

Abstract
A short and efficient total synthesis of the alkaloid isosolenopsin and its enantiomer has been achieved. In the key step, a ω-transaminase catalyzed the regioselective mono-amination of the diketone pentadecane-2,6-dione which was obtained in a single step via Grignard reaction. Initial low conversions in the biotransformation could be overcome by optimisation of the reaction conditions employing suitable cosolvents. In the presence of 20 vol% DMF or n-heptane best results were obtained employing two enantio-complementary ω-transaminases originating from Arthrobacter between 30-40 °C; under these conditions conversions of >99% and perfect stereocontrol (ee > 99%) were achieved. Diastereostelective chemical reduction (H2/Pd/C) of the biocatalytic product gave the target compound. The linear three step synthesis provided the natural product isosolenopsin in diastereomerically pure form (ee > 99%, d.r. = 99:1) with an overall yield of 64%.
Keywords: piperidines, alkaloids, reductive amination, ω-transaminases, diastereoselectivity
Introduction
Functionalised chiral piperidines are popular key elements in a vast number of synthetic protocols and are among the most present skeletal fragments in natural products.[1] Moreover, due to the wide range of their useful pharmacological properties extensive efforts have been devoted to their (stereoselective) synthesis.[2] In particular simple 2,6-disubstituted piperidines constitute an important class of natural products due to their biological activity and abundant occurrence in nature. Prominent examples are the secreted fire ant venom alkaloids from Solenopsis invitica; selected members of this family were found to display cytotoxic, insectical, hemolytic, antibacterial, antifungal and necrotic properties.[3] Furthermore, physiological investigations revealed that e.g. isosolenopsin A (1b) selectively inhibits designated neuronal nitric oxide synthases (nNOS)[4] whereas solenopsin A (2b) inhibits the regulator protein phosphatidylinositol-3-kinase (PI3K); the latter is involved in controlling apoptosis, proliferation and angiogenesis.[5]
These diverse biological activities and their apparent simple structure (figure 1) have made them attractive targets to study and showcase new synthetic methods. Several racemic and asymmetric total syntheses have been reported based on chiral pool precursors, catalytic (asymmetric) transformations in addition to electrochemical and auxiliary controlled methods.[6] Even though several routes are very elegant and involve highly sophisticated key steps, long reaction sequences and the use of an orthogonal protecting-group-strategy hampered their overall efficiency. Hence, more capable and environmental begin methods are desired.
Figure 1.
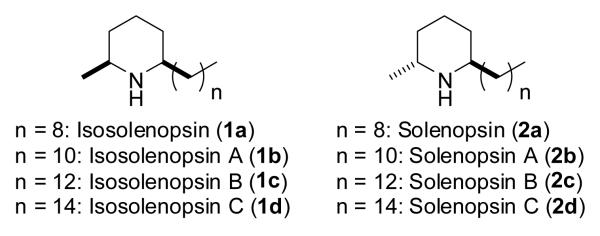
Secreted fire ant venom alkaloids and their structure.
Recently, we demonstrated that 2,6-diketones 3 serve as useful precursors for the synthesis of the optically pure 2,6-disubstituted piperidine scaffold 8 via enzymatic reductive amination (Scheme 1).[7] A ω-transaminase (ω-TA)[8-10] catalyzed the regio- and stereoselective mono-amination of 3 at the sterically less demanding (ω-1)-keto moiety yielding preferentially amino-ketone 4 which underwent spontaneous ring-closure to give the corresponding Δ1-piperideines 6 in enantiomerically pure form. Based on this concept we now report an extension of this method for the total synthesis of isosolenopsin [(2S,6R)-1a] and its enantiomer (2R,6S)-1a.[11]
Scheme 1.

Chemoenzymatic asymmetric synthesis of 2,6-disubstituted piperidines involving regio- and stereoselective mono-amination.
Results and Discussion
Various enantio-complementary ω-TAs were tested for the transformation of diketone 3a to the corresponding Δ1-piperideines 6a and 7a under designated reaction conditions employing alanine as amine donor (scheme 2). The equilibrium was shifted towards the product side via removal/recycling of the formed pyruvate with an alanine-dehydrogenase (AlaDH).[12] Unfortunately only low conversions were achieved probably due to the low solubility of diketone 3a in pure buffer solution. Subsequently, various water miscible organic solvents [MeCN, THF, DMF, 1,2-dimethoxyethane (DME), DMSO] and immiscible solvents (EtOAc, toluene) were assayed at 10 vol%. Although most of the investigated ω-TAs gave only insufficient conversions (< 5%), four showed conversions of around 25% rendering them targets for a more detailed study. Notably, regioisomer 7a was not detected for any enzyme investigated (see supporting information).
