

Abstract
Reelin-Disabled-1 (Dab1) signaling has a well-established role in regulating neuronal migration during brain development. Binding of Reelin to its receptors induces Dab1 tyrosine phosphorylation. Tyrosine-phosphorylated Dab1 recruits a wide range of SH2 domain-containing proteins and activates multiple signaling cascades, resulting in cytoskeleton remodeling and precise neuronal positioning. In this review, we summarize recent progress in the Reelin-Dab1 signaling field. We focus on Dab1 alternative splicing as a mechanism for modulating the Reelin signal in developing brain. We suggest that correct positioning of neurons in the developing brain is at least partly controlled by alternatively-spliced Dab1 isoforms that differ in the number and type of tyrosine phosphorylation motifs that they contain. We propose a model whereby different subsets of SH2 domain-containing proteins are activated by different Dab1 isoforms, resulting in coordinated migration of neurons.
Keywords: neuronal migration, Reelin, Disabled-1, alternative splicing, tyrosine phosphorylation, SH2 domain
Introduction
The precise positioning of neurons within the brain remains an important question in developmental neurobiology. There is accumulating evidence supporting key roles for molecules such as Cdk5, Lis1, Reelin and n-cofilin in regulating neuronal migration, resulting in the formation of laminated structures [1–5]. The Reelin-Disabled-1 (Dab1) signaling pathway in particular has a well-established role in regulating the “inside-out” lamination of the cerebral cortex [5–7]. Recent advances provide important insight into how Reelin induces the activation of intracellular signaling cascades and rearrangement of cytoskeleton to guide neuronal migration. Yet, how these intracellular signaling events are orchestrated in a temporal and spatial manner and how Reelin coordinates the movement of individual subpopulation of neurons to achieve ordered assembly in the brain remains poorly understood. Here, we review recent publications that address Reelin signaling in the regulation of neuronal migration. We focus on the role of developmentally-regulated Dab1 alternative splicing in modulating the activity of the Reelin pathway to coordinate neuronal migration.
Neuronal migration during cortical development
In the cerebral cortex, neuronal migration begins when the first wave of early-born neurons leaves the ventricular zone (VZ) to establish the preplate [8–9]. The second wave of early-born neurons then split the preplate to form the superficial marginal zone (MZ) and the deeper subplate. Late-born neurons subsequently migrate through the subplate and pass early-born neurons, leading to the “inside-out” lamination of neurons in the cortical plate (CP), with early-born neurons located in deeper layers, and late-born neurons located in more superficial layers (Figure 1A) [9–10].
Figure 1. Migration of cortical neurons in wild-type and reeler mice.

(A) Inside-out lamination of cortical neurons in wild-type mice. At early developmental stages, layer VI neurons split the preplate (PP) to form the subplate (SP) and pial surface (PS). Late-born neurons continue to migrate and bypass older neurons, resulting in the inside-out formation of the cortical plate, with older neurons located in the inner layers, and the younger neurons located in outer layers. (B) Inverted cortical neuronal layers in reeler mice. Layer VI fails to split the preplate, leading to accumulation of neurons underneath the preplate. The inability of late-born neurons to bypass the old neurons results in the inversion of neuronal layers in the cortical plate.
Cortical neurons usually adopt two main migratory modes to reach their final destination: radial glia-independent somal translocation and radial glia-guided locomotion [11–12]. Translocating neurons usually extend long leading processes attached to the pial surface (PS) and move continuously at a relatively fast speed. In contrast, locomoting neurons have shorter processes and move in a slower saltatory (jerky) manner [12]. At early stages of development (~E10.5–12.5 in mouse), the prevalent mode of neuronal cell migration is somal translocation, which underlies subplate formation and layer VI migration in the cerebral cortex [11]. Radial glia-guided locomotion appears to be the main mode of cell migration for late-born neurons and is responsible for the “inside-out” lamination that characterizes the later stages of cortical development (~E13.5 and onwards in mouse) [12–14]. Notably, these two migratory modes are not mutually exclusive as locomoting neurons switch to somal translocation once their leading processes reach the PS during the final phase of migration. The dynamic nature of neuronal migration allows cells to constantly explore and respond in a timely manner to environmental cues, resulting in the ordered assembly of cells required for brain function.
An overview of the Reelin-Disabled-1 (Dab1) signaling pathway
Falconer described the reeler mouse phenotype more than fifty years ago [15]. These mutant mice are characterized by ataxia, tremors and a reeling gait. A striking feature of reeler mice is that neurons are aberrantly positioned in laminated brain structures. In the cerebral cortex, neurons are unable to split the preplate and bypass their predecessors, resulting in accumulation of neurons underneath the preplate and inversion of cortical layers (“outside-in” pattern) (Figure 1B) [16–18]. In 1995, Reelin, the gene mutated in reeler mice, was cloned [5]. Interestingly, spontaneous or targeted mutations of several other genes, including Disabled-1 (Dab1), very low density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2), generate a phenotype indistinguishable from that of reeler mice [5, 19–21]. Reelin encodes a secreted glycoprotein expressed in Cajal-Retzius cells of the marginal zone (MZ), whereas products of VLDLR, ApoER2 and Dab1 are expressed in migrating neurons and radial glia [19, 21–22]. Further studies demonstrated that products of these genes comprise a critical signaling pathway that regulates the migration of cortical, hippocampal and cerebellar neurons, with little or no effect on the migration of striatal neurons and cortical interneurons. Reelin binding to VLDLR and ApoER2 receptors induces the tyrosine phosphorylation of adaptor protein Dab1 and activation of downstream cascades, resulting in accurate neuronal positioning [6, 23–24]. Previous reviews have described the complex interplay of the signaling molecules involved in Reelin function [7, 25–27]. In this review, we focus on the role of the adaptor protein Dab1, and its alternative splicing, in Reelin signaling-regulated neuronal migration.
