
De Gassart et al10.1073/pnas.0708874105XXYYYYY103. |
Fig. 7. Ubiquitination of peptide-loaded MHC class II is regulated during MoDC maturation. (A) Ubiquitination regulates MHC class II traffic during human MoDC maturation. LPS-stimulated MoDCs were stained for HLA-DR (green) and LAMP-1 (red). In iDCs, MHC II is localized in LAMP-1+ compartments and is redistributed to the cell surface on maturation. (B) Ii chain immunoprecipitation (BU45 and PIN-1 antibodies) (Left) was performed on maturing MoDCs before immunoblotting with anti-ubiquitin, anti-MHC II, and anti-Ii antibodies. Immunoprecipitated mature MHC class II (L243 antibody) (Right) from unbound fraction of first IP are shown as control. The asterisks represent nonspecific bands.

Fig. 8. Expression levels of eGFP-MARCH chimeras in MoDC. At day 6 of differentiation, MoDCs were nucleoporated with MARCH-eGFP (I and II) mRNA and eGFP as control. FACS analysis of eGFP expression 5 h after transfection indicates relatively low expression levels, but high transfection efficiency. Thin line, nontransfected cells; thick line, transfected cells.
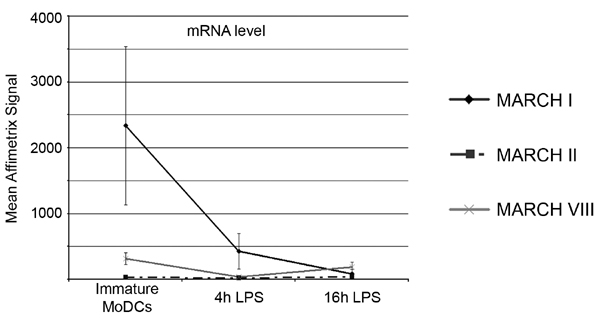
Fig. 9. Affymetrix GeneChip technology, array, hybridizations, and data analysis. Total RNA was extracted from MoDCs at different times after LPS stimulation by using the RNeasy Miniprep kit (Qiagen). For each condition, 100 ng of total RNA was used to synthesize double-stranded cDNA by using two successive reverse transcription reactions according to standard Affymetrix protocols (GeneChip Two-Cycle Target Labeling; Affymetrix). Linear amplification with T7-RNA polymerase and biotin labeling were performed by in vitro transcription after standard Affymetrix procedures. The resulting biotin-labeled cDNA was fragmented and hybridized to the Affymetrix Human Genome U133 2.0 oligonucleotide 14,500-gene microarray chip for 16 h at 45°C. After hybridization, the probe array was washed and stained on a fluidics station and immediately scanned on an Affymetrix GCS 3000 GeneArray Scanner. Data generated from the scan were then analyzed by using the MicroArray Suite software (MAS 5.0; Affymetrix). The data derived from four independents experiments were normalized by using the GC-RMA algorithm. The mean and the SDs related to the normalized gene expression signals are reported in the graph. Data obtained by this process confirm qPCR experiments shown in Fig. 4B. Indeed, MARCH I mRNA is efficiently down-regulated during maturation, whereas MARCH II and VIII mRNA level stay stable.

Fig. 10. Anti-MARCH I antibody and silencing of MARCH I by RNAi strategy validation. (A) HeLa cells, not expressing endogenous MARCH I, were transfected with MARCH I-eGFP. Cell lysates were separated on SDS/PAGE and immunoblotted in parallel with either an anti-GFP antibody or an anti-MARCH I antibody. NT, nontransfected cells. Both antibodies recognize the same band at the expected molecular weight of 58 kDa (MARCH I + eGFP) exclusively in MARCH I-eGFP-transfected cells. (B) HeLa cells were transfected with both MARCH I-eGFP construct and nonrelevant or MARCH I-specific RNAi. WB analysis of cell lysates with anti-eGFP and anti-MARCH I antibodies showed a strong decrease of MARCH I-eGFP expression level in MARCH I RNAi-transfected cells. Actin was shown as a loading control. MARCH I- RNAi-dependent extinction of MARCH I-eGFP fluorescence was also monitored by FACS.

Fig. 11. MARCH I acts at the early endocytic level. (A) Immunofluorescence confocal analysis of MARCH I in HeLa-CIITA-transfected cells indicate that MARCH I colocalizes with TfR+ compartments (red) but not HLA-DM (blue). Five hours after mRNA transfection, HeLa-CIITA cells were fixed, permeabilized, stained with anti-TfR and HLA-DM, and subjected to examination with a confocal laser-scanning microscope. (B) mRNA-transfected HeLa-CIITA cells were cultured in presence of Vacuolin-1 (5 mM) for 4 h, fixed, and permeabilized for staining and confocal analysis. Vacuolin-1 treatment induces homotypic fusion between early compartments, allowing a better characterization of MARCH I distribution in early endosomes. (C) HeLa-CIITA MARCH I-transfected cells were incubated with (black curve) or without (dark gray curve) dynasore and bafilomycin for 4 h, before surface MHC class II staining (L243 antibody) and FACS analysis. Dynasore and bafilomycin abolished MARCH I-induced down-regulation of surface MHC II molecules.