Scheme 2.
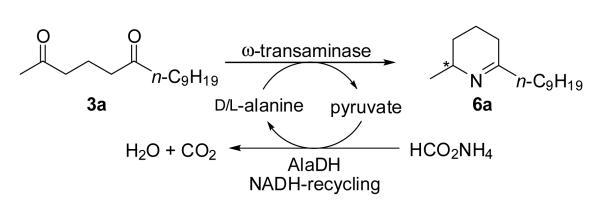
Mono-amination of diketone 3a to yield Δ1-piperideine 6a. Conditions: lyophilized E. coli cells containing the overexpressed ω-TA (20 mg), diketone 3a (12 mg, 50 mm), PLP (1 mm), NAD+ (1 mm), ammonium formate (150 mm), d- respective l-alanine (500 mm), AlaDH (12 U), FDH (11 U), organic co-solvent and KPi buffer (100 mm, pH 7.0), 30 °C, 24 hours in an Eppendorf orbital shaker (700 rpm).
Focussing on the four most promising ω-TAs identified, originating from Arthrobacter citreus,[13c] Arthrobacer sp.[13d] Pseudomonas fluorescens,[13a,13b] and Hyphomonas neptunium[9f], the influence of the concentration of water miscible- (DMF, DME, DMSO) and immiscible solvents (toluene) at concentrations ranging from 5-40 vol% was investigated [for the analytical scale: 25 mm 3a (6 mg/mL); 1.00 mL total volume]. The first two enzymes are (S)-selective, while the latter two are (R)-selective. The conversion obtained depended on the solvent and on the enzyme employed (Figure 2): While the enzymes from P. fluoresecens and H. neptunium led to conversions of 25% at maximum, the two enantio-complementary ω-TAs from Arthrobacer turned out to be superior. For example, the (S)-selective ω-TA from A. citreus gave conversions up to 60% if DMF was applied. Notably, increasing DMF-concentrations led to higher conversions whereas a reversed trend was observed for toluene or DME as co-solvent. In the case of toluene, this is probably due to the lower availability of the organic substrate in the aqueous phase. An analogous observation was made for the ω-TA from (R)-Arthrobacter sp. For this enzyme, piperideine 6a was obtained with 60% conversion applying 5 vol% of toluene and 80% conversion in the presence of 40 vol% DMF.
Figure 2.

Conversions for the mono-amination of diketone 3a in the presence of various organic co-solvents employing (R)- and (S)-selective ω-TAs. Conditions: diketone 3a (6 mg/mL, 25 mm), lyophilized E. coli cells containing overexpressed ω-TA (20 mg), PLP (1 mm), NAD+ (1 mm), ammonium formate (150 mm), d- respective l-alanine (500 mm), AlaDH (12 U), FDH (11 U), organic co-solvent, K-phosphate buffer (100 mm, pH 7.0), 30 °C, 24 hours in an Eppendorf orbital shaker (700 rpm).
Having identified two suitable enzymes with opposite stereopreference [Arthrobacter citreus for the (S)- and Arhrobacter sp. for the (R)-enantiomer] and co-solvents, further optimisation of temperature and pH was conducted. The biocatalytic monoamination of 3a was tested at different pH-values (ranging from 6.5 to 9.9) at varied DMF concentrations (20 and 40 vol%) (Figure 3). Subsequently, the conversion was studied as a function of temperature at varied DMF concentrations (20 and 40 vol%) (Figure 4). Thereby highest conversion was obtained for the asymmetric reductive amination of diketone 3a at 20 vol% DMF, pH 6.5, 30 °C with the ω-TA originating from (R)-Arthrobacter sp., whereas 20 vol% DMF (pH 7.0) and elevated temperatures of 40 °C were best suitable for the (S)-selective ω-TA of Arthrobacter citreus.
Figure 3.

