparative HPLC (C4 column) and lyophilized to obtain 3 mg of 9 as red solid. Yield: 11%
LC-MS
(C4) 782.1 [M+H]+, 804.3 [M+Na]+; Rt = 7.50 min. 1H NMR (400 MHz, CD3OD) d = ppm 8.50 (d, J = 8.9 Hz, 1H), 6.28 (dd, J = 8.9, 0.8 Hz, 1H), 5.47-5.44 (m, 1H), 5.19-5.09 (m, 1H), 5.08-5.00 (m, 1H), 4.64-4.51 (m, 1H), 4.16 (dd, J = 8.6, 4.9 Hz, 1H), 4.10-4.01 (m, 2H), 3.83 (dd, J = 7.5, 5.6 Hz, 2H), 3.49-3.46 (m, 1H), 3.13 (dd, J = 3.3, 1.6 Hz, 1H), 3.06 (dd, J = 14.4, 8.7 Hz, 1H), 2.99 (dd, J = 14.0, 4.6 Hz, 1H), 2.78 (dd, J = 13.7, 7.8 Hz, 1H), 2.27-2.12 (m, 2H), 2.07-1.98 (m, 8H), 1.70 (s, 3H), 1.63 (s, 3H), 1.57 (s, 3H), 1.36 (s, 9H) ppm HRMS (ESI) m/z calc. for C34H51N7O8S3 781.2961, found 782.3036 [M+H]+
1. Pylypenko O, Rak A, Durek T, Kushnir S, Dursina BE, Thomae NH, Constantinescu AT, Brunsveld L, Watzke A, Waldmann H, et al. (2006) EMBO J 25:13-23.
2. Musha T, Kawata M, Takai Y (1992) J Biol Chem 267:9821-9825.