
Liverman et al. 10.1073/pnas.0703196104. |
Fig. 7. Induction of actin filaments by V. para depends on VopL. HeLa cells were either mock-infected (A and B) or infected with POR-2 (C and D), POR-2DVopL (E and F), POR-2DVopL+pLM1877-VopL (KO+VopL) (G and H), or POR-2DVopL+pLM1877 (KO+Vector) (I and J) at a multiplicity of infection of 25 for 3 h. Infected cells were analyzed by confocal microscopy using rhodamine-phalloidin to stain actin and Hoechst to stain nuclei and bacteria. Images are shown at ´100 magnification. (K) Secretion of VopL by the V. para strains detected by analysis of secreted proteins that were TCA-precipitated from culture supernatants and analyzed on immunoblots using a rabbit anti-VopL antibody.

Fig. 8. Transfection of VopL into NIH 3T3 cells induces formation of actin fibers. Shown are images from confocal microscopy using rhodamine-phalloidin to stain for actin in NIH 3T3 cells transfected with peGFP-N1 (GFP) and vector (A) or pSFFV-VopL-FLAG (B). (C) Quantitation of transfected cells with increased actin fibers in a double blind study.

Fig. 9. VopL activity is unaffected by dominant negative Rac1 and CDC42. Shown are images from confocal microscopy using rhodamine-phalloidin to stain for actin in HeLa cells transfected with peGFP-N1 (GFP) and vector (A), pSFFV-VopL-FLAG (B), pcDNA3-(HA)3-Rac1 T17N (Rac1DN) (C), Rac1DN+VopL (D), pcDNA3-(HA)3-CDC42 T17N (CDC42DN) (E), or CDC42DN+VopL (F). (G) Quantitation of transfected cells with increased actin fibers in a double blind study.
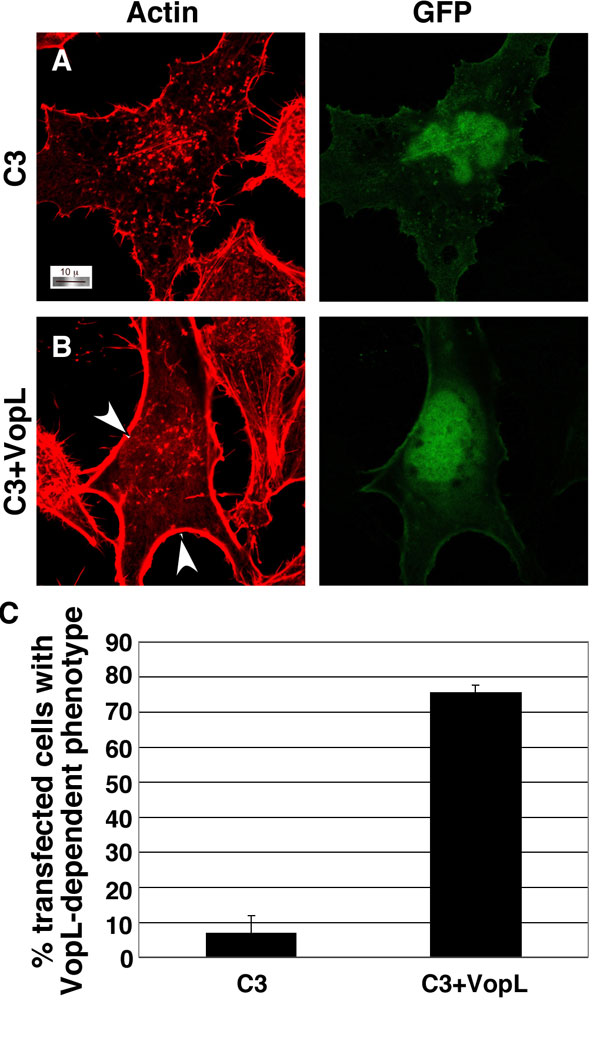
Fig. 10. VopL activity is affected by coexpression of C3 exoenzyme with VopL in HeLa cells. Shown are images from confocal microscopy using rhodamine-phalloidin to stain for actin in HeLa cells transfected with peGFP-N1 (GFP) and pCMV5-C3 exoenzyme alone (A) or C3 exoenzyme + pSFFV-VopL-FLAG (B). White arrowheads indicate strong actin staining observed around the periphery of cells transfected with both C3 and VopL. (C) Quantitation of transfected cells with the VopL-dependent phenotype in a double blind study.