Influence of pH on the conversion of diketone 3a at varying DMF concentrations: (●) 20 vol% DMF (R)-Arthrobacter sp., (■) 40 vol% DMF (R)-Arthrobacter sp., (▲) 20 vol% DMF A. citreus, (▼) 40 vol% DMF A. citreus. Conditions: diketone 3a (25 mm), lyophilized E. coli cells containing the overexpressed ω-TA (20 mg), PLP (1 mm), NAD+ (1 mm), ammonium formate (150 mm), d-/l-alanine (500 mm), AlaDH (12 U), FDH (11 U), 20-40 vol% DMF, KPi buffer (100 mm), 30 °C, 24 h, 700 rpm.
Figure 4.

Influence of temperature on the conversion of diketone 3a at varying DMF concentrations: (◇) 40 vol% DMF (R)-Arthrobacter sp., (■) 20 vol% DMF (R)-Arthrobacter sp., (•) 20 vol% DMF A. citreus, (▲) 40 vol% DMF A. citreus. Conditions: diketone 3a (25 mm), lyophilized E. coli cells containing the overexpressed ω-TA (20 mg), PLP (1 mm), NAD+ (1 mm), ammonium formate (150 mm), d-/l-alanine (500 mm), AlaDH (12 U), FDH (11 U), 20-40 vol% of DMF, KPi buffer (100 mm, pH 7.0), 20-50 °C, 24 h, 700 rpm.
Although, conversions above 95% were achieved employing the reaction conditions mentioned above, n-heptane was also investigated for curiosity due to the similarity in polarity of the solvent to the starting material 3a (Figure 5). For solubility reasons the substrate still had to be pre-dissolved in DMF (5 vol% final conc.). Despite the formation of a biphasic system, the reaction resulted in full conversion (>99%) at 20 vol% of n-heptane for the ω-TA from (R)-Arhtrobacter sp. and 85% conversion for the (S)-ω-TA from Arthrobacter.
Figure 5.

Reductive amination of diketone 3a in an aqueous two phase system: (■) (R)-Arhtobacter sp., (●) A. citreus. Conditions: diketone 3a (25 mm) dissolved in 5 vol% DMF, lyophilized E. coli cells containing the overexpressed ω-TA (20 mg), PLP (1 mm), NAD+ (1 mm), ammonium formate (150 mm), d-/l-alanine (500 mm), AlaDH (12 U), FDH (11 U), 5-40 vol% n-heptane, KPi buffer (100 mm, pH 7.0), 30 °C, 24 h, 700 rpm.
The synthetic potential was finally demonstrated in the total synthesis of both enantiomers of isosolenopsin [(2S,6R)-1a and (2R,6S)-1a]: Starting with a Grignard reaction, the required diketone 3a was obtained in a single step from commercially available dihydropyrane-2-one 9 in 65% yield (Scheme 3). The subsequent biotransformation was conducted under the optimised conditions at increased scale (36 mg, 0.15 mmol, 25 mm); the ω-TA of Arthrobacter sp. gave access to the (R)- while the ω-TA of Arthrobacter citreus to the (S)-enantiomer of piperideine 6a. Both reactions yielded the corresponding cyclic imine (R)- and (S)-6a, respectively, in optically pure form (ee > 99%) at full conversion (>99% in both cases); notably, the reaction times were prolonged (48 hours) in order to ensure full conversion and hence simplify extensive work up procedures. Due to the instability of imine 6a, it was directly subjected to the diastereoselective reduction using hydrogen with Pd/C as catalyst after extraction. Under these conditions the second stereocenter was established under perfect substrate control, affording the natural product (2S,6R)-1a and (2S,6R)-1a in diastereomerically pure form [d.r. (syn/anti) = 99% as deduced by GC and NMR analysis] in a excellent yield between 94-98% (over two steps). Thus, starting from pyranone 9, the natural alkaloids (+) and (−)-1a were obtained optically pure form in 64% overall yield.
Scheme 3.

Chemoenzymatic synthesis of both enantiomers of isosolenopsins (2S,6R)-1a and (2R,6S)-1a under the optimised reaction conditions. Reagents and conditions: a) Diketone 3a (36 mg, 0.15 mmol, 25 mm) dissolved in DMF (20 vol%), ω-TA from Arthrobacter citreus, PLP (1 mm), NAD+ (1 mm), l-alanine (20 eq.), ammonium formate (150 mm), 11 U FDH, 12 U AlaDH, 48 h, 40 °C, 700 rpm; b) same as for (a) but with the ω-TA from (R)-Arthrobacter sp. at 30 °C with d-alanine and 20 vol% n-heptane, 5 vol% DMF; c) Pd/C, H2 (1 atm.), 4 hours, 22 °C; precipitation with 5 eq. etherical HCl solution.
Conclusions
An efficient chemoenzymatic total synthesis of the alkaloid isosolenopsin and its enantiomer has been achieved via a regioselective mono-amination of the corresponding diketone in the key step. Optimisation of the biocatalytical reaction conditions through medium engineering allowed overcoming initial low conversions. Among various organic solvents tested, n-heptane and DMF turned out to be best suitable at 30 °C for the (R)-selective ω-TA from Arthrobacter sp. and at 40 °C for the (S)-selective ω-TA from Arthrobacter citreus. The asymmetric reductive mono-amination of diketone 3a proceeded finally with perfect regio- and stereoselectivity at full conversions. This strategy afforded the natural product in optically pure form with an overall yield between 64-65% over three steps.
Experimental Section
All starting materials were obtained from commercial suppliers and used as received unless stated otherwise. The reactions were carried out with standard Schlenk techniques under N2 atmosphere in oven-dried (120 °C) glassware. Preparative chromatographic separations were performed by column chromatography on Merck silica gel 60 (0.063–0.200 mm). TLC was carried out with precoated aluminium sheets (TLC Silica gel 60 F254, Merck) with detection by staining with cerium molybdenum solution or visualisation by UV. Optical rotation was measured at 20 °C on a Perkin–Elmer Polarimeter 341. GC-MS spectra were recorded with an Agilent 7890A GC-system, equipped with an Agilent 5975C mass selective detector and a HP-5 MS column (30 m × 0.25 mm × 0.25 μm; hydrogen as carrier gas [flow = 0.55 mL/min]). 1H and 13C NMR spectra were recorded at 20 °C on a 300 Bruker NMR unit spectrometer; chemical shifts are given in ppm relative to Me4Si (1H: Me4Si = 0 ppm) or relative to the resonance of the solvent (1H: CDCl3 = 7.26; 13C: CDCl3 = 77.0 ppm). Formate dehydrogenase from Candida boidinii (2.2 Umg−1) [one unit will oxidize 1.0 μmole of formate to CO2 per min at pH 7.6 at 37 °C], catalog no. FDH 002) was purchased from Codexis. Lyophilised E. coli cells containing overexpressed ω-TA were prepared as previously reported.[9a,b,g] Purified recombinant l-alanine dehydrogenase was prepared as described in literature (15 μL of the crude solution are equal to 12 U).[9b]
Pentadecane-2,6-dione (3b)
CuI (340 mg, 1.78 mmol) was added to a stirred solution of n-C9H19MgBr [8.82 mL, 8.82 mmol (1 n in Et2O)] in Et2O (30 mL), MS 3Å (50 mg) in one portion. The mixture was stirred for 30 minutes at 21 °C and cooled to −78 °C afterwards. Dihydro-2H-pyran-2-one 9 (1.00 g, 8.92 mmol) was added drop wise via syringe over a time period of 15 minutes. The reaction mixture was kept at −78 °C, whereby another 0.5 equivalents of Grignard-reagent (4.46 mL, 4.46 mmol) were added after four and after eight hours again. When tlc control revealed full conversion of the starting material (10 hours), the mixture was treated with aqueous HCl solution (1 n, 30 mL) and allowed warming to room temperature. The reaction mixture was extracted afterwards four times with small portions of EtOAc and the organic layer was dried over MgSO4. Subsequent filter flash chromatography (eluent: petroleum ether/EtOAc = 90:10) of the concentrated solution afforded the diketone in 65% yield (1.39 g, 5.79 mmol) as a colorless powder. Spectroscopic data are in agreement with those previously published.[14] Melting point: 65-66 °C. RF (PE/EtOAc = 80:20) = 0.49. 1H-NMR (300 MHz, CDCl3): δH = 0.84 (t, 3J15,14 = 6.7 Hz, 3 H, 15-H), 1,22 (brs, 12 H, 9-H, 10-H, 11-H, 12-H, 13-H and 14-H), 1.50 (tt, 3J8,7 = 7.3 Hz, 3J8,9 = 6.6 Hz, 2 H, 8-H), 1.80 (tt, 3J4,3 = 7.2 Hz, 3J4,5 = 7.1 Hz, 2 H, 4-H), 2.09 (s, 3 H, 1-H), 2.30 (t, 3J7,8 = 7.5 Hz, 2 H, 7-H), 2.37 (t, 3J3,4 = 7.2 Hz, 2 H, 3-H), 2.40 (t, 3J5,4 = 7.2 Hz, 2 H, 5-H). 13C-NMR (75 MHz, CDCl3): δC = 14.2 (C-15), 17.8 (C-4), 22.7 (C-14), 23.9 (C-8), 29.3 (C-11), 29.3 (C-12), 29.5 (C-10), 29.5 (9), 30.0 (C-1), 31.9 (C-13), 41.5 (C-3), 42.6 (C-5), 42.9 (C-7), 208.5 (C-2), 210.9 (C-6). GC-MS (EI, 70 eV): m/z (%) = 240 [M+] (1), 155 [C10H19O+] (15), 128 [C7H12O+] (82), 85 [C5H9O+] (65), 43 [C2H3O+](100).
Optimisation of the reaction conditions (analytical scale)
Lyophilized cells of E. coli containing the corresponding overexpressed ω-TA (20 mg) were rehydrated in a potassium phosphate buffer (100 mm at varying pH as indicated in figure 2-5) containing PLP (1.0 mm), NAD+ (1.0 mm), ammonium formate (150 mm), FDH (11 U), Ala-DH (12 U) and d-respective l-alanine (500 mm) at room temperature for 30 minutes. The substrate 3a was added afterwards (6 mg, 25 mm, dissolved in the corresponding organic solvent as indicated in figure 2-5) and the reductive amination was performed at defined temperature as indicated (figure 2-5) in an Eppendorf orbital shaker (700 rpm, vertical position) for 24 hours. After the period of time, the reaction was aborted by the addition of saturated Na2CO3 (200 μL) and vigorous shaking. The mixture was extracted with EtOAc (3 × 500 μL). Combined organic layers were dried over MgSO4 and an aliquot withdrawn for further analysis. Conversions were determined by GC analysis on an achiral phase: column HP-5 (Agilent); carrier gas = hydrogen; temperature program: 100 °C to 250 °C with slope 10 °C·min−1; Rt (6a) = 8.7 min, Rt (3a) = 10.6 min. Determination of enantiomeric excess by GC analysis on a chiral phase: column DEX-CB (Agilent; CP-Chirasil); carrier gas = helium, temperature program: 60 °C (1 minute isotherm) to 90 °C with slope 5.0 °C·min−1 then to 180 °C with slope 2 °C·min−1; Rt [(2R)-6a] = 37.23 min, Rt [(2S)-6a] = 37.41 min.
Biocatalytical synthesis of Δ1-piperideine (R)-6a (preparative scale)
Lyophilized cells of E. coli containing overexpressed ω-TA from (R)-Arthrobacter sp. (125 mg) were rehydrated in a K-phosphate buffer (4.8 mL , pH 6.5, 100 mm) containing PLP (1 mm), NAD+ (1 mm), ammonium formate (150 mm), FDH (11 U), Ala-DH (12 U) and d-alanine (500 mm) at 22 °C for 30 minutes. The substrate 3a [36 mg (dissolved in 300 μL DMF), 0.15 mmol, 25 mm], n-heptane (1.2 mL) was added afterwards and the reaction was shaken for 48 hours at 30 °C. Saturated aqueous Na2CO3 solution was added (1 mL) afterwards and the reaction mixture was extracted with EtOAc (4 × 5 mL). Combined organic layers were dried over Na2SO4, filtrated and converted without further purification. The conversion and optical purity was determined as described (vide supra). GC-MS (EI+, 70 eV): m/z (%) = 223 [M+] (2), 124 [C8H14N+] (25), 111 [C7H13N+] (100), 96 [C6H10N+] (61)
Biocatalytical synthesis of Δ1-piperideine (S)-6a (preparative scale)
Lyophilized cells of E. coli containing overexpressed ω-TA from Arthrobacter citreus (125 mg) were rehydrated in a KPi buffer (5.7 mL, pH 6.5, 100 mm) containing PLP (1 mm), NAD+ (1 mm), ammonium formate (150 mm), FDH (11 U), Ala-DH (12 U) and l-alanine (500 mm) at 22 °C for 30 minutes. The substrate 3a [36 mg (dissolved in 1.2 mL DMF), 0.15 mmol, 25 mm] was added afterwards and the reaction was shaken for 48 hours at 40 °C. Saturated Na2CO3 solution was added (0.50 mL) afterwards and the reaction mixture extracted with EtOAc (4 × 5 mL). Combined organic layers were dried over Na2SO4, filtrated and converted without further purification. The conversion and optical purity was determined as described (vide supra).
Diastereoselective reductions: synthesis of (−)-Isosolenopsin [(2S,6R)-1a] (representative procedure)
The crude solution of the biotransformation containing the corresponding cyclic imine (S)-6a was treated with palladium on activated charcoal (10% wt). The mixture was stirred vigorously and a stream of hydrogen was bubbled through the solution for 4 hours. After full conversion of the starting material as verified by GC and GC-MS, the solution was filtrated thorough a small plug of Celite 545 and cooled to 0 °C. Etherical HCl solution was added dropwise (~5 equiv.), the solvent removed and the precipitate collected. If necessary, the product was recrystallised from pure CHCl3 or purified via filter flash chromatography (Et2O + 1 vol% 2-PrNH2). (+)-Isosolenopsin [(2S,6R)-1a] was obtained as colourless solid in 98% yield (38.5 mg, 0.15 mmol) over two steps. Analytical data are full in agreement with those previously published.[6a] Melting point: 172 °C. RF [(Et2O + 1vol% 2-PrNH2) = 0.35. 1H-NMR (300 MHz, CDCl3): δH = 0.85 (t, 3J14,13 = 6.9 Hz, 3 H, 14-H), 1.15-1.50 (m, 15 H), 1.55 (d, 3JMe,1 = 6.3 Hz, 3 H, Me), 1.55-2.05 (m, 6 H), 2.06-2.20 (m, 1 H), 2.88 (mc, 1 H, 1-H), 3.06 (mc, 1 H, 6-H), 9.01 (brs, 1 H, NH2+), 9.39 (brs, 1 H, NH2+); 13C-NMR (75 MHz, CDCl3): δC = 14.2 (C-14), 19.6 (Me at C-1), 22.8, 23.0, 25.8, 27.6, 29.4, 29.5, 29.6, 29.7, 30.9, 32.0, 33.4, 54.7 (C-5), 58.8 (C-1); GC-MS (EI+, 70 eV): m/z (%) = 224 [M+] (1), 98 (C6H12N+) (100); [α]D20 = −9.0 (c 1.0, CHCl3, ee >99%, de >98%); Lit.:[6a] [α]D25 = +11.1 (c 1.02, CHCl3) for the opposite enantiomer.
Synthesis of (+)-Isosolenopsin [(2R,6S)-1a]
The product was prepared analogue the procedure described for its enantiomer albeit starting with the crude solution obtained from the biotransformation with (R)-Arthrobacter sp. (vide supra): [α]D20 = +8.7 (c 0.96, CHCl3, ee >99%, de >98%).
Supplementary Material
Acknowledgments
This work has been supported by the Austrian BMWFJ, BMVIT, SFG, Standortagentur Tirol and ZIT through the Austria FFG-COMET-Funding Program. H. L acknowledges DK “Molecular Enzymology” and financial support by the FWF (Project W9). Financial support by NAWI-Graz is acknowledged. We thank Barbara Grischek for her ongoing support in all projects.
