Preparation of Atto488 labeled wheat germ agglutinin (WGA)
To a vial containing ~0.3 mg of Atto488-NHS ester (ATTO-TEC, Siegen, Germany) was added 0.5 ml of WGA solution (2 mg/ml in PBS, Cambridge BioScience, Cambridge United Kingdom) and 50 µl of sodium bicarbonate buffer (1 M, pH 8.3) to adjust the solution pH to ~8.3. The resulting solution was thoroughly mixed and then gently stirred at room temperature for 1 h after which the labeling reaction was terminated by addition of 100 µl of hydroxylamine solution (1.5 M, pH 8.5). The mixture was then loaded onto a gel filtration column in PBS buffer (pH 7.2). The labeled wheat germ agglutinin (WGA) was collected as the first-eluted, pale-yellow band. The degree of labeling was determined by measuring the UV-vis absorbance at 280 and 501 nm, based on the molecular extinction coefficients of 78,000 cm-1M-1 for WGA at 280 nm and 90,000 cm-1M-1 for Atto488 at 501 nm. The average labeling was 7.9 Atto488 fluorophores per WGA molecule. Alexa647-labeled WGA was purchased from Molecular Probes (Invitrogen).
Preparation of Alexa488 and Alexa647 dual-labeled WGA
To a vial of Alexa488-TFP ester (Molecular Probes, Invitrogen) was added 1.5 mg of Alexa647-labeled WGA (Molecular Probes, Invitrogen) dissolved in 300 µl of sodium bicarbonate buffer (50 mM, pH 8.3). The resulting solution was thoroughly mixed and then gently stirred at room temperature for 45 min, after which the labeling reaction was terminated by addition of 150 µl of hydroxylamine solution (1.5 M, pH 8.5). The mixture was then loaded onto a gel filtration column and eluted with PBS buffer (pH 7.2). The labeled WGA was collected as the first-eluted, blue band. The degree of labeling was determined by measuring the UV-vis absorbance at 280, 494 and 650 nm, based on the molecular extinction coefficients of 78,000 cm-1M-1 for WGA at 280 nm, 71,000 cm-1M-1 for Alexa488 at 494 nm, and 239,000 cm-1M-1 for Alexa647 at 650 nm. The average labeling was 4.2 Alexa647 and 1.4 Alexa488 fluorophores per WGA molecule.
WGA labeling of cells
For the WGA experiments, an analogous procedure to Fab labeling was followed. Approximately 2.5 x 105 DO11.10 cells were pelleted at 600´g for 2 min at room temperature, and the resulting supernatant was completely removed with a syringe. Cells were resuspended in 10 ml of 100 pM Atto488-WGA and 10 ml of 100 pM Alexa647-WGA for the single-labeling experiments; for the dual-labeling experiments, 20 ml of 100 pM WGA labeled with both Alexa488 and Alexa647 dyes was used. All samples were labeled with WGA prepared with 10% ethanol from a high-concentration stock to prevent artifactual association due to protein clumping. Cells were incubated for 30 min at 4°C with regular agitation and were subsequently washed twice with ice-cold buffer to remove unbound label and resuspended in 37°C buffer to take measurements. Control experiments showed that this treatment resulted in no changes in cell viability or membrane integrity (data not shown). Laser powers of ~ 2 mW in conjunction with time binning of 10 ms gave good signal to noise without any indication of photobleaching.
Characterization of Fab fragments
In summary, each Fab molecule was labeled on average with a single fluorophore. F23.1 Fab bound stoichiometrically to the TCR without cross-linking it or interfering with antibody-induced TCR triggering.
T cells were loaded with a dye (Fluo-4) that fluoresces in the presence of Ca2+ ions, which are released from intracellular stores on cell activation. By using cross-linked antibodies in solution, it was possible to follow the transient increase in intracellular [Ca2+] by FACS (Fig. 5). Importantly, this activation was observed even when the cells were labeled to saturation with Alexa647-labeled F23.1 (TCR) Fabs. It is thus unlikely that the binding of the Fab alters the stoichiometry or behavior of the TCR complex.