Fig. 11. Mutation of the WH2 domains affects VopL activity in HeLa cells. Shown are confocal images of rhodamine-phalliodin-stained (Rh-Ph) HeLa cells cotransfected with peGFP-N1 (GFP) as a marker of transfected cells and vector (A), VopL-Flag (B), VopL-WH2-1* (C), VopL-WH2-2* (D), VopL-WH2-3* (E), VopL-WH2*1&2 (WH2-1* and WH2-2*) (F), or VopL-WH2 ´ 3* (G). WH2* indicates one or more mutated domain(s). All constructs are expressed from pSFFV. Images are shown at ´100 magnification.

Fig. 12. VopL-nucleated filaments grow at their barbed ends. Data are from pyrene actin polymerization assays in the presence of actin (1 mM, 20% pyrene), 5 nM VopL-WH2C, and mouse capping protein a1b2 (CP) from 0.2 nM to 5 nM.
SI Text
SI Materials and Methods
Bioinformatics.
Proteins from Vibrio parahaemolyticus (V. para) were examined by using the BLAST (www.ncbi.nlm.nih.gov/BLAST) and SMART (smart.embl-heidelberg.de) protein databases. The WH2 alignment was created in MacVector using the Clustal W alignment function and BLOSUM 30 matrix.Cell Culture and Transfections.
HeLa and NIH 3T3 cells were obtained from American Type Culture Collection and passaged under standard conditions. Approximately 1.5-2.0 ´ 105 cells were plated into six-well plates containing glass coverslips. Cells were transfected with FuGENE (Roche Diagnostics) according to the manufacturer's instructions. A total of 2.2 mg of DNA was transfected per well. A total of 1 mg of pSFFV-VopL or pSFFV-VopL WH2 mutant was transfected per well, and 100 ng of the RhoA, Rac1, or CDC42 and 200 ng of peGFP-N1 expression vectors were transfected per well where indicated. C3 exoenzyme expression vector is transfected at 500 ng per well. All transfections were carried out for 24 h (except for the transfections with C3 exoenzyme, which were carried out for 17 h) before being fixed and stained in preparation for confocal microscopy.Vectors and Strains.
VopL was cloned into pSFFV (1) by PCR (Vent polymerase; New England Biolabs) by using specific primers (Flag-tagged C-terminal) and Vibrio genomic DNA (Dneasy Tissue Kit; Qiagen). pSFFV-VopL-Flag was then used as the template for cloning VopL or VopL-WH2C (90-483 aa) (without the FLAG tag) into the recombinant protein expression vector, pGEX-rTEV, which produces an N-terminal GST fusion protein (2). All WH2 mutants (SI Figs. 11 C-G and 12) are point mutants that were generated by using the Stratagene QuikChange mutagenesis kit according to the manufacturer's instructions and using pSFFV-VopL-Flag as the template. Four amino acids in each WH2 domain were changed to alanine, as illustrated in Fig. 4I. Expression plasmids containing dominant negative (pcDNA3-(HA)3-RhoA T19N) RhoA, Rac1 T17N, and CDC42 T17N were obtained from the Guthrie cDNA Resource Center. pEGFP-N1 was obtained from BD Biosciences Clontech. pLM1877 was obtained from L. McCarter (3). pCMV5-C3 was a kind gift from P. C. Sternweis. V. para strains POR-1, POR-2, and POR-3 were obtained from T. Iida and T. Honda (4, 5). Routine culture of all Vibrio strains is carried out in either LB plus 3%NaCl (MLB) or HI with 3% NaCl (MHI) at 30°C.Gene Disruption.
POR-2DVopL was constructed by using a method adapted from Datsenko and Wanner (6) and using vectors (pLAFR and pPHIJI) and an Escherichia coli strain containing pRK2013, a tra donor plasmid, kindly provided by L. McCarter. Briefly, VopL and 1Kb of upstream and downstream flanking genomic sequence was cloned into pLAFR (Tetr) by using specific primers and V. para genomic DNA. The entire VopL coding sequence was replaced with a chloramphenicol resistance cassette by lambda red recombination in E. coli. Triparental matings were then performed between the E. coli strain harboring the VopL deletion, an E. coli strain containing the tra donor pRK2013, and POR-2 at 37°C overnight on heart infusion agar plates (HI agar). Transconjugants were selected on minimal media containing 20 mg/ml tetracycline (Tet) and 3% NaCl, then passaged once on MHI agar with Tet, then five times on MHI agar with 10 mg/ml chloramphenicol (Cm) to allow for allelic exchange of the chloramphenicol cassette for VopL. The pLAFR plasmid carrying the genomic copy of VopL was eliminated by conjugating in an incompatible plasmid, pPHIJI (Genr). The positive (Genr, Cmr) clones were confirmed by PCR and TTSS-dependent secretion assays. Positive clones were cured of the pPHIJI plasmid by successive passage without Gen at 30°C.Confocal Microscopy.