References
- [1]a).Bates RW, Sa-Ei K. Tetrahedron. 2002;58:5957–5978. [Google Scholar]; b) Asano N, Nash RJ, Molyneux RJ, Fleet GWJ. Tetrahedron: Asymmetry. 2000;11:1645–1680. [Google Scholar]
- [2]a).Girling PR, Kiyoi T, Whitning A. Org. Biomol. Chem. 2011;9:3105–3121. doi: 10.1039/c0ob00996b. [DOI] [PubMed] [Google Scholar]; b) Buffat MGP. Tetrahedron. 2004;60:1701–1729. [Google Scholar]; c) Bailey PD, Millwood PA, Smith PD. Chem. Commun. 1998:633–640. [Google Scholar]; d) Baliah V, Jeyaraman R, Chandrasekaran L. Chem. Rev. 1983;83:379–423. [Google Scholar]
- [3]a).Breakman JC, Daloze D, Pasteels JM. In: Alkaloids: Biochemistry, Ecology, and Medicinal Applications. Roberts MF, Wink M, editors. Plenum Press; 1998. pp. 349–374. [Google Scholar]; b) Breakman JC, Daloze D, Braz J. Chem. Soc. 1996;7:251–256. [Google Scholar]; c) Jones TH, Blum MS. Tetrahedron. 1982;38:1949–1958. [Google Scholar]
- [4].Yi GB, McClendon D, Desaiah D, Goddard J, Lister A, Moffitt J, Vander Meer RK, deShazo R, Lee KS, Rockhold RW. Int. J. Toxicol. 2003;22:81–86. doi: 10.1080/10915810305090. [DOI] [PubMed] [Google Scholar]
- [5].Arbiser JL, Kau T, Konar M, Narra K, Ramchandran R, Summers SA, Vlahos CJ, Ye K, Perry BN, Matter W, Fischl A, Cook J, Silver PA, Bain J, Cohen P, Whitmire D, Furness S, Govindarajan B, Bowen JP. Blood. 2007;109:560–565. doi: 10.1182/blood-2006-06-029934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Selected examples since 2005: Gouault N, Roch ML, Campos Pinto G. de, David M. Org. Biomol. Chem. 2012;10:5541–5546. doi: 10.1039/c2ob25685a. Reddy CR, Latha B. Tetrahedron: Asymmetry. 2011;22:1849–1854. Leijohndal K, Borén L, Braun R, Bäckvall J-E. J. Org. Chem. 2009;74:1988–1993. doi: 10.1021/jo8025109. Kumar RSC, Sreedhar E, Reddy GV, Babu KS, Rao JM. Tetrahedron: Asymmetry. 2009;20:1160–1163. González-Gómez JC, Foubelo F, Yus M. Synlett. 2008:2777–2780. Takahata H, Saito Y, Ichnose M. Org. Biomol. Chem. 2006;4:1587–1595. doi: 10.1039/b601489e. Dobbs AP, Guesné SJJ. Synlett. 2005:2101–2103. Wang X, Dong Y, Sun J, Xu X, Li R, Hu Y. J. Org. Chem. 2005;70:1897–1900. doi: 10.1021/jo0480444. Girard N, Gautier C, Malassene R, Hurvois J-P, Moinet C, Toupet L. Synlett. 2004:2005–2009. Wang X, Dong Y, Sun J, Xu X, Li R, Hu Y. J. Org. Chem. 2005;70:1897–1900. doi: 10.1021/jo0480444.
- [7]a).Simon RC, Grischek B, Zepeck F, Steinreiber A, Belaj F, Kroutil W. Angew. Chem. Int. Ed. 2012;51:6713–6716. doi: 10.1002/anie.201202375. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124:6817–6820. [Google Scholar]; b) Simon RC, Zepeck F, Kroutil W. Chem. Eur. J. 2013;19:2859–2865. doi: 10.1002/chem.201202793. [DOI] [PubMed] [Google Scholar]
- [8].Selected review articles: Mathew S, Yun H. ACS Catal. 2012;2:993–1001. Malik MS, Park E-S, Shin J-S. Appl. Microbiol. Biotechnol. 2012;94:1163–1171. doi: 10.1007/s00253-012-4103-3. Tufvesson P, Lima-Ramos J, Jensen JS, Al-Haque N, Neto W, Woodley JM. Biotechnol. Bioeng. 2011;108:1479–1493. doi: 10.1002/bit.23154. Koszelewski D, Tauber K, Faber K, Kroutil W. Trends Biotechnol. 2010;28:324–332. doi: 10.1016/j.tibtech.2010.03.003. Turner NJ, Truppo M. In: Chiral Amine Synthesis: Methods, Developments and Applications. Nugent TC, editor. Wiley-VCH; Weinheim: 2010. pp. 431–459. Hailes HC, Dalby PA, Lye GJ, Baganz F, Micheletti M, Szita N, Ward JM. Curr. Org. Chem. 2010;14:1883–1893. Ward J, Wohlgemuth R. Curr. Org. Chem. 2010;14:1914–1927.