In a single-molecule, free solution study, bright fluorescence bursts were observed from the Fabs that were comparable to those from the free dye under the same conditions (data not shown). This experiment indicates that there is no evidence of significant quenching of the Alexa488 or Alexa647 fluorophore when attached to the Fab. To gain more information about the distribution of fluorophore labels on the Fabs fragments, complexes were formed by adding an anti-Fab antibody to cross-link the Alexa488- and Alexa647-tagged Fab fragments, which were then analyzed in solution by using two-color coincidence detection (TCCD). The ratio histogram of this solution-based experiment is shown in Fig. 6.
Plotting the histogram of logarithms of the ratio of the Alexa488 to Alexa647 fluorescence counts allows the curve to be fitted by Gaussian functions corresponding to populations with different ratios of Alexa488 to Alexa647 intensities . The areas, positions, and widths of the Gaussian peaks in the fit shown in Fig. 6 are defined as functions of the underlying parameters of interest as follows:
Peak | Area | Centre | Standard deviation |
· |
|
|
|
· |
|
|
|
· |
|
|
|
· |
|
|
|
In the formulae above, ![]() is total number of events (29,665);
is total number of events (29,665); ![]() is the fraction of singly labeled antibodies;
is the fraction of singly labeled antibodies; ![]() is the effective brightness of each fluorophore on channel A;
is the effective brightness of each fluorophore on channel A; ![]() is the effective brightness of each fluorophore on channel B;
is the effective brightness of each fluorophore on channel B; ![]() is the number of fluorophores on multiply labeled antibodies;
is the number of fluorophores on multiply labeled antibodies; ![]() is the square of the factor by which the widths exceed the shot-noise limit.
is the square of the factor by which the widths exceed the shot-noise limit.
For the fitting procedure, ![]() and
and ![]() were fixed and
were fixed and![]() ,
, ![]() ,
, ![]() , and
, and ![]() were varied to minimize the Chi-squared difference between the data and the model. The fitted population fractions are independent of the choice of
were varied to minimize the Chi-squared difference between the data and the model. The fitted population fractions are independent of the choice of ![]() because the magnitudes of the brightnesses vary during the fitting to compensate for any changes made to this parameter. In order to make the fitted brightnesses approximately accurate, however, the reasonable estimate of
because the magnitudes of the brightnesses vary during the fitting to compensate for any changes made to this parameter. In order to make the fitted brightnesses approximately accurate, however, the reasonable estimate of ![]() was made, so that the widths are set at double those due to shot noise. The fitted values of the parameters of interest were as follows:
was made, so that the widths are set at double those due to shot noise. The fitted values of the parameters of interest were as follows:
A / ms-1 | B / ms-1 | N | F |
26.2 | 22.2 | 3.32 | 0.920 |
Thus, this quantitative analysis of the histogram indicates that 92% of the molecules had one fluorophore while there was a small population of 8% with a mean number of 3.3 fluorophores. This suggests that the labeling process is nonrandom, resulting in most of the antibodies having one specific lysine labeled, since the other lysines react more slowly (a Poisson distribution of labels with a mean labeling level of 1 would only have 59% of molecules with one fluorophore, taking account of the fact that a molecule with no fluorophore is not detectable). This difference in lysine reactivity has also been observed for other proteins .
The binding and dissociation characteristics of the Fab antibodies were characterized under the conditions of the single-molecule experiments, that is, Fab binding at 4°C to reduce dissociation, and experimental measurements at the physiological temperature of 37°C. As shown in Fig. 7, the association constant of the Alexa647-labeled TCRβ Fab is of the same order as that of the full antibody, and was found to be (3.8 ± 0.5) Χ 108 M at 4°C. The off-rate at 37°C was (3.5 ± 0.2) × 10-4 s-1 (Fig. 7). In the absence of sodium azide, which blocks internalization of the proteins, the apparent rate of Fab disappearance from the surface is faster due to internalization of the Fabs. A report has measured the TCR internalization rate as being between 1 × 10-4 s-1 and 3.8 × 10-4 s-1 , giving an estimated apparent rate of dissociation of between 4.5 × 10-4 s-1 to 7.3 × 10-4 s-1. For this reason, single-molecule data were only recorded for the first 20 min after placing the cells on the microscope, ensuring that the surface-protein-bound fraction of Fabs is greater than approximately 50%.