Confocal microscopy was performed on a Zeiss LSM 510 laser scanning microscope (using the ´100 objective, a 0.7-mm optical slice, 1,024 ´ 1,024 resolution or 2,048 ´ 2,048 resolution) and the accompanying software package. Images were prepared by using ImageJ software for Macintosh and Adobe Photoshop. HeLa cells were fixed in 4% paraformaldehyde and stained with rhodamine-phalloidin (Molecular Probes) and Hoechst (Sigma) using standard techniques. Immunocytochemistry was performed by using standard techniques. Mouse monoclonal anti-vinculin, mouse anti-flag, and FITC-conjugated goat anti-mouse and rabbit IgG antibodies are all from Sigma. Rabbit anti-myosin IIA antibodies are from Covance. Polyclonal rabbit anti-VopL antibodies were generated by Strategic Biosolutions (Newark, DE) using purified recombinant VopL, and Alexa Fluor 680 goat anti-rabbit IgG is from Invitrogen. Funding for the confocal microscope was provided by National Institutes of Health Shared Instrumentation Grant 1-S10-RR19406.Secretion Assay.
Overnight cultures of V. para strains were diluted back to 0.05 OD600 in MLB. Cultures were then grown for 4.5 h at 37°C. Secreted proteins were precipitated from the filtered supernatant of 6 OD600 equivalents of the culture and supplemented with 66 mg of BSA as a load control. TCA was added to a final concentration of 10%, and proteins were incubated on ice. After 1 h, the protein was pelleted at 4°C at 14,000 rpm in a microcentrifuge and washed in ice-cold acetone. Pellets were allowed to air-dry and resuspended in SDS sample buffer. Samples were analyzed by Western blot with anti-VopL and Coomassie staining.Infection Assay.
HeLa cells were plated at a density of 0.5 ´ 105cells per milliliter on coverslips in 35-mm dishes. Vibrio strains were grown in LB overnight at 30°C, diluted back to 0.2 OD600, and grown at 30°C for 2 h. Bacteria were pelleted and resuspended in DMEM. HeLa cells were infected with Vibrio at a multiplicity of infection of 25 for 3 h. At the end of infection HeLa cells were washed with PBS and fixed in 4% paraformaldehyde in PBS. Cells were permeabilized in PBS with 0.1% Triton X-100 and stained with Hoechst and rhodamine-phalloidin. Slides were viewed on the confocal microscope.Protein Purification.
Bacterial cell pellets from 1 liter of cells were thawed, resuspended in 20 mM Tris, pH 8.0/100 mM NaCl/10 mM DTT/1 mM EDTA/1% CHAPS/10% glycerol/1 mg/ml antipain/1 mM benzamidine/1 mg/ml leupeptin/0.5 mg/ml pepstatin/1 mM PMSF, and lysed by three passes through a cell disrupter (Avestin, Ottawa, Canada) at 10,000 psi. Cleared lysates were incubated with 12 ml of glutathione Sepharose beads (Amersham Pharmacia) for 30 min. The beads were then washed with 120 ml of 20 mM Tris, pH 8.0/800 mM NaCl/10 mM DTT/1 mM MgCl2/5 mM ATP/0.1% CHAPS/10% glycerol/1 mM benzamidine at 25°C; GST-VopL was eluted with 20 mM Tris, pH 8.0/100 mM NaCl/10 mM DTT/15 mM L-glutathione/0.1% CHAPS/10% glycerol/1 mM benzamidine. GST was cleaved from the recombinant protein at 25°C for 3 h by using His-Tev protease (Invitrogen) and monitored by SDS/PAGE. VopL was separated from GST and His-Tev proteins by gel filtration chromatography using a HiLoad Superdex 200 column (Amersham Pharmacia) as a final purification step. Purification of VopL mutant proteins were similar to the wild type, except that the VopL-WH2C construct (90-483 aa) was subjected to cation exchange and anion exchange chromatographies before gel filtration. TEV-cleaved VopL-WH2C was buffer-exchanged against 50 mM sodium phosphate, pH 6.0/5 mM DTT/1 mM EDTA/1 mM benzamidine and loaded onto a Mono S 10/100 column (GE Healthcare) equilibrated in the same buffer. VopL-WH2C was eluted with a 0-500 mM NaCl gradient over 10 column volumes (CV). Fractions containing VopL-WH2C were pooled and desalted into 20 mM Tris, pH 8.0/5 mM DTT/1 mM EDTA/1 mM benzamidine before loading onto a Mono Q 10/100 column (GE Healthcare). VopL-WH2C was eluted with a 0-100 mM NaCl gradient over 5 CV. Bovine Arp2/3 complex, human WASP VCA, actin from rabbit skeleton muscle, and mouse capping protein were obtained as described previously (7-9).Quantitation of Transfections.