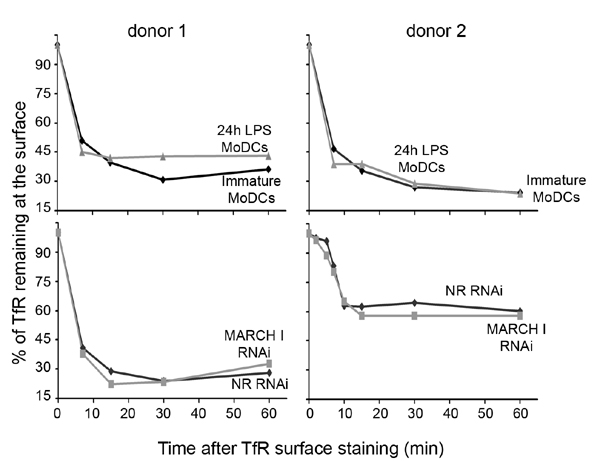
Fig. 12. TfR endocytosis is not affected by LPS-induced maturation or MARCH I down-regulation in MoDCs. Anti-TfR antibody was surface bound at 4°C on MoDCs, as in Fig. 5. After incubation at 37°C for indicated times, cells were stained with anti-mouse-Cy5 secondary antibody and analyzed by FACS. Graphs represent the percentage of TfR molecules remaining at the surface in immature and 24-h LPS-activated MoDCs (Upper) and in nonrelevant and MARCH I siRNA-transfected MoDCs (Lower). Results obtained with two different blood donors are shown.
SI Materials and Methods
Cell Culture.
All cell lines were maintained in RPMI 1640 (Invitrogen), supplemented with 10% FCS (HyClone; PERBIO) and penicillin/streptomycin at 100 ng/ml (>1,000 units/ml). Fresh human leukapheresis products were obtained from the EFS. PBMCs were isolated by Ficoll-Paque PLUS (Amersham Biosciences) and washed four times with RPMI, and CD14+ cells were immunomagnetically purified with AutoMACS system following the protocol of the manufacturer (Miltenyi Biotec). Purified monocytes were analyzed by using FACSCalibur (Becton Dickinson) and resulted to be ≈96% CD14+ cells. To promote differentiation into iDC, the purified CD14+ cells (0.5 ´ 106 cells per ml) were plated in six-well plates (2 ´ 106 cells per ml) and cultured in RPMI 1640 medium supplemented with 10% FCS, nonessential amino acids, penicillin/streptomycin at 100 ng/ml (>1,000 units/ml), recombinant human GM-CSF, and 20 ng/ml (>100 units/ml) IL-4 for 5 days (both from PeproTech). At days 2 and 4, half of the volume of the medium was replaced by fresh medium supplemented with GM-CSF and IL-4. For DC maturation, 5 mg/ml LPS (Escherichia coli type 026:B6; Sigma-Aldrich) was added to the cells starting at day 5 and for the indicated periods of times.Immunocytochemistry.
DCs were harvested and let adhere on 1% Alcian blue-treated coverslips for 10 min at 37°C, fixed with 3% PFA in PBS for 10 min at room temperature, permeabilized with 0.1% saponin in PBS/5% FCS/100 mM glycine for 10 min at room temperature, and stained 1 h with indicated primary antibody. All Alexa secondary antibodies were from Molecular Probes. Immunofluorescence and confocal microscopy (using microscope model LSM 510; Carl Zeiss MicroImaging) were performed as described previously in ref. 12.Immunoprecipitation.
A total of 3 ´ 106 MoDCs was lysed at 4°C in 1% Triton X-100, 10 mM Tris·HCl, pH 7.6, 100 mM NaCl, 5 mM MgCl2, which was freshly supplemented with 20 mM N-ethylmaleimide (Sigma), complete protease inhibitor mixtures (Roche Molecular Biochemicals), and 5 mM MG132 (Sigma). Nuclei were removed by centrifugation. Soluble cell lysates were mixed with 4 mg of specific antibody and rocked overnight. Protein G-Sepharose beads (Amersham Biosciences) were added and samples were rocked for 2 more hours. Beads were washed four times with lysis buffer. Immunoprecipitated material was eluted in SDS sample buffer [2.5% SDS, 25 mM Tris·HCl (pH 6.8), 3% glycerol] for 5 min at 100°C and separated for SDS/PAGE. When indicated, immunoprecipitated proteins were eluted in 1% SDS at 100°C for subsequent immunoprecipitation. For this goal, the eluate was diluted 10 times in lysis buffer, incubated with specific antibody for 2 h at 4°C, and protein G-Sepharose beads were then added for 2 more hours. After four additional washes, beads were boiled in SDS sample buffer. Western blot was performed according to standard techniques.Cloning of Human MARCH cDNA and in Vitro Transcription of RNA.