Dab1 is a crucial cellular adaptor in Reelin signaling
Dab1 contains three main domains: an N-terminal protein interaction/phosphotyrosine binding (PI/PTB) domain that binds to Reelin receptors [21], an internal tyrosine-rich region [28], and a C-terminal serine/threonine-rich region. The tyrosine-rich region consists of five highly conserved tyrosine residues (Y185, Y198, Y200, Y220, Y232). These residues correspond to two consensus Src family kinase recognition sites (YQxI, Y185 and Y198) and two consensus Abl/Crk recognition sites (YxVP, Y220 and Y232) [29]. At least three of the tyrosine residues, Y198, Y220 and Y232, can be phosphorylated in response to Reelin stimulation [28, 30–31]. Mice expressing a mutant Dab1 form with substitutions at all five tyrosine residues exhibit a phenotype similar to that observed in reeler and Dab1−/− mice [32], demonstrating that Dab1 tyrosine phosphorylation is essential for Reelin signaling.
Tyrosine-phosphorylated Dab1 acts as a hub to recruit different Src homology 2 (SH2) domain-containing proteins, including the p85 regulatory subunit of phosphatidylinositide-3-kinase (PI3K), cellular adaptors CrkL, Crk, Nckβ and SOCS (suppressor of cytokine signaling) [30, 33–37]. Different tyrosine residues appear to interact with distinct SH2 domains: Y220 and Y232 are required for the recruitment of SH2 domains from Crk, CrkL and Nckβ adaptor proteins [30, 35–37], whereas Y185 and Y198 are necessary for the recruitment of PI3K and SOCS SH2 domains [33–34]. Tyrosine-phosphorylated Dab1 also interacts with the microtubule associated protein, Lis1, albeit in a SH2 domain-independent manner [38]. These interactions activate different downstream signaling cascades involved in both activation and termination of Reelin signaling to ensure accurate neuronal positioning (Figure 2).
Figure 2. The Reelin-Disabled 1 signaling pathway.

Binding of Reelin to its receptors, VLDLR and ApoER2, induces SFK activation and Dab1 tyrosine phosphorylation. Phosphorylated Dab1 acts as a hub to recruit different downstream SH2 domain-containing proteins including Crk, Nckβ, p85 (PI3K) and SOCS. Dab1-Crk interaction activates the downstream C3G-Rap1 pathway. Activated Rap1 then regulates the membrane distribution of N-cadherin. Recruitment of Nckβ to Dab1 is likely involved in actin remodeling through p130Cas. The association of PI3K p85 subunit with Dab1 activates the PI3K-Akt pathway, which in turn modulates the phosphorylation of microtubule binding proteins, tau and MAP1B, leading to microtubule remodeling. Activation of PI3K-Akt may engage LIMK1 and n-cofilin, resulting in stabilization of actin polymerization. In contrast, Dab1-SOCS association down-regulates Reelin signaling by degrading phosphorylated Dab1 through the ubiquitin proteasome system, resulting in termination of Reelin signaling. The interaction between tyrosine-phosphorylated Dab1 and Lis1 also causes microtubule remodeling. Reelin-Dab1 signaling and LKB-STRAD-Stk25 play opposing roles in regulating the rearrangement of the Golgi apparatus and neuronal polarity.
Dab1 tyrosine phosphorylation activates downstream signaling to promote neuronal migration and regulate neuronal polarization
Dab1-Crk-C3G-Rap1-cadherin pathway
Reelin promotes Dab1 (at Y220 and Y232 tyrosine phosphorylation sites)-Crk interaction, induces tyrosine phosphorylation of C3G (a guanine nucleotide exchange factor for Rap1) and activates the small GTPase Rap1 [30, 34, 36–37]. Ablation of CrkII−/−CrkL−/− in mouse cortex generates a reeler-like phenotype without affecting Reelin-induced Dab1 tyrosine phosphorylation [39]. However, two Dab1 phosphorylation-dependent downstream events, C3G and PI3K activation, are abolished [39], in support of a direct role for Crk proteins downstream of Dab1. Neurons in C3G-deficient mice fail to split the preplate and do not form the CP, a phenotype reminiscent of reeler [40], suggesting that C3G is critical for the regulation of neuronal migration. Recent studies have highlighted a role for Rap1 in neuronal migration, especially in the regulation of somal translocation of early-born neurons and multipolar migration of late-born neurons [41–42]. Reelin appears to specifically activate Rap1 in the intermediate zone (IZ) and increases the membrane localization of the cell adhesion molecule, N-cadherin, to regulate the polarity of multipolar neurons. Mechanisms underlying the activation of Rap1 in the IZ and the redistribution of N-cadherin in the cell membrane of the IZ, as well as how cadherin-mediated signaling guides the radial orientation of migrating neurons, remain to be elucidated.
Dab1-PI3K-Akt pathway
Tyrosine-phosphorylated Dab1 (likely at Y185 and Y198) also recruits the p85 regulatory subunit and activates the PI3K-Akt pathway [43–45]. Inhibition of PI3K disrupts normal cortical plate formation in slice cultures, suggesting that PI3K is required for aspects of cortical development that are dependent on Reelin [44, 46]. Reduced phosphorylation of microtubule-associated protein, MAP1B and microtubule-stabilizing protein tau in response to Reelin stimulation is dependent on the activity of the PI3K-Akt-glycogen synthase kinase (GSK) 3β pathway [44, 47].
The observation that tau hyperphosphorylation in Dab1-null mice occurs in a strain-specific manner [48] led to the identification of Stk25, a Ste20-like serine/threonine kinase, as a genetic modifier of Dab1 mutant phenotypes [49]. Interestingly, Reelin-Dab1 signaling and Stk25 have antagonistic roles in regulating neuronal polarization and dendritic Golgi deposition [50]. For example, inactivation of Stk25 in hippocampal neurons inhibits axon specification, whereas absence of Dab1 in these same neurons promotes axon production. Conversely, overexpression of Stk25 induces Golgi condensation and the formation of multiple axons, effects that can be neutralized by Reelin treatment. Reelin-stimulated dendritic Golgi extension can also be suppressed by Stk25 overexpression [50].
Stk25 functions downstream of liver kinase B1 (LKB1) and interacts with STE20-related kinase adapter protein alpha (STRADα) and GM130, thus forming a LKB1-STRADα-Stk25-GM130 axis that regulates Golgi distribution and neuronal polarization [50]. The balance between Reelin-Dab1 signaling and the LKB1-STRADα-Stk25-GM130 axis may therefore determine the final outcome of neuronal polarization, thereby controlling the direction of the migratory path. At this time, it is not known whether these two pathways regulate neuronal polarization in parallel or converge through physical interactions with scaffold proteins involved in Dab1 phosphorylation.
Reelin-induced PI3K/Akt pathway activation also regulates the activity of the actin-depolymerizing protein, n-cofilin (non-muscle cofilin) [51]. Reelin activates the serine/threonine kinase LIM kinase 1 (LIMK1) which then induces n-cofilin phosphorylation on serine 3, thus abolishing its ability to depolymerize actin [51–53]. In the cerebral cortex, phosphorylation of n-cofilin mainly occurs in the leading processes of migrating neurons when they move toward the Reelin-expressing marginal zone [51], indicating that Reelin-induced n-cofilin phosphorylation may stabilize and attach these neuronal processes to the MZ. Another downstream target of PI3K in the Reelin-Dab1 signaling pathway is Rho GTPase cdc42. Reelin activates cdc42 in a PI3K-dependent manner to promote growth cone motility and filopodia formation in cortical neurons [54]. Whether phosphorylation of n-cofilin and activation of Cdc42 are required for Reelin-mediated terminal translocation and how these processes are coordinated during migration remain unclear.
Dab1 tyrosine phosphorylation triggers the proteasome degradation pathway to stop neuronal migration
A striking feature of mice deficient in key components of Reelin signaling, including Reelin (reeler mice), VLDLR and ApoER2 (Vldlr−/−Apoer2−/− mice), Dab1 (Dab1Y5F mice) and Fyn/Src (Src−/−Fyn−/− mice), is the accumulation of Dab1 protein [6, 20–21, 32, 55–56]. All these genetic deficiencies result in either reduced levels or absence of Dab1 tyrosine phosphorylation, suggesting that Dab1 tyrosine phosphorylation is important in regulating Dab1 levels and may provide a negative feedback to terminate the activity of Reelin signaling.
Studies have shown that Reelin-induced Dab1 tyrosine phosphorylation on Y185 and Y198 (YQXI sites) recruits the SH2 domain-containing SOCS proteins (adaptors for cullin-based E3 ubiquitin ligase), resulting in polyubiquitination and degradation of Dab1 protein via the cullin-5 (Cul5) E3 ligase [33, 57–58]. Depletion of Cul5 or expression of a degradation-resistant Dab1 mutant in the cerebral cortex leads to over-migrated neurons positioned at the surface of the cortical plate [33, 59]. These studies suggest that developmental stage-specific tyrosine phosphorylation of Dab1 triggers Cul5-mediated proteasome degradation, thereby preventing “over-migration” of cortical neurons.
Detach and stop, detach and go - models for Reelin signaling in cortical development
Despite significant advances in our understanding of Reelin signaling during cortical development, the exact roles that Reelin signaling plays in the regulation of neuronal migration remain elusive. Early on, the predominant expression of Reelin in Cajal-Retzius cells in the marginal zone of the cortex led researchers to postulate that Reelin stops the migration of neurons at the final migratory stages [7]. Since Reelin and Dab1 have both been shown to promote detachment of neurons from radial glia [60–61], a “detach and stop” model was proposed to explain the role of Reelin in the regulation of neuronal migration [26, 60, 62]. However, recent studies do not support a role for Reelin signaling in stopping neuronal migration. In fact, ectopic expression of Reelin in the VZ rescues the subplate split phenotype in the reeler cortex and cortical slice cultures [63–64]. Ectopic Reelin also induces neuronal aggregation generating an “inside-out” pattern for cortical neurons in the developing cerebral cortex [65]. Moreover, depletion of Dab1 by knockdown or tissue-specific genetic ablation in the cortex delays neuronal migration [42, 66], whereas introduction of wild-type Dab1 into Dab1−/− cortex promotes the migration of neurons out of the ventricular zone [59]. Together, these studies indicate that Reelin-Dab1 signaling promotes rather than inhibits neuronal movement.
Another model, called “detach and go”, has recently been proposed to explain how Reelin regulates neuronal cell migration [67]. In this model, Reelin stimulates the translocation of layer VI cortical neurons at early developmental stages, which allows layer VI cells to split the subplate in a radial glia-independent manner. Late-generated neurons can still use radial-glia dependent locomotion to move toward the cortical plate. Once their leading processes reach the Reelin-containing MZ, Reelin induces detachment of neurons from the radial glia and promotes their translocation so that they bypass their predecessors, thus forming the “inside-out” pattern in the cortical plate [67]. This model is supported by several recent studies. First, expression of an activated form of Dab1 that is resistant to proteosome degradation in the cerebral cortex causes “overmigration” of neurons rather than migration delay. Second, real-time imaging analysis shows that Reelin-Dab1 directly promotes somal translocation of both early-born (layer VI) and late-born neurons [41–42]. Third, functional Reelin fragments have been found to diffuse throughout the cortex, suggesting that early- and late-born neurons are exposed to Reelin prior to reaching the MZ [64]. As well, Reelin-induced downregulation of functional Reelin receptors and cell surface re-distribution of N-cadherin were observed in the IZ [41, 68], indicating that Reelin can regulate biochemical characteristics in migrating neurons before they reach the Reelin-enriched MZ.
However, the “detach and go” model cannot explain several aspects of neuronal migration. For example, if Reelin acts from a distance, migrating neurons (layer II–V) would be expected to respond to Reelin and undergo translocation before their processes reach the Reelin-containing MZ. What prevents the neurons from translocating in response to Reelin and what triggers the Reelin-specific migratory switch at late migratory stages are unknown. Furthermore, the developing cerebral cortex is a heterogeneous environment. Neurons located within different regions of the brain migrate in an asynchronous manner. It’s not clear why different populations of neurons demonstrate distinct responses to the Reelin signal and how their movements are coordinated at different developmental stages. Recent studies point to an additonal layer of Reelin signaling regulation through alternative splicing of Dab1. We propose that Dab1 alternative splicing fine-tunes Reelin-induced cellular responses and coordinates neuronal movement in different populations of neurons [69–72].