Quantitation of molecule density on DO11.10 T cells
In order to estimate the number of molecules present at the T cell surface, a flow cytometry assay was used that employed Quantibrite-PE beads (Becton Dickinson, Oxford, United Kingdom) labeled with known levels of R-phycoerythrin (PE) to calibrate the measured fluorescence of cell-bound PE-labeled antibodies to antigen density. The fluorescence values of the beads were acquired on a Cyan ADP flow cytometer in the FL2 channel, which were then used to construct a calibration curve that linearly related number of PE molecules to the amount of fluorescence (Fig. 8A). DO11.10 T cells expressing human CD86 (described in the main text) were incubated with either anti-mouse TCRβ or anti-human CD86 PE-labeled antibodies at 100 μg/ml for 60 min on ice. This ensured saturation of antigen sites and predominantly monovalent antibody binding. DO11.10 T cells expressing human CD28 were labeled with an anti-human CD28 PE-labeled antibody in a similar manner. The measured PE fluorescence (Fig. 8B) was then converted to the number of PE molecules per cell. Assuming univalent binding and 1:1 antibody:PE labeling (which is invariably the case), the number of PE molecules can be equated to antigen density. For the molecules in this study, these values were:
FCS Measurements
The autocorrelation function of Rhodamine 6G (3 nM) excited by the 488-nm laser (100 mW), with a bin time of 10 ms, was used to estimate the laser beam diameter 15 mm above the coverslip. Tween-20 (0.01%) was also added to reduce surface adhesion of the dye molecules. The autocorrelation function was fitted to a 3D diffusion model (12),
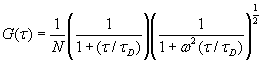 [2]
[2]
where N is the number of molecules in the probe volume, τD = r02/4D is the characteristic diffusion time during which a molecule resides in the observation volume with a lateral (r0) to axial (z0) dimension ratio, ω (ω = r0 / z0) and diffusion constant D (2.9 ±0.7 ´ 10-10 m2s-1 for Rhodamine 6G (13)). In this experiment the 488-nm laser beam diameter was measured to be 632 nm when focused 15 mm into solution.
The red beam size was estimated by using the blue beam waist obtained from the autocorrelation (316 nm) and the mean TCR burst time for the red and blue channels (598 ms and 574 ms respectively, derived from the threshold-independent Bayesian analysis described below). By calculating the ratio of the blue beam waist and burst time to the red,![]() , a red beam diameter of 645 nm was obtained, showing that the red (645 nm) and blue (632 nm) probe areas are of similar size in solution.
, a red beam diameter of 645 nm was obtained, showing that the red (645 nm) and blue (632 nm) probe areas are of similar size in solution.
The autocorrelation function of the fluorescence time trajectories of Alexa488 and Alexa647 Fabs bound to the TCR complex were measured to calculate the TCR diffusion constants. The autocorrelation function was fitted to a standard 2D diffusion equation used in FCS (14),
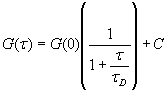 [3]
[3]
where ![]() (N is the number of molecules in the probe volume),
(N is the number of molecules in the probe volume), ![]() is the characteristic diffusion time through the laser beam waist
is the characteristic diffusion time through the laser beam waist ![]() with diffusion constant D and C is the constant offset due to slow fluorescence fluctuations. All correlation functions were fitted by using a nonlinear least-squares fit on the basis of the Levenberg-Marquardt algorithm within Origin 7.0 (OriginLab).
with diffusion constant D and C is the constant offset due to slow fluorescence fluctuations. All correlation functions were fitted by using a nonlinear least-squares fit on the basis of the Levenberg-Marquardt algorithm within Origin 7.0 (OriginLab).
Detection efficiency
Analysis of the fluorophore labeling indicates that this is fairly uniform with 92% of the Fabs carrying a single fluorophore and the remaining 8% carrying three or more. We considered the possibility that the red and blue events detected were predominantly due to this smaller fraction of brighter molecules. However, the number of brightly dual-labeled molecules on the surface is so low that it cannot explain the measured rate of coincident events. For example, in the CD28 experiment there would only be 32 detectable dual-labeled dimers on the whole cell surface. This explanation is clearly inconsistent with our data.
Our data are instead consistent with all the Fabs being detectable, since the number of dual-labeled molecules on the cell is then much higher. For example, there will be 2,000 dual-labeled CD28 molecules on the cell surface and 13,000 dual-labeled CD3e molecules. As discussed in more detail below, at higher molecule densities, higher thresholds are used to detect fluorescence bursts so that the red and blue event rates and mean occupancy are comparable in all experiments as shown in Table 1 in the main text. The use of higher thresholds has the effect of reducing the effective probe area, which is calculated for each of the Fab experiments in the next section, using the assumption that all the molecules are labeled and detectable. The values for the effective probe areas thus obtained appear plausible, which strongly supports the conclusion that all of the Fabs are detectable.
Threshold dependence on surface labeling
In our experiments the estimated maximum value of Q is 70 ´ 10-3 for a completely dual-labeled sample. For a homodimer (e.g. for CD28 and for TCR complexes labeled with anti-CD3e Fabs), Q will be half this maximum value since the red-red and blue-blue complexes do not give coincident signals (i.e., a value of 35 ´ 10-3 would be expected). However, the maximum value of Q also depends on the time bin used in the experiment (11). For the present Fab experiments, we needed to select a short time bin so that single-molecule analysis was possible, and this was achieved as indicated by the consistent low mean occupancy for all the experiments (Table 2). This has the effect of slightly reducing the maximum Q for the homodimers below the expected value of 35 ´ 10-3.
The thresholds used for all the experiments are listed in Table 2 and it is clear that the thresholds are higher for the experiments where the protein expression level is higher. We have calculated the number of detectable red- and blue-labeled molecules in the probe area (i.e., the occupancy) and since we have also measured the number of proteins on the cell surface, we can determine the effective red and blue probe diameters in all experiments. For the CD28 experiment, the effective red and blue probe diameters are 295 nm and 250 nm, respectively, slightly less than half the laser beam diameter, which seems plausible. The effective probe diameter had to be reduced to 95 nm in the TCRb/CD3e and CD3e/ CD3e experiments due to the larger amounts of surface labeling. It is also clear that the threshold in the TCRb/CD3e experiment is significantly higher than that for the other experiments (Table 2), since this experiment has the highest number of labeled proteins. In this situation, the maximum value of Q for oligomers is further reduced (11). This is because at very high rates of entry of red- and blue-labeled molecules into the laser foci the chance background event-rate becomes high enough to mask real coincident events, reducing the measured value of Q (11). This is the likely explanation as to why the value of Q for the TCRb/CD3e experiment is not twice that of the CD3e/CD3e experiment.
| Average red threshold | Average blue threshold | No. of detectable red molecules in effective probe area | No. of detectable blue molecules in effective probe area | Red effective probe diameter, nm | Blue effective probe diameter, nm |
Single WGA | 24.0 ± 1.3 | 31.2 ± 1.7 | 0.25 | 0.20 | N.D. | N.D. |
Dual WGA | 49.3 ± 2.3 | 38.6 ± 1.8 | 0.20 | 0.21 | N.D. | N.D. |
CD86/CD86 | 33.4 ± 3.0 | 44.9 ± 3.9 | 0.26 | 0.18 | 240 | 200 |
CD28/CD28 | 25.9 ± 2.1 | 59.4 ± 4.7 | 0.20 | 0.16 | 290 | 255 |
TCR b/CD3e | 125.6 ± 3.5 | 150.7 ± 4.3 | 0.27 | 0.26 | 95 | 95 |
CD3 e/CD3e | 53.4 ± 2.7 | 96.4 ± 4.0 | 0.28 | 0.24 | 100 | 90 |
TCR b/TCRb | 66.4 ± 1.7 | 113 ± 2.5 | 0.27 | 0.25 | 135 | 130 |
Table 2.
TCCD thresholds for T cell surface proteins expressed at different densities and labeled with singly or dually fluorescently labeled WGA, or with pairs of fluorescently labeled Fab fragments. The expression levels for CD86, CD28 and the TCR were used to estimate the effective red and blue probe diameter, within which molecules are able to be excited above the average threshold level. Values are calculated for a spherical cell of diameter 15 μm by using the values for the number of detectable blue and red molecules in the probe area and assuming two CD3e constitutively associated with each TCRb.
Alternative data analysis using Bayesian methodology
Typically, in single-molecule spectroscopy, peaks are detected among the noise by a simple thresholding method where the same threshold is applied to all the data files (15). However, in the case of cells, there is a variable level of background making it difficult to apply this method. Attempting to remove the background by fitting splines or using a moving average filter, then continuing with a single global threshold was not successful. Therefore, a more general Bayesian based approach was employed. The use of Bayesian algorithms for modeling the background of different spectra is not a new concept (16-18) but the following is one of the first applications in the single molecule field; for another example see ref. 19. The Bayesian methodology used herein is borrowed from nuclear spectroscopy (20) and simultaneously models the background, the noise and the signal of interest. This technique has been proven to work well in removing the background of nuclear spectra (20) and more recently in X-ray powder diffraction studies (21).
The Bayesian analysis methodology is based around Bayes' rule,
![]() , [4]
, [4]
which allows one to relate the probability that an event a has occurred given than an event b occurred to the probability that event b occurred given that event a occurred. The first stage in this Bayesian analysis is to determine where the 'real' peaks in the data are (i.e., those corresponding to real events rather than background fluctuations and noise), and what their structure is. To achieve this in the Bayesian methodology a model (M) is proposed with parameters (θ), and the probability of the parameters given the data (x) and the model is calculated, the posterior probability:
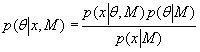 [5]
[5]
and given that ![]() is independent of the parameters, we may simplify this by writing:
is independent of the parameters, we may simplify this by writing:
![]() [6]
[6]
The model used in our analysis is as follows. The TCRs follow a random 2D diffusion path around the cell surface and the bursts of fluorescence correspond to their transit through the Gaussian laser probe volume. Consequently, the peaks in the data were modeled as Gaussians, and the area of this Gaussian shaped peak is defined as the burst size. Little is known about the structure of the random autofluorescence background, but it was expected to be smooth and slowly varying and was modeled as a piecewise spline with an unknown number of knot points. The noise was assumed to be additive, white (i.e., uncorrelated), Gaussian and a good approximation to the expected Poisson statistics (22).
The parameters that maximize the posterior probability are estimated using a Reversible Jump Markov Chain Monte Carlo (RJMCMC) algorithm (23). RJMCMC is an algorithmic framework that allows sampling from complex distributions over both model order and parameter values. Having obtained a large collection of samples one is free to obtain the posterior mean estimate of the parameter values (the average over the sampled values) or the maximum a posteriori estimator (that which maximizes the posterior probability density among the samples that were obtained). Although the latter approach can be seen to be less robust in certain circumstances, it has a clearer physical interpretation and was employed here. In our RJMCMC algorithm, the following eight moves were employed:
- Birth of a new peak: a new peak is proposed.
- Death of an existing peak: removing an existing peak.
- Splitting of a peak: splitting of an existing peak into two peaks.
- Merging of peaks: merging of two existing peaks into a single peak.
- Birth of a new knot: a new knot is proposed.
- Death of an existing knot: removing an existing knot.
- Update move for peaks: peak parameters are tweaked but no peak is added or removed.
- Update move for knot: knot parameters are tweaked but no new knot is added or removed.
The RJMCMC technique is run numerous times until an obvious most probable distribution is achieved. It is these most probable parameters that are then used to 'fit' the data. A representative result of this method is shown in Fig. 9 (Top).