Total RNA was extracted from immature MoDCs by using the RNeasy Mini kit (Qiagen) and reverse-transcribed by using SuperScript II Reverse Transcriptase (Invitrogen). MARCH cDNAs were amplified by PCR by using specific primers (sequences available on request). Each full-length MARCH sequence was then cloned in-frame with eGFP into the pEGFP-N1 vector (Clontech). Then, full-length c-DNAs sequences of MARCH-eGFP fusions were excised by restriction enzyme digestion and cloned into the pGEM4Z-A64 vector (kindly provided by E. Gilboa, University of Miami, Miami, FL) containing a synthetic poly(A) tail. MARCH-eGFP mRNAs were generated by in vitro transcription (IVT) using the mMESSAGEmMACHINE T7 kit (Ambion) after SpeI vector linearization and according to the protocol provided by the manufacturer. Purification of IVT products were performed with RNeasy Mini kit (Qiagen). Quantity and purity of generated IVT products were determined by UV spectrophotometry and by formaldehyde/agarose gel electrophoresis. RNA was stored at -80°C in small aliquots.Cell Transfection.
mRNA HeLa-CIITA transfections were performed by using Lipofectamine 2000 Reagents (Invitrogen) following the manufacturer's instructions. For mRNA transfections, immature DCs were harvested at day 6 of culture and resuspended in the specified Amaxa electroporation buffer (Human Dendritic Cell Nucleofector kit; Amaxa) to a final concentration of 3 ´107 cells/ml. A total of 15 mg of in vitro-transcribed mRNA was mixed with 0.1 ml of cell suspension, transferred to a 2.0-mm electroporation cuvette, and nucleofected with an Amaxa Nucleofector apparatus as indicated by the manufacturer. RNA quantity, cell concentration, and buffer volume were kept constant throughout all experiments. After electroporation, cells were immediately transferred in complete medium, and cultured in six-well plates at 37°C for 5 h until analysis. For RNA interference, MoDCs were nucleofected at day 4 with 0.2 nM nontargeted, MARCH I, or MARCH VIII targeted synthetic 21-mer siRNAs (HP GenomeWide siRNA; Qiagen) and cultured in complete medium for 20 h.Antibody Uptake.
MoDCs were harvested, washed once in cold PBS, and stained with unlabeled antibodies (L243 for MHC II and OKT9 for TfR) for 30 min at 4°C in PBS/1% FCS. After extensive washes, cells were incubated at 37°C in complete medium for indicated times, harvested, washed in ice-cold PBS, surface stained with fluorescent secondary antibodies for 15 min at 4°C. Cells were fixed in 2% PFA prior FACS analysis by using a FACSCalibur apparatus (Beckman Coulter).For immunoprecipitation, cells were treated as above. After being incubated at 37°C for the given time, cells were washed in PBS and lysed in previously described 1% Triton X-100 lysis buffer. Protein G-Sepharose beads were added and samples were immunoprecipitated for 2 h at 4°C, before SDS/PAGE. For immunofluorescence analysis, MoDCs were allowed to adhere on Alcian blue-coated coverslips and surface stained with unlabeled antibody at 4°C for 30 min. After washes, coverslips were placed in warm medium for the indicated time, and then fixed in 3% PFA for conventional staining with secondary antibodies.
Pharmacological Treatments.
Cells were mRNA transfected and cultured for 4 h in complete medium, supplemented with different drugs at the following concentrations: 5 mM for MG132 (Sigma), 10 mM for LY294002 (Calbiochem), 100 nM for wortmannin (Sigma), 250 nM for Bafilomycin A1 (Calbiochem), 0.5 mg/ml for brefeldin A (Sigma), 40 mM for dynasore, and 5 mM for Vacuolin-1 (both a kind gift of T. Kirchhausen, Harvard Medical School, Boston, MA).mRNA Quantification by Real-Time RT-PCR.
Total RNA was extracted at indicated stage of MoDC maturation by using the RNeasy Mini kit (Qiagen). To exclude the amplification of genomic DNA, an on-column DNase digestion was performed by using the RNase-Free DNase Set (Qiagen). One to 5 mg was retrotranscribed by using SuperScript II reverse transcriptase (Invitrogen) and random (pDN6) primers. First-strand c-DNA templates were then used for PCR amplification of short (50- to 100-bp) fragments from MARCH I, MARCH II, and MARCH VIII with appropriate primers (sequences available on request). PCR was carried out in complete SYBR Green PCR buffer (PE Biosystems) by using 200 nM of each specific primer. A total of 20 ml of PCR mix was added to 5 ml of cDNA template, and the amplification was tracked via SYBR Green incorporation by using a Stratagene sequence detection system. cDNA concentration in each sample were normalized by using GAPDH. A nontemplate control was also routinely performed. A dissociation curve was generated at the end of each PCR cycle to verify that a single product was amplified. Relative quantification of target cDNA was determined by calculating the difference in cross-threshold (CT) values after normalization to GAPDH signals, according to the Pfaffl's method and the automated Excel-based program available (REST).