Alternative splicing regulates Dab1 function
Alternative splicing modulates Dab1 tyrosine phosphorylation
Multiple alternatively-spliced Dab1 isoforms resulting from alternative promoter usage, alternative cassette exons and mutually exclusive alternative exons have been identified (Table 1) [70, 72–74]. Interestingly, the majority of Dab1 splicing events affect exons encoding the tyrosine phosphorylation motifs. These exons are set up in such a way that individual Dab1 tyrosine phosphorylation sites (Y185QXI, Y198QXI and Y220QVP) are encoded by two adjacent exons (exons 6&7, exons 7&8 and exons 8&9; Figure 3). For example, nucleotides encoding the tyrosine and +1 position (relative to tyrosine, Y185Q, Y198Q and Y220Q) are located at the 3′ ends of exons 6, 7 and 8, respectively, whereas nucleotides encoding the +2 and +3 residues are located at the 5′ ends of their respective downstream exons (7, 8 and 9).
Table 1.
alternatively-spliced Dab1 isoforms
| Species | Alternative splicing events | Sequence difference relative to the canonical Dab1 | Ref. |
|---|---|---|---|
| Chicken | Exclusion of exons 7&8 and inclusion of exon 9b | Deletion of 35 aa containing Y198, Y200 and Y220, insertion of 19 aa | 31, 65 |
| Pig | Exclusion of exons 7&8 and inclusion of exons 9b&9c | Deletion of 35 aa containing Y198, Y200 and Y220, insertion of 33 aa | 71 |
| Lizard | Inclusion of exon 9b | Insertion of 19 aa | 67 |
| Zebrafish | Exclusion of exons 8&9 | Deletion of 42 aa containing Y200, Y220 and Y232 | 68 |
| Human | Alternative promoter usage | NA | 67 |
| Human | Exclusion of exons 7&8 and inclusion of exons 9b&9c | Deletion of 35 aa containing Y198, Y200 and Y220, insertion of 33 aa | 70 |
| Mouse | Exclusion of exon 7 | Deletion of 13 aa containing Y198 | 63 |
| Mouse | Exclusion of exon 8 | Deletion of 22 aa containing Y200 and Y220 | 63 |
| Mouse | Exclusion of exons 7&8 | Deletion of 35 aa containingY198, Y200 and Y220 | 63 |
| Mouse | Inclusion of exons 9b&9c | Insertion of 33 aa | 63, 66, 67, 69 |
| Mouse | Exclusion of exons 7&8 and inclusion of exons 9b&9c | Deletion of 35 aa containing Y198, Y200 and Y220, insertion of 33 aa | 63 |
| Mouse | Exclusion of exon 8, inclusion of exons 9b&9c | Deletion of 22 aa containing Y200 and Y220, insertion of 33 aa | 63 |
| Mouse | Alternative 5′ splice site of intron 9 | Deletion of 2 aa (valine and serine) | 63 |
| Mouse | Alternative 5′ splice site of intron 9, exclusion of exons 7&8 | Deletion of 2 aa (valine and serine), deletion of 35 aa containing Y198, Y200 and Y220 | 63 |
| Mouse | Alternative 5′ splice site of intron 9, inclusion of exons 9b&9c | Deletion of 2 aa (valine and serine), insertion of 33 aa | 63 |
| Mouse | Alternative 5′ splice site of intron 9, exclusion of exons 7&8, inclusion of exons 9b&9c | Deletion of 2 aa (valine and serine), deletion of 35 aa containingY198, Y200 and Y220, insertion of 33 aa | 63 |
| Mouse | Alternative 5′ splice site of intron 9, exclusion of exons 7&8 | Deletion of 2 aa (valine and serine), deletion of 35 aa containingY198, Y200 and Y220 | 63 |
| Mouse | Alternative 5′ splice site of intron 9, inclusion of exons 9b&9c | Deletion of 2 aa (valine and serine), insertion of 33 aa | 63 |
| Mouse | Alternative 5′ splice site of intron 9, exclusion of exons 7&8, inclusion of exons 9b&9c | Deletion of 2 aa (valine and serine), deletion of 35 aa containing Y198, Y200 and Y220 and insertion of 33 aa | 63 |
| Mouse | Inclusion of exon 7a | Addition of 18 aa with a stop codon | 69 |
| Mouse | Inclusion of exon 9a | Addition of 31 aa with a stop codon | 69 |
Figure 3. Mixing-and-matching tyrosine phosphorylation sites in Dab1 by alternative splicing.

Schematic representation of exon-intron structure (exons 6 to 9) of the Dab1 gene. White boxes represent constitutive exons, whereas purple and gold boxes indicate alternative exons. The amino acid sequences of the four consensus Dab1 tyrosine phosphorylation sites are shown. Dab1 contains two YQXI motifs (Y185QTI and Y198QYI, framed in magenta) and two YXVP motifs (Y220QVP and Y232DVP, framed in green). Exclusion of exon 7 (ΔEx 7) removes Y198Q but links Y185Q to Y199I200, thereby reconstituting a Y185QYI motif identical to the original Y198QYI. Exclusion of exon 8 (ΔEx 8) removes Y220Q but connects Y198Q with V222P223, converting Y198QXI into a Y198QVP site. Exclusion of exons 7 and 8 (ΔEx 7&8) removes both Y198Q and Y220Q and rejoins Y185Q to V222P223, resulting in loss of both YQXI motifs but retention of two YXVP motifs.
We have recently found that the unique organization of Dab1 exons allows alternative splicing events that result in precise “mixing-and-matching” of Dab1 tyrosine phosphorylation sites during brain development (Figures 3 and 4) [69]. For example, Dab1 exons 7 and/or 8 are excluded at early developmental stages, resulting not only in deletion but also in the reconstruction of distinct tyrosine phosphorylation motifs in different Dab1 isoforms [69–70, 75–76]. When exon 7 is excluded, Y198Q is removed. However, Y185Q is linked to Y200I201 as a consequence of joining exon 6 to exon 8, thereby reconstituting a Y185QYI motif that is identical to the original Y198QYI motif. In contrast, exclusion of exon 8 leaves the Y198Q199 intact but removes Y220Q221, thereby converting the original Y198QYI motif to a Y198QVP motif (Y198Q199 connected to V222P223). When both exons 7 and 8 are excluded, Y185Q186 are directly connected to V222P223, generating an isoform that retains two YxVP motifs but no YQxI motifs. Developmentally-regulated alternative splicing thus produces multiple Dab1 variants, with preferential loss (partial or complete) of YQXI motifs observed at early developmental stages. In contrast, canonical Dab1 isoforms which contain the complete set of tyrosine phosphorylation sites are predominantly expressed at later stages of development [69].
Figure 4. Schematic representation of Dab1 variants.

Dab1 exons are shown (exons are not drawn to scale). The start codon is located in exon 2, whereas the stop codon is located in exon 14. Exons 3–6 encode the PTB domain, while exons 6–9 encode the four critical tyrosine phosphorylation motifs. Exclusion of exons 7 and/or 8 in mice or exons 8 and 9 in zebrafish results in partial deletion of tyrosine phosphorylation motifs in Dab1. Inclusion of exons 9b/9c results in an in-frame 33 aa insertion. Inclusion of an alternative exon after exon 7 in Dab1217 results in deletion of two tyrosine motifs and introduction of an in-frame stop codon generating a 217 aa product. Inclusion of an alternative exon after exon 9 in Dab1271 introduces an in-frame codon generating a 271 aa product.
What might be the significance of expressing Dab1 isoforms with truncated Dab1 tyrosine phosphorylation sites at early developmental stages? A direct consequence of expressing such Dab1 isoforms would be altered phosphotyrosine-associated interactions resulting from modified tyrosine phosphorylation status. Previous studies have suggested that the YQXI and YQXP motifs in Dab1 have distinct functions in Reelin signaling [33–34]. YQXI motifs are important for both turning on and turning off downstream pathways by activating PI3K-Akt and triggering Cul5-mediated proteasome degradation of Dab1 [33–34]. YQVP motifs appear to be critical in recruiting Crk and Nck adaptors to form a signaling scaffold complex [30, 34–37].
We have recently shown that alternative splicing of exons 7 and/or 8 in Dab1 controls Dab1 phosphorylation and Dab1-SH2 domain interactions [69]. Exclusion of both exons 7 and 8 abolishes Dab1 tyrosine phosphorylation and known Dab1-SH2 interactions [Src, Crl, p85 (PI3K), Nckβ, SOCS]. Exclusion of either exon 7 or 8 decreases the levels of Dab1 tyrosine phosphorylation and significantly reduces its binding affinity to p85 (PI3K), Src and SOCS SH2 domains. Intriguingly, skipping of exon 7 or 8 does not significantly affect Dab1-Crk SH2 domain interaction, whereas skipping of exon 7 but not exon 8 inhibits Dab1-Nckβ SH2 domain interaction. As different SH2 domains are connected to different downstream effectors, these results indicate that different Dab1 isoforms may direct Reelin signaling to distinct downstream cascades, leading to differential responses to Reelin stimulation.
Dab1 isoforms are expressed in different subsets of cells at different developmental stages [69–71]. Differential interactions of Dab1 isoforms with distinct sets of SH2 domains may therefore have an important impact on the temporo-spatial coordination of neuronal movement in response to Reelin stimulation. The fact that Dab1 isoforms with reduced numbers of tyrosine phosphorylation sites fail to recruit the full spectrum of SH2 domains suggests that downstream signaling may be attenuated or inactivated in cells expressing these isoforms (Figure 5). This elegant mechanism for modulating Dab1 function may prevent premature activation of Reelin signaling in migrating cells before they reach their correct destination. As development proceeds and neurons continue to migrate, alternative splicing produces the canonical Dab1 isoform, which can fully respond to Reelin and transmit the signal to the complete spectrum of downstream effectors. Therefore, only cells expressing the canonical Dab1 may be capable of translocating and bypassing their predecessors to form the inside-out cortical pattern. We propose that this dynamic and rapid isoform transition ensures differential competence and proper response of neurons to Reelin at different developmental and migratory stages, allowing a prompt, efficient and exact response to migration cues in a heterogeneous environment.
Figure 5. Fine-tuning Reelin signaling by Dab1 alternative splicing during development - a model.
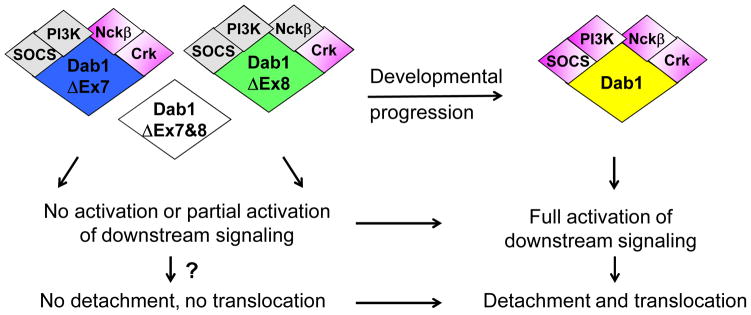
Early in development, alternative splicing produces Dab1 isoforms that exclude exons 7 and/or 8. These isoforms show absent or reduced associations with SH2 domain-containing proteins, leading to absent or attenuated activation of downstream pathways. As a result, cells expressing these isoforms may have a compromised ability to detach and translocate in response to Reelin. As development proceeds, inclusion of exons 7 and 8 in Dab1 generates the canonical Dab1 isoform that recruits the full set of SH2 domain-containing proteins, resulting in full activation of downstream signaling. Cells expressing the canonical Dab1 isoform are able to detach and translocate in response to Reelin, resulting in the inside-out lamination that is characteristic of the cortical plate. Grey indicates reduced interaction; magenta indicates normal interaction.
