

Abstract
Macromolecular crowding decreases the diffusion rate, shifts the equilibrium of protein-protein and protein-substrate interactions, and changes the protein conformational dynamics. Collectively, these effects contribute to enzyme catalysis. Here we describe how crowding may bias the conformational change and dynamics of enzyme populations and in this way affect catalysis. Crowding effects have been studied using artificial crowding agents and in-vivo-like environment. These studies revealed a correlation between protein dynamics and function in the crowded environment. We suggest that crowded environments can be classified into uniform crowding and structured crowding. Uniform crowding represents random crowding conditions created by synthetic particles with a narrow size distribution. Structured crowding refers to the highly coordinated cellular environment, where proteins and other macromolecules are clustered and organized. In structured crowded environments, the perturbation of protein thermal stability may be lower; however, it may still be able to effectively dynamically modulate function. Dynamic, allosteric enzymes could be more sensitive to cellular perturbations if their free energy landscape is flatter around the native state; on the other hand, if their free energy landscape is rougher, with high kinetic barriers separating deep minima, they could be more robust. Above all, cells are structured; and this holds both for the cytosol and for the membrane environment. The crowded environment is organized, which limits the search; and the crowders are not necessarily inert. More likely, they too transmit allosteric effects, and as such play important functional roles. Overall, structured cellular crowding may lead to higher enzyme efficiency and specificity.
Keywords: Macromolecular crowding, conformational selection, protein dynamics, allostery, enzymemics, energy landscape
1. Introduction
Even though it is still highly debated whether enzyme dynamic motions contribute to decrease the chemical reaction barrier, the consensus picture emerging from experimental and computational studies indicates that enzyme conformational transitions are highly organized with stepwise conformational selection in catalysis, which increases enzyme specificity and efficiency [1, 2]. The close coupling of enzyme conformational dynamics and catalysis can be viewed as the outcome of the enzyme’s adaptation to aqueous-based life. Stable, folded macromolecules in crystal structures have well defined three-dimensional structures, which are not as dynamic as those in aqueous solution. In solution, enzymes exist as conformational ensembles and their populations follow thermodynamic distributions [3–5]. Local energy fluctuations in water range between 10 to 20 kcal/mol [6], which is enough to perturb a well folded enzyme to a vast number of states. Enzyme dynamics in catalysis are not only real; they are inevitable [2].
Enzyme dynamics may involve small or large amplitude conformational changes. Large conformational change may involve nanoscale movements of domains or loops [7]. One example is the dengue virus NS2B-NS3 protease, for which the functionally important C-terminal segment of the NS2B cofactor dissociates in the open state via a large structural change to produce the closed state and confer activity [8] through a conformational selection mechanism [9]. Large conformational changes typically relate to function, for example, to accommodate the conformational reorganization required to stabilize the transition states [10] or to allow allosteric regulation [11, 12]. The enzyme’s dynamic conformational changes are also necessary for signal transduction across long distances in the cell [7, 13].
The enzyme’s native playground is in a highly crowded cellular environment. In the E. coli cell, proteins and nucleic acids can occupy 20–30% of the total volume [14]. Macromolecular crowding decreases the diffusion rate [14, 15], shifts the equilibrium of protein-protein association and of protein-substrate interaction, and changes the protein’s conformational dynamics. These effects collectively contribute to enzyme catalysis. Enzymes respond differently to crowding agents; the activity of some decreases and of others increases, and the extents differ as well. For example, while macro- molecular crowding has minimal effects on the kinetics and function of yeast hexokinase in physiological solutions [16], it can significantly increase the thermal stability of catalase; and the overall structure becomes more rigid [17]. Crowding effects are also concentration-dependent. For the monomeric multi-copper oxidase, Saccharomyces cerevisiae Fet3p, at low amounts of crowding agent, both the Km (substrate binding) and the kcat (catalytic efficiency) increase; whereas at higher crowding levels, both parameters decrease [18]. Enzyme conformations also change with crowding conditions. The functional properties of the tryptophan synthase α2β2 complex in the presence of the crowding agents dextran 70 and ficoll 70 indicate that the rates of the conformational transitions which are associated with catalysis and regulation are reduced, and an open and less catalytically active conformation is stabilized [19]. Given these complex scenarios, one important question is whether the crowing effects simply reduce the extent of the enzyme dynamics or whether enzyme catalysis can benefit from a crowded environment. In this article, we review experimental data, focusing on the relationship between marcomolecular crowding and conformational dynamics as they relate to enzyme catalysis in a crowded environment. Experimental data present different crowding outcomes: this could be because of different experimental conditions, type of molecular crowder, and the concentration of the enzyme and its substrates. While bearing in mind these possible caveats, the results may also reflect the different types of systems and cofactors, enzyme conformations and chemical reactions. Overall, the emerging picture suggests coupling between conformational dynamics and structured cellular crowding which could increase the enzyme specificity.
2. Correlation between crowding effects and enzyme conformational dynamics and activity
The effects of crowding on internal protein dynamics can be well illustrated by the HIV-1 protease. The distance between a pair of flaps in the HIV-1 protease can vary from 5 Å in the closed form to 22 Å in the open form. Molecular dynamics simulations indicated that flap opening is significantly suppressed in a highly crowded environment [20, 21]. It is reasonable to expect that the decrease in the open state and the reduction of the enzyme-ligand diffusion encounter rate are likely to slow the in vivo enzymatic activity [20]. Unfortunately, there is no published experimental study which would allow a comparison with these theoretical predictions for the HIV-1 protease. However, experimental studies of the reverse proteolysis, the formation of peptide bond, show that crowding can enhance protease-catalyzed synthesis of model peptides [22]. Extensive studies of α-chymotrypsin also revealed a correlation between protein dynamics and crowding effects.
It is well established that conformational dynamics of α- chymotrypsin correlate with its activity. α-chymotrypsin undergoes a reversible conformational change from an inactive chymotrypsino- gen-like structure at high pH to an active conformation at neutral pH. Molecular dynamics simulations revealed that bovine (active) and rat (inactive) chymotrypsin explore different regions of the conformational space, indicating that conformational dynamics is more important than the sequence differences for activation [23]. Consistent with the kinetic parameters observed by fