Inoue et al. 10.1073/pnas.0700901104. |
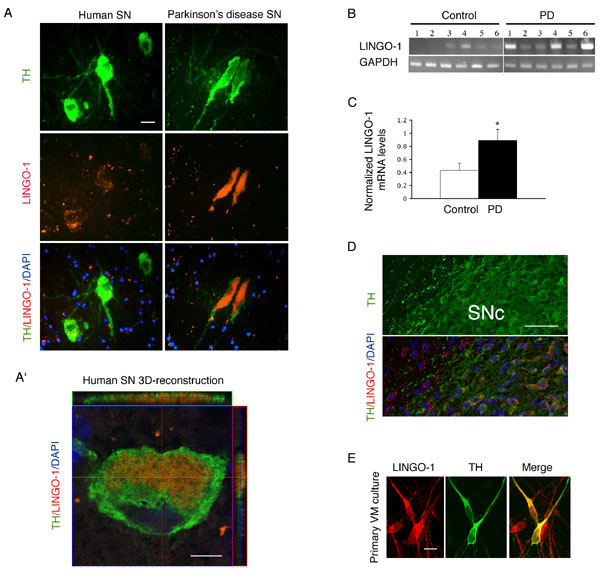
Fig. 4. Expression of LINGO-1 in dopamine (DA) neurons of the ventral midbrain (VM). (A) In situ hybridization of LINGO-1 in the substantia nigra (SN) of PD patients (Right) (n = 6) and aged-matched controls (human SN; Left) (n = 6). Green, stained with anti-TH antibody; red, LINGO-1 mRNA; blue, DAPI nuclear stain. Merging showing overlaps of the LINGO-1 and TH cells. (A') Localization of LINGO-1 mRNA (red) and TH (green) in an orthogonal reconstruction of a confocal optical series. (B and C) Semiquantitative RT-PCR showing a significant elevation in LINGO-1 levels in the PD SN compared with controls. (D) In situ hybridization of LINGO-1 in adult rat midbrain sections. Green, stained with anti-tyrosine hydroxylase (TH) antibody. Yellow color reflects the merging of the LINGO-1 and TH stains. (E) Immunohistochemistry staining of LINGO-1 and TH in embryonic day 15 rat VM primary cultures. Green, stained with anti-TH; red, stained with anti-LINGO-1; yellow, merging LINGO-1 and TH staining. (Scale bars: 20 mm, A; 5 mm, A'; 50 mm, D; 10 mm, E).
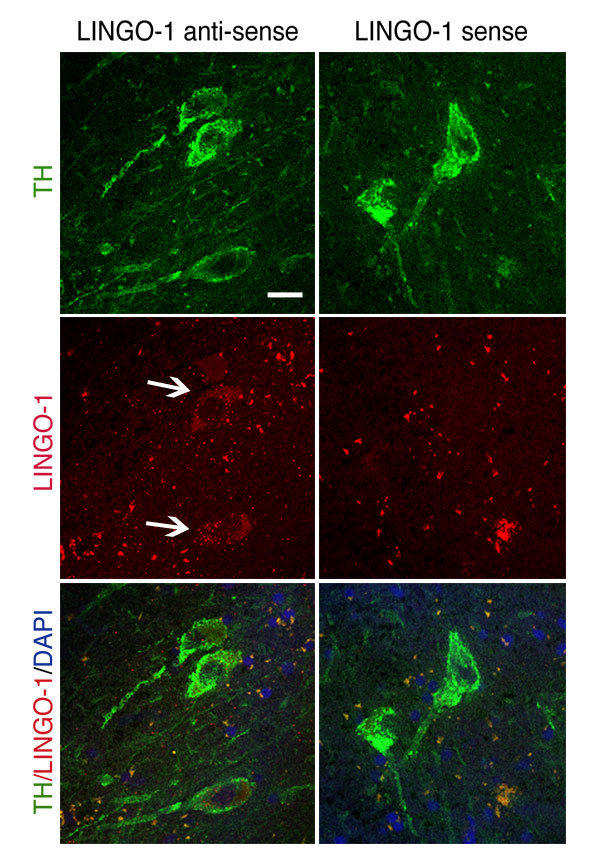
Fig. 5. (Left) LINGO-1 antisense hybridization of TH-labeled neurons in SN of human cases (controls). (Right) The LINGO in situ sense control probe did not label any cellular structure or neuron. (Scale bar: 20 mm.)
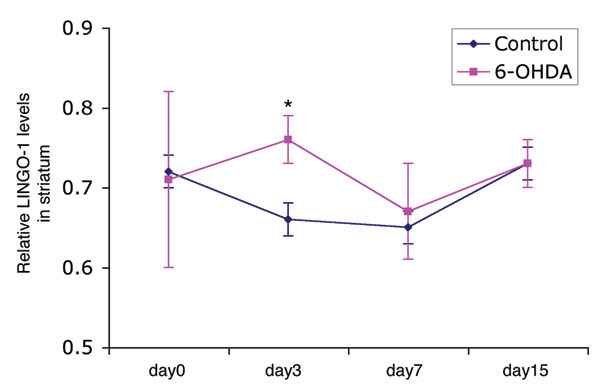
Fig. 6. Changes of LINGO-1 protein levels after 6-OHDA lesion. LINGO-1 is significantly up-regulated in the lesioned side (6-OHDA) compared with the contralateral side (control) 3 days after striatal 6-OHDA administration in wild-type mice (n = 3 each time point; unpaired Student's t test: *, P < 0.05).

Fig. 7. DAT levels in LINGO-1 KO and WT mice (n = 6 each group). DAT levels are not different in KO and WT mice (unpaired Student's t test: P > 0.05).

Fig. 8. LINGO-1 suppresses p-EGFR and EGFR levels. Shown are COS7 cells infected with LINGO-1 lentivirus as 0, 1, and 5 MOI per cell for 2 days and treated with or without EGF for 5 min. EGFR levels were markedly reduced regardless of EGF stimulation. Phosphorylated EGFR was down-regulated with the EGF treatment.

Fig. 9. 1A7 suppresses inhibitory effects of LINGO-1 on EGFR activity. Shown is Western analysis of PC12 cells transduced with LINGO-1 for 2 days and treated with a LINGO-1 blocking antibody (1A7) for 6 h. We observed that p-Akt levels were higher in the 1A7-treated cells than in the controls.
Fig. 10. Schematic drawing depicts possible mechanisms involved in the interactions of LINGO-1 and EGFR. (A) Normally, EGFRs become dimerized and phosphorylated upon ligand binding and stimulate the PI3K-Akt neuronal survival pathway. (B) Binding of LINGO-1 to EGFR might accelerate EGFR internalization and degradation, thus decreasing the availability of EGFR. (C) LINGO-1 may directly inhibit EGFR phosphorylation and reduce PI3-K-Akt signaling pathway.
SI Methods
In Situ
Hybridization.Rat brain or human SN tissues were embedded by Tissue-Tek OCT (Sakura Finetechnical, Japan). The frozen sections were prepared and processed and were probed with digoxigenin-labeled LINGO-1 antisense and sense RNA as previously described (1). Sections were stained by using the TSA plus fluorescence and anti-digoxigenin conjugated antibodies kit (PerkinElmer, Wellesley, MA) following the manufacturer's instructions. Sections were then stained with anti-TH (Chemicon, Temecula, CA) and DAPI (Sigma, St. Louis, MO). To control for any autofluorescence caused by neuromelanin, neuromelanin was removed by a bleaching procedure by incubating sections in 0.25% potassium manganese peroxide (30 min) and in 5% oxalic acid (5 min) as previously described (2). The LINGO-1 expression pattern was not affected by bleaching out the neuromelanin (SI Fig. 7).
Semiquantitative RT-PCR.
mRNA extracted (Absolutely RNA miniprep kit; Stratagene) from human SN tissues was subjected to RT-PCR for LINGO-1 by using forward primer (AGAGACATGCGATTGGTGA) and reverse primer (AGAGATGTAGACGAGGTCATT).
HPLC Assay for Dopamine and MPP+ Levels.
For dopamine and metabolites, dissected striata were sonicated and centrifuged in chilled 0.1 M perchloric acid (PCA, ~100 ml per milligram of tissue). The supernatants were taken for measurements of dopamine as described previously (3). Briefly, 15 ml of the supernatant was isocratically eluted through an 80 ´ 4.6 mm C18 column (ESA, Chelmsford, MA) with a mobile phase containing 0.1 M LiH2PO4, 0.85 mM 1-octanesulfonic acid, and 10% (vol/vol) methanol and detected by a two-channel Coulochem II electrochemical detector (ESA). Concentrations of dopamine were expressed as nanograms per milligram of protein. Protein concentration of tissue homogenates was determined by using the Bradford method (Bio-Rad, Hercules, CA) and PerkinElmer Bio Assay Reader (Norwalk, CT). For measuring MPP+, striatal tissues were sonicated and centrifuged in 0.1 M PCA and an aliquot of supernatant was injected onto a META 250 ´ 4.6 C18 column (ESA). Samples were eluted isocratically with 20 mM boric acid-sodium borate buffer (pH 7.75) containing 3 mM tetrabutylammonium hydrogensulfate, 0.25 mM 1-heptanesulfonic acid, and 10% isopropanol. MPP+ were detected with a fluorescence detector set by excitation at 295 nm and emission at 375 nm (4).
VM Primary Cultures.
Primary cultures of DA neurons were obtained from embryonic day 15 Sprague-Dawley rat (Charles River, Wilmington, MA) VM as described (5). Briefly, tissue was mechanically dissociated with polished Pasteur pipettes in cold DMEM (Invitrogen, Carlsbad, CA) containing heat-inactivated horse serum (10%), glucose (6.0 mg/ml), penicillin (1000 units/ml), streptomycin (10 mg/ml; Sigma), and glutamine (2 mM; Gibco). A total of 2 ´ 105 cells were resuspended in the medium and seeded on a coverslip precoated with 15 mg/ml polyL-ornithine (Sigma) and 1 mg/ml fibronectin (Sigma) of each well of 24-well trays (Becton Dickinson, San Jose, CA). Unattached primary VM cultured cells were aspirated after 1 h, and 1 ml of fresh medium containing each group of virus was added at 5 MOI per cell. For the TH neurite outgrowth assay, lentivirus transduced (DN-LINGO-1, FL-LINGO-1, or GFP) cells were cultured for 24 h and then fixed with 4% paraformaldehyde in PBS (pH 7.4) for 30 min at room temperature. To investigate the protective effects of LINGO-1 antagonist in MPP+-treated cultures, LINGO-1 antagonists (10 mg/ml for both LINGO-1-Fc and 1A7) were added 8 h before MPP+ (10 mM) exposure at day 4. Cultures were fixed 48 h later. For Western blot analysis for Akt, pAkt, HA tag, and b-actin, 2.5 ´ 106 VM cells were seeded in six-well culture plates and then followed the same procedure as described above. Each experiment was repeated in triplicate.
Western Blotting.
The cultured VM cells, or mouse tissues, were lysed in lysis buffer (50 mM Tris·HCl, pH 7.5/150 mM NaCl/1 mM EDTA/1% Triton X-100/1% sodium deoxycholate/0.1% SDS) containing protease inhibitors and phosphatase inhibitors (Sigma) as previously described (1). The supernatants were electrophoresed on either 4-20% or 10% SDS/PAGE gels (Bio-Rad). The membranes were probed with anti-phospho-Akt, anti-Akt (Cell Signaling Technology, Beverly, MA), anti-EGFR, and anti-DAT antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoblotting for b-actin served as loading control.
Immunoprecipitation.
HEK 293 cells (100-mm dishes) were transfected with LINGO-1, EGFR, and LINGO-1/EGFR. The cells were harvested after 48 h and lysed in 1 ml of RIPA buffer (50 mM Tris, pH 7.2/1% Triton X-100/0.5% sodium deoxycholate/0.1% SDS/150 mM NaCl/10 mM MgCl2/5% glycerol) for 30 min at 4°C. After centrifugation at 14,000 ´ g for 15 min, the supernatants were incubated with protein A/G-Sepharose beads (Santa Cruz Biotechnology) at 4°C for 1 h. A total of 200 ml of the precleared lysates were then incubated with either anti-EGFR at 4°C for 1 h (Santa Cruz Biotechnology) or anti-LINGO-1 antibody (Biogen, Cambridge, MA) followed by treatment with protein A/G-Sepharose beads. The beads were washed three times with buffer (50 mM Hepes, pH 7.5/150 mM NaCl/1.5 mM MgCl2/1 mM EGTA/1% Triton X-100/5% glycerol), boiled in Laemmli sample buffer, subjected to 4-20% SDS/PAGE, and analyzed by Western blotting with anti-EGFR antibody (Santa Cruz Biotechnology) or the anti-LINGO-1 antibodies 1A7 and 2F3 (Biogen). The EGFR and LINGO-1 were visualized by using HRP-conjugated secondary antibodies. LINGO-1 KO or WT VM tissues were lysed in RIPA buffer and precleared by protein A/G plus-Sepharose beads as described above. One milligram of the VM cell extracts was then subjected for immunoprecipitations with an anti-LINGO-1 antibody (Upstate Biotechnology, Lake Placid, NY) at 4°C overnight, followed by 1 h of incubation with protein A/G plus-Sepharose beads.
Histological and Stereological Procedures.
Animals were killed with CO2. The brains were quickly removed and fixed with 10% neutral buffered formalin for 48 h. Brains were equilibrated in 30% sucrose in PBS for cryoprotection and sectioned serially. Routine ABC immunohistochemistry was performed on randomly selected series of sections that contain every sixth of the total VM. Sections were incubated with anti-TH antibody (Chemicon) overnight at 4°C, followed by incubation with biotinylated secondary antibody (1:300; Vector Laboratories, Burlingame, CA) and with streptavidin-biotin complex each for 1 h at room temperature. Staining was visualized by incubation in 3,3'-diaminobenzidine solution with nickel enhancement (Vector Laboratories). Omission of the primary antibody served as control. Stereological analysis (6, 7) was performed by investigators blind to group treatments. Stereology was performed on the stained sections by using an integrated Axioskop 2 microscope (8) (Carl Zeiss, Thornwood, NY) and Stereo Investigator image capture equipment and software (MicroBrightField, Williston, VT). TH-positive cell bodies were counted by using an optical fractionator probe. The midbrain sections were viewed at a ´4 objective, and SN and VTA were outlined by using the third cranial nerve as a landmark to define the vertical border between SN and VTA and by using the medial lemniscus as their dorsal border. At a random start, a counting frame (110 ´ 110 mm) was used to count TH positive cells at 150-mm steps in the x-y axis via the ´40 objective. Tissue had been sectioned at 40 mm. Fixation times and other histological procedures were adjusted to minimize tissue shrinkage. Tissue thickness was measured before stereology design, and the dissector height was set at 12 mm for unbiased stereological estimation of the total cell number (7). Additionally, tissue thickness was measured on each section at every fifth counting site to obtain a more accurate estimation of tissue thickness and reduce error as previously described (7). Using a sampling ratio of 1:6, we counted six to seven sections per animal. The number of sampling sites and other stereological parameters were experimentally determined to obtain an overall Gundersen coefficient of variance <0.1 for each case, thereby satisfying all criteria for objective stereological cell counts (6). The DAPI-stained nuclei in the VM cultures were counted by using an optical fractionator of the Stereo Investigator (the counting frame was 300 ´ 300 mm, and the grid size was 4000 ´ 4000 mm). The estimated cell number per coverslip was obtained by counting eight fields by using randomized rotation.
Immunofluorescence.
Routine fluorescent immunohistochemistry was performed for VM primary cultures (5). In brief, coverslips were pretreated with 10% normal goat serum (The Jackson Laboratory, Bar Harbor, ME) and 0.1% Triton X-100 in 0.1 M PBS for 30 min at room temperature, followed by incubation with primary antibodies at 4°C overnight, and then with appropriate secondary antibodies conjugated with distinct fluorescence at room temperature for 1 h. A primary antibody against TH (1:300; Chemicon) was used. A secondary antibody conjugated with Alexa Fluor 488 (1:500; Molecular Probes, Eugene, OR) was used. Omission of primary antibodies or antibodies preincubated with excess antigens was used as control. Fluorescent signal was examined by a confocal imaging system (LSM510 META; Carl Zeiss). For analysis of neurite extension or cell numbers, several fields from each coverslip were captured by using an integrated Axioskop 2 microscope (Carl Zeiss). Stereo Investigator image capture equipment and software (MicroBrightField) was then used to make cell counts and measurements of neurite length by investigators blinded to coverslip identities.
Human Samples.
Postmortem tissues were obtained with consent from the Harvard Brain Tissue Resource Center (Belmont, MA) (see SI Table 1).
1. Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, et al. (2004) Nat Neurosci 7:221-228.
2. Schneider JA, Bienias JL, Gilley DW, Kvarnberg DE, Mufson EJ, Bennett DA (2002) J Histochem Cytochem 50:99-106.
3. Yang L, Calingasan NY, Chen J, Ley JJ, Becker DA, Beal MF (2005) Exp Neurol 191:86-93.
4. Cleren C, Starkov AA, Calingasan NY, Lorenzo BJ, Chen J, Beal MF (2005) Neurobiol Dis 20:701-708.
5. Lin L, Rao Y, Isacson O (2005) Mol Cell Neurosci 28:547-555.
6. Slomianka L, West MJ (2005) Neuroscience 136:757-767.
7. West MJ, Slomianka L, Gundersen HJ (1991) Anat Rec 231:482-497.
8. Sanchez-Pernaute R, Ferree A, Cooper O, Yu M, Brownell AL, Isacson O (2004) J Neuroinflammation 1:6.