Additional splicing events regulate Dab1 activity and neuronal migration
Alternative inclusion of Dab1 exons 9b/9c has also been described (Figure 4) [72–73]. Neuronal-specific splicing factors, Nova1 and Nova2, have been shown to inhibit the inclusion of exons 9b and 9c in Dab1 [72, 77–78]. Inclusion of exons 9b/9c leads to in-frame insertion of 33 aa downstream of the tyrosine enriched region. Although the encoded 33 aa contains no known motif, their aberrant inclusion in Dab1 results in neuronal migration defects [72]. Ablation of Nova2 in mice increases the levels of exons 9b/9c-containing Dab1 transcripts and causes neuronal migration defects in late-born, but not early-born neurons [72], suggesting a specific role for this particular Dab1 isoform in early-born neurons. Introduction of the canonical Dab1 into Nova2−/−mice rescues the migration defects, in support of an antagonistic role for Dab1 and exons 9b/9c-containing Dab1 isoforms. One possibility is that insertion of the exon 9b/9c-encoded region causes conformational changes in the protein, masking PTB and tyrosine residues and preventing their access to interacting partners. Further analysis using crystallography may help to address this question.
It is unclear, however, why Dab1 and exons 9b/9c-containing Dab1 isoforms counteract each other, as both forms are tyrosine phosphorylated and recruit the same sets of SH2 domains [69, 72]. Notably, splicing of exons 9b/9c and exons 7/8 is often mutually exclusive [69, 76], suggesting coordinated regulation of the exclusion and inclusion of these exons during development. It would be of interest to investigate whether Nova also promotes exons 7/8 inclusion along with exons 9b/9c exclusion, and whether isoforms that exclude exons 7/8 are increased in Nova2−/− mice along with isoforms that include exons 9b/9c. Finally, divergent roles for VLDLR and ApoER2 in regulating cortical neuron migration have been described, with VLDLR predominantly regulating migration of early-born neurons, and ApoER2 primarily regulating the migration of late-born neurons [79]. The migration defects in Nova2−/− mice are reminiscent of the phenotype observed in ApoER2−/− mice. Therefore, it will be important to investigate whether exons 9b/9c-containing Dab1 isoforms preferentially associate with VLDLR, whereas exons 9b/9c-excluding Dab1 isoforms preferentially associate with ApoER2, thus contributing to different aspects of neuronal migration.
Conclusions and perspective
Remarkable progress has been made in our understanding of the role of Reelin signaling in neuronal migration at both the cellular and molecular levels. Recent findings in Dab1 alternative splicing have revealed a new level of complexity for the coordination of the Reelin signal during development. We propose that Reelin modulates cellular effects and migratory behaviors in different populations of neurons and at different migratory stages through Dab1 alternative splicing, thereby coordinating neuronal movements in a spatio-temporal manner. Future experiments will involve examination of the precise cellular distribution of individual Dab1 isoforms in the developing brain in order to directly link individual Dab1 isoforms to specific downstream effectors of Dab1 based on their co-expression. In utero electroporation experiments to express different Dab1 isoforms in the brain combined with real-time imaging to track cell migratory behavior will help to decipher the physiological roles of Dab1 isoforms. In addition, other than the involvement of Nova splicing factors, mechanisms governing the complex Dab1 splicing pattern during development remain to be elucidated.
Neuronal migration is a highly complex and dynamic process, which requires the integration of inputs and outputs from numerous signaling pathways. How these signaling events are orchestrated to regulate specific migratory behavior such as polarity, process outgrowth, process trailing and nuclear movement, is not well understood. Reelin signaling has been linked to multiple pathways implicated in neuronal migration including Cdk5, Notch, and Lis1 [38, 80–82]. It would be important to address whether there is cross-regulation between different pathways involving alternatively-spliced Dab1 isoforms as converging and diverging points. Finally, as Reelin signaling and Dab1 isoforms have also been implicated in non-neuronal tissue [83], it will be important to address the full spectrum of Dab1 isoform activities by carrying out investigations in non-neuronal tissues.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research - Funding Reference Number 114993.
References
- 1.Bellenchi GC, et al. N-cofilin is associated with neuronal migration disorders and cell cycle control in the cerebral cortex. Genes Dev. 2007;21(18):2347–57. doi: 10.1101/gad.434307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chae T, et al. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18(1):29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 3.Ohshima T, et al. Migration defects of cdk5(−/−) neurons in the developing cerebellum is cell autonomous. J Neurosci. 1999;19(14):6017–26. doi: 10.1523/JNEUROSCI.19-14-06017.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai JW, Bremner KH, Vallee RB. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat Neurosci. 2007;10(8):970–9. doi: 10.1038/nn1934. [DOI] [PubMed] [Google Scholar]
- 5.D’Arcangelo G, et al. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374(6524):719–23. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 6.Howell BW, Herrick TM, Cooper JA. Reelin-induced tryosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999;13 (6):643–8. doi: 10.1101/gad.13.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–39. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- 8.Pearlman AL, et al. New directions for neuronal migration. Curr Opin Neurobiol. 1998;8(1):45–54. doi: 10.1016/s0959-4388(98)80007-x. [DOI] [PubMed] [Google Scholar]
- 9.Gupta A, Tsai LH, Wynshaw-Boris A. Life is a journey: a genetic look at neocortical development. Nat Rev Genet. 2002;3(5):342–55. doi: 10.1038/nrg799. [DOI] [PubMed] [Google Scholar]
- 10.Nadarajah B, et al. Neuronal migration in the developing cerebral cortex: observations based on real-time imaging. Cereb Cortex. 2003;13(6):607–11. doi: 10.1093/cercor/13.6.607. [DOI] [PubMed] [Google Scholar]
- 11.Nadarajah B, Parnavelas JG. Modes of neuronal migration in the developing cerebral cortex. Nat Rev Neurosci. 2002;3(6):423–32. doi: 10.1038/nrn845. [DOI] [PubMed] [Google Scholar]