Quantitation of transfections was performed by counting at least 150 transfected cells per slide, from three independent experiments per experimental condition, by three individuals. At least 10 digital images of transfected cells from each experimental condition within each experiment were randomized and counted by each individual independently to decide if the cell appeared (i) like a control cell (Fig. 1C) or (ii) like a cell with increased number of stress fibers (Fig. 1G). In the case of C3 transfected cells, using the same experimental design, each individual independently decided if the cell appeared (i) like a C3-transfected cell (SI Fig. 10A) or (ii) not like a C3-transfected cell (SI Fig. 10B). The results were totaled for each person, converted into percentage of cells displaying VopL-induced actin fibers, and averaged, and they are presented with standard deviation.Pyrene Actin Assembly Assays.
Actin polymerization assays were performed as described previously (10). Briefly, pyrene fluorescence at 407 nm (lex = 365 nm) was monitored over time. If not specified, each assay contained 4 mM actin (5% pyrene) and various concentrations of wild-type or mutant VopL proteins. Ca2+-actin was exchanged for Mg2+-actin by addition of EGTA and MgCl2 to a final concentration of 1 mM each and preincubated for 2 min before the start of each assay. Polymerization assays were performed in KMEI buffer (10 mM imidazole, pH 7.0/50 mM KCl/1 mM MgCl2/1 mM EGTA). Polymerization assay in the presence of 5 nM bovine Arp2/3 and 500 nM human WASP was used as a control. In reactions with mouse capping protein (CP) shown in Fig. 5 C and E and SI Fig. 12 CP was preincubated with VopL before the addition of Mg2+-actin. Identical results were also obtained when CP was added 60 seconds after actin addition (data not shown).Barbed-End Elongation Assays.
Phalloidin-stabilized actin filaments were prepared as described previously (11). Each assay contained 0.5 mM actin (40% pyrene) and 1 mM actin filaments, with or without 3 nM VopL-WH2C. Actin filaments were preincubated with VopL-WH2C in KMEI buffer for 3 min, and Ca2+-actin was exchanged for Mg2+-actin by addition of EGTA and MgCl2 to a final concentration of 1 mM before the start of each assay.1. Palmer LE, Pancetti AR, Greenberg S, Bliska JB (1999) Infect Immun 67:708-716.
2. Chook YM, Blobel G (1999) Nature 399:230-237.
3. Boles BR, McCarter LL (2000) J Bacteriol 182:1035-1045.
4. Park KS, Ono T, Rokuda M, Jang MH, Iida T, Honda T (2004) Microbiol Immunol 48:313-318.
5. Park KS, Ono T, Rokuda M, Jang MH, Okada K, Iida T, Honda T (2004) Infect Immun 72:6659-6665.
6. Datsenko KA, Wanner BL (2000) Proc Natl Acad Sci USA 97:6640-6645.
7. Leung DW, Rosen MK (2005) Proc Natl Acad Sci USA 102:5685-5690.
8. Cooper JA, Pollard TD (1982) Methods Enzymol 85:182-210.
9. Palmgren S, Ojala PJ, Wear MA, Cooper JA, Lappalainen P (2001) J Cell Biol 155:251-260.
10. Higgs HN, Blanchoin L, Pollard TD (1999) Biochemistry 38:15212-15222.
11. Harris ES, Li F, Higgs HN (2004) J Biol Chem 279:20076-20087.