- [9].Selected examples of ω-TA in the asymmetric reductive amination: Steffen-Munsberg F, Vickers C, Thontowi A, Schätzle S, Tumlirsch T, Humble MS, Land H, Berglund P, Bornscheuer UT, Höhne M. ChemCatChem. 2013;5:150–153. Mutti FG, Fuchs CS, Pressnitz D, Turrini NG, Sattler JH, Lerchner A, Skerra A, Kroutil W. Eur. J. Org. Chem. 2012:1003–1007. Mutti FG, Fuchs CS, Pressnitz D, Sattler JH, Kroutil W. Adv. Synth. Catal. 2011;353:3227–3233. Schätzle S, Steffen-Munsberg F, Thontowi A, Höhne M, Robins K, Bornscheuer U. Adv. Synth. Catal. 2011;353:2439–2445. Truppo MD, Rozzell JD, Turner NJ. Org. Process Res. Dev. 2010;14:234–237. Höhne M, Schätzle S, Jochens H, Robins K, Bornscheuer UT. Nat. Chem. Biol. 2010;6:807–813. doi: 10.1038/nchembio.447. Fuchs M, Koszelewski D, Tauber K, Kroutil W, Faber K. Chem. Commun. 2010;46:5500–5502. doi: 10.1039/c0cc00585a. Koszelewski D, Göritzer M, Clay D, Seisser B, Kroutil W. ChemCatChem. 2010;2:73–77. Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Javis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ. Science. 2010;329:305–309. doi: 10.1126/science.1188934. Koszelewski D, Lavandera I, Clay D, Guebitz GM, Rozzell D, Kroutil W. Angew. Chem. Int. Ed. 2008;47:9337–9340. doi: 10.1002/anie.200803763. Angew. Chem. 2008;120:9447–9480. Koszelewski D, Lavandera I, Clay D, Rozzell D, Kroutil W. Adv. Synth. Catal. 2008;350:2761–2767. selected examples of ω-TA in the kinetic resolution: Cho B-K, Park H-Y, Seo J-H, Kim J, Kang T-J, Lee B-S, Kim B-G. Biotechnol. Bioeng. 2008;99:275–284. doi: 10.1002/bit.21591. Bea H-S, Park H-J, Lee S-H, Yun H. Chem. Commun. 2011;47:5894–5896. doi: 10.1039/c1cc11528f. Hanson RL, Davis BL, Goldberg SL, Johnston RM, Parker WL, Tully TP, Montana MA, Patel RN. Org. Process Res. Dev. 2008;12:1119–1129.
- [10].Screening assays for ω-TA: Sehl T, Simon RC, Hailes HC, Ward JM, Schell U, Pohl M, Rother D. J. Biotechnol. 2012;159:188–194. doi: 10.1016/j.jbiotec.2011.12.023. Hopwood J, Truppo MD, Turner NJ, Lloyed RC. Chem. Commun. 2011;47:773–775. doi: 10.1039/c0cc02919j. Truppo MD, Rozzell JD, Moore JC, Turner NJ. Org. Biomol. Chem. 2009;7:395–398. doi: 10.1039/b817730a.
- [11].Determination of the absolute configuration of cis- and trans-Solenopsins: Leclercq S, Thirionet I, Broeders F, Daloze D, Vander Meer R, Braekman JC. Tetrahedron. 1994;50:8465–8478.
- [12].Methods to shift the equilibrium: Truppo MD, Turner NJ, Rozzell JD. Chem. Commun. 2009:2127–2129. doi: 10.1039/b902995h. Höhne M, Kühl S, Robins K, Bornscheuer UT. ChemBioChem. 2008;9:363–365. doi: 10.1002/cbic.200700601. Shin J-S, Kim B-G. Biotechnol. Bioeng. 1999;65:206–2011. Yun H, Yang Y-H, Cho B-K, Hwang B-Y, Kim B-G. Biotechnol. Lett. 2003;25:809–814. doi: 10.1023/a:1023500406897. Cassimjee KE, Branneby C, Abedi V, Wells A, Berglund P. Chem. Commun. 2010;46:5569–5571. doi: 10.1039/c0cc00050g.
- [13]a).Kawano S, Ito N, Yasohara Y. 2007. WO 2007/139055.; b) Ito N, Kawano S, Hagesawa J, Yasohara Y. Biosci. Biotechnol. Biochem. 2011;75:2093–2095. doi: 10.1271/bbb.110240. [DOI] [PubMed] [Google Scholar]; c) Pannuri S, Kamat SV, Garcia ARM. Cambrex North Brunswick Inc.; 2006. WO 2006/063336 A2. [Google Scholar]; d) Yamada Y, Iwasaki A, Kizaki N. Kaneka Corporation; 2000. EP 0987332 A1. [Google Scholar]
- [14].Abe K, Okumura H, Tsugoshi T, Nakamura N. Synlett. 1984:603–605. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.