- 12.Nadarajah B, et al. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4(2):143–50. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- 13.Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128(1):29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 14.Hatanaka Y, et al. Distinct migratory behavior of early- and late-born neurons derived from the cortical ventricular zone. J Comp Neurol. 2004;479(1):1–14. doi: 10.1002/cne.20256. [DOI] [PubMed] [Google Scholar]
- 15.Falconer D. Two mutant mutants, Tremler and Reeler, with neurological actions in the house mouse. J Genet. 1951;50:192–201. doi: 10.1007/BF02996215. [DOI] [PubMed] [Google Scholar]
- 16.Caviness VS., Jr Time of neuron origin in the hippocampus and dentate gyrus of normal and reeler mutant mice: an autoradiographic analysis. J Comp Neurol. 1973;151(2):113–20. doi: 10.1002/cne.901510203. [DOI] [PubMed] [Google Scholar]
- 17.Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Res. 1982;256(3):293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- 18.Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4(6):496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- 19.Howell BW, et al. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389(6652):733–7. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 20.Sheldon M, et al. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389(6652):730–3. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- 21.Trommsdorff M, et al. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97(6):689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 22.D’Arcangelo G, et al. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci. 1997;17(1):23–31. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D’Arcangelo G, et al. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24 (2):471–9. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 24.Hiesberger T, et al. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24(2):481–9. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 25.Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7(11):850–9. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- 26.Chai X, et al. Reelin acts as a stop signal for radially migrating neurons by inducing phosphorylation of n-cofilin at the leading edge. Commun Integr Biol. 2009;2(4):375–7. doi: 10.4161/cib.2.4.8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forster E, et al. Emerging topics in Reelin function. The European journal of neuroscience. 2010;31(9):1511–8. doi: 10.1111/j.1460-9568.2010.07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keshvara L, et al. Identification of reelin-induced sites of tyrosyl phosphorylation on disabled 1. J Biol Chem. 2001;276(19):16008–14. doi: 10.1074/jbc.M101422200. [DOI] [PubMed] [Google Scholar]
- 29.Songyang Z, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72(5):767–78. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 30.Ballif BA, et al. Activation of a Dab1/CrkL/C3G/Rap1 pathway in Reelin-stimulated neurons. Curr Biol. 2004;14(7):606–10. doi: 10.1016/j.cub.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 31.Katyal S, et al. Hierarchical disabled-1 tyrosine phosphorylation in Src family kinase activation and neurite formation. J Mol Biol. 2007;368(2):349–64. doi: 10.1016/j.jmb.2007.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howell BW, et al. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol. 2000;10(15):877–85. doi: 10.1016/s0960-9822(00)00608-4. [DOI] [PubMed] [Google Scholar]
- 33.Feng L, et al. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 2007;21(21):2717–30. doi: 10.1101/gad.1604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng L, Cooper JA. Dual functions of Dab1 during brain development. Mol Cell Biol. 2009;29(2):324–32. doi: 10.1128/MCB.00663-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pramatarova A, et al. Nck beta interacts with tyrosine-phosphorylated disabled 1 and redistributes in Reelin-stimulated neurons. Mol Cell Biol. 2003;23(20):7210–21. doi: 10.1128/MCB.23.20.7210-7221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen K, et al. Interaction between Dab1 and CrkII is promoted by Reelin signaling. J Cell Sci. 2004;117(Pt 19):4527–36. doi: 10.1242/jcs.01320. [DOI] [PubMed] [Google Scholar]
- 37.Huang Y, et al. Tyrosine phosphorylated Disabled 1 recruits Crk family adapter proteins. Biochem Biophys Res Commun. 2004;318(1):204–12. doi: 10.1016/j.bbrc.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 38.Assadi AH, et al. Interaction of reelin signaling and Lis1 in brain development. Nat Genet. 2003;35(3):270–6. doi: 10.1038/ng1257. [DOI] [PubMed] [Google Scholar]
- 39.Park TJ, Curran T. Crk and Crk-like play essential overlapping roles downstream of disabled-1 in the Reelin pathway. J Neurosci. 2008;28(50):13551–62. doi: 10.1523/JNEUROSCI.4323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voss AK, et al. C3G regulates cortical neuron migration, preplate splitting and radial glial cell attachment. Development. 2008;135(12):2139–49. doi: 10.1242/dev.016725. [DOI] [PubMed] [Google Scholar]
- 41.Jossin Y, Cooper JA. Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat Neurosci. 2011;14(6):697–703. doi: 10.1038/nn.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franco SJ, et al. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron. 2011;69(3):482–97. doi: 10.1016/j.neuron.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beffert U, et al. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J Biol Chem. 2002;277(51):49958–64. doi: 10.1074/jbc.M209205200. [DOI] [PubMed] [Google Scholar]
- 44.Bock HH, et al. Phosphatidylinositol 3-kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J Biol Chem. 2003;278(40):38772–9. doi: 10.1074/jbc.M306416200. [DOI] [PubMed] [Google Scholar]
- 45.Ballif BA, Arnaud L, Cooper JA. Tyrosine phosphorylation of Disabled-1 is essential for Reelin-stimulated activation of Akt and Src family kinases. Brain Res Mol Brain Res. 2003;117(2):152–9. doi: 10.1016/s0169-328x(03)00295-x. [DOI] [PubMed] [Google Scholar]
- 46.Jossin Y, Goffinet AM. Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol Cell Biol. 2007;27(20):7113–24. doi: 10.1128/MCB.00928-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gonzalez-Billault C, et al. A role of MAP1B in Reelin-dependent neuronal migration. Cerebral cortex. 2005;15(8):1134–45. doi: 10.1093/cercor/bhh213. [DOI] [PubMed] [Google Scholar]
- 48.Brich J, et al. Genetic modulation of tau phosphorylation in the mouse. J Neurosci. 2003;23(1):187–92. doi: 10.1523/JNEUROSCI.23-01-00187.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuki T, et al. Identification of Stk25 as a genetic modifier of Tau phosphorylation in Dab1-mutant mice. PloS one. 2012;7(2):e31152. doi: 10.1371/journal.pone.0031152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsuki T, et al. Reelin and stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell. 2010;143(5):826–36. doi: 10.1016/j.cell.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chai X, et al. Reelin stabilizes the actin cytoskeleton of neuronal processes by inducing n-cofilin phosphorylation at serine3. J Neurosci. 2009;29(1):288–99. doi: 10.1523/JNEUROSCI.2934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arber S, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393(6687):805–9. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 53.Yang N, et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393(6687):809–12. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 54.Leemhuis J, et al. Reelin signals through apolipoprotein E receptor 2 and Cdc42 to increase growth cone motility and filopodia formation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30(44):14759–72. doi: 10.1523/JNEUROSCI.4036-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rice DS, et al. Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development. 1998;125(18):3719–29. doi: 10.1242/dev.125.18.3719. [DOI] [PubMed] [Google Scholar]
- 56.Kuo G, et al. Absence of Fyn and Src causes a reeler-like phenotype. J Neurosci. 2005;25(37):8578–86. doi: 10.1523/JNEUROSCI.1656-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arnaud L, Ballif BA, Cooper JA. Regulation of protein tyrosine kinase signaling by substrate degradation during brain development. Mol Cell Biol. 2003;23 (24):9293–302. doi: 10.1128/MCB.23.24.9293-9302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bock HH, et al. Apolipoprotein E receptors are required for reelin-induced proteasomal degradation of the neuronal adaptor protein Disabled-1. J Biol Chem. 2004;279(32):33471–9. doi: 10.1074/jbc.M401770200. [DOI] [PubMed] [Google Scholar]
- 59.Simo S, Jossin Y, Cooper JA. Cullin 5 regulates cortical layering by modulating the speed and duration of Dab1-dependent neuronal migration. J Neurosci. 2010;30(16):5668–76. doi: 10.1523/JNEUROSCI.0035-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dulabon L, et al. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron. 2000;27(1):33–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- 61.Sanada K, Gupta A, Tsai LH. Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron. 2004;42(2):197–211. doi: 10.1016/s0896-6273(04)00222-3. [DOI] [PubMed] [Google Scholar]
- 62.Tabata H, Nakajima K. Neurons tend to stop migration and differentiate along the cortical internal plexiform zones in the Reelin signal-deficient mice. J Neurosci Res. 2002;69(6):723–30. doi: 10.1002/jnr.10345. [DOI] [PubMed] [Google Scholar]
- 63.Magdaleno S, Keshvara L, Curran T. Rescue of ataxia and preplate splitting by ectopic expression of Reelin in reeler mice. Neuron. 2002;33(4):573–86. doi: 10.1016/s0896-6273(02)00582-2. [DOI] [PubMed] [Google Scholar]
- 64.Jossin Y, et al. The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J Neurosci. 2004;24(2):514–21. doi: 10.1523/JNEUROSCI.3408-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kubo K, et al. Ectopic Reelin induces neuronal aggregation with a normal birthdate-dependent “inside-out” alignment in the developing neocortex. J Neurosci. 2010;30(33):10953–66. doi: 10.1523/JNEUROSCI.0486-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olson EC, Kim S, Walsh CA. Impaired neuronal positioning and dendritogenesis in the neocortex after cell-autonomous Dab1 suppression. J Neurosci. 2006;26(6):1767–75. doi: 10.1523/JNEUROSCI.3000-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cooper JA. A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 2008;31(3):113–9. doi: 10.1016/j.tins.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 68.Uchida T, et al. Downregulation of functional Reelin receptors in projection neurons implies that primary Reelin action occurs at early/premigratory stages. J Neurosci. 2009;29(34):10653–62. doi: 10.1523/JNEUROSCI.0345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao Z, et al. Splice-Mediated Motif Switching Regulates Disabled-1 Phosphorylation and SH2 Domain Interactions. Molecular and cellular biology. 2012;32(14):2794–808. doi: 10.1128/MCB.00570-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Katyal S, Godbout R. Alternative splicing modulates Disabled-1 (Dab1) function in the developing chick retina. Embo J. 2004;23(8):1878–88. doi: 10.1038/sj.emboj.7600185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao Z, et al. The early isoform of disabled-1 functions independently of Reelin-mediated tyrosine phosphorylation in chick retina. Molecular and cellular biology. 2010;30(17):4339–53. doi: 10.1128/MCB.00545-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yano M, et al. Nova2 regulates neuronal migration through an RNA switch in disabled-1 signaling. Neuron. 2010;66(6):848–58. doi: 10.1016/j.neuron.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bar I, et al. The gene encoding disabled-1 (DAB1), the intracellular adaptor of the Reelin pathway, reveals unusual complexity in human and mouse. J Biol Chem. 2003;278(8):5802–12. doi: 10.1074/jbc.M207178200. [DOI] [PubMed] [Google Scholar]
- 74.Costagli A, et al. Identification of alternatively spliced dab1 isoforms in zebrafish. Dev Genes Evol. 2006;216(6):291–9. doi: 10.1007/s00427-005-0052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Katyal S, et al. Disabled-1 alternative splicing in human fetal retina and neural tumors. PloS one. 2011;6(12):e28579. doi: 10.1371/journal.pone.0028579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Long H, et al. Identification of alternatively spliced Dab1 and Fyn isoforms in pig. BMC neuroscience. 2011;12:17. doi: 10.1186/1471-2202-12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ule J, et al. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444(7119):580–6. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 78.Ule J, et al. Nova regulates brain-specific splicing to shape the synapse. Nat Genet. 2005;37(8):844–52. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 79.Hack I, et al. Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development. 2007;134(21):3883–91. doi: 10.1242/dev.005447. [DOI] [PubMed] [Google Scholar]
- 80.Ohshima T, Mikoshiba K. Reelin signaling and Cdk5 in the control of neuronal positioning. Mol Neurobiol. 2002;26(2–3):153–66. doi: 10.1385/MN:26:2-3:153. [DOI] [PubMed] [Google Scholar]
- 81.Hashimoto-Torii K, et al. Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron. 2008;60(2):273–84. doi: 10.1016/j.neuron.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sibbe M, et al. Reelin and Notch1 cooperate in the development of the dentate gyrus. J Neurosci. 2009;29(26):8578–85. doi: 10.1523/JNEUROSCI.0958-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Khialeeva E, Lane TF, Carpenter EM. Disruption of reelin signaling alters mammary gland morphogenesis. Development. 2011;138(4):767–76. doi: 10.1242/dev.057588. [DOI] [PMC free article] [PubMed] [Google Scholar]