After the Bayesian analysis, the data were thresholded by using burst size and the total coincident events were determined by counting the number of bursts that occur within a time window of 50 ms of each other. Other time windows were considered (0 and 25 ms), but this made no difference to the percentage coincidence seen and only reduced the number of events counted. To obtain the percentage of statistical background coincident events, the coincidence from random pairs (Alexa488 and Alexa647) of data files was calculated. This method involves determining the coincidence from nonmatching randomly chosen Alexa488 and Alexa647 data sets. A sufficient number of random pairs of files were run to determine the statistical coincidence with low error bars. We have validated this method for diffusion studies in solution where it is possible to make measurements of the background level directly and compare this to the random file method. We found that the agreement was excellent (24). The percentage of statistical background events was then subtracted from the total percentage of coincident events to obtain the percentage of true coincident events. The results of this analysis are shown in Fig. 10. The TCRb/TCRb coincidence levels are significantly below those obtained for TCRb/CD3e and CD3e/CD3e experiments for all thresholds, in complete agreement with the analysis based on derivation of Q.
References
1. Staerz UD, Rammensee HG, Benedetto JD, Bevan MJ (1985) J Immunol 134:3994-4000.
2. Luhder F, Huang Y, Dennehy KM, Guntermann C, Muller I, Winkler E, Kerkau T, Ikemizu S, Davis SJ, Hanke T, et al. (2003) J Exp Med 197:955-966.
3. Tomonari K (1988) Immunogenetics 28:455-458.
4. Knapp W (1989) Leucocyte typing IV: white cell differentiation antigens: 4th International conference on human leucocyte differentiation antigens: Papers. (Oxford Univ Press, London).
5. Demaison C, Parsley K, Brouns G, Scherr M, Battmer K, Kinnon C, Grez M, Thrasher AJ (2002) Hum Gene Ther 13:803-813.
6. Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D (1997) Nat Biotechnol 15:871-875.
7. Ren XJ, Li HT, Clarke RW, Alves DA, Ying LM, Klenerman D, Balasubramanian S (2006) J Am Chem Soc 128:4992-5000.
8. Buechler JA, Vedvick TA, Taylor SS (1989) Biochemistry 28:3018-3024.
9. Anderson LK, Caspersson M, Baltzer L (2002) Chem-Eur J 8:3687-3697.
10. Liu HY, Rhodes M, Wiest DL, Vignali DAA (2000) Immunity 13:665-675.
11. Clarke RW, Orte A, Klenerman D (2007) Analyt Chem 79:2771-2777.
12. Hess ST, Huang SH, Heikal AA, Webb WW (2002) Biochemistry 41:697-705.
13. Gell C, Brockwell DJ, Beddard GS, Radford SE, Kalverda AP, Smith DA (2001) Single Mol 2:177-181.
14. Elson EL, Magde D (1974) Biopolymers 13:1-27.
15. Li HT, Ying LM, Green JJ, Balasubramanian S, Klenerman D (2003) Anal. Chem. 75:1664-1670.
16. Denison DGT, Mallick BK, Smith AFM (1998) J R Stat Soc Ser B-Stat Methodol 60:333-350.
17. Fischer R, Hanson KM, Dose V, von der Linden W (2000) Phys Rev E 61:1152-1160.
18. DiMatteo I, Genovese CR, Kass RE (2001) Biometrika 88:1055-1071.
19. Kou SC, Sunney Xie X, Liu JS (2005) J R Stat Soc Ser C-Appl Stat 54:469-496.
20. Razul SG, Fitzgerald WJ, Andrieu C (2003) Nucl Instrum Methods Phys Res Sect A-Accel Spectrom Dect Assoc Equip 497:492-510.
21. Razul SG, Fitzgerald WJ (2003) (University of Cambridge, Department of Engineering, Cambridge).
22. Burzykowski T, Szubiakowski J, Ryden T (2003) Chem Phys 288:291-307.
23. Green PJ (1995) Biometrika 82:711-732.
24. Orte A, Clarke R, Balasubramanian S, Klenerman D (2006) Anal Chem 78:7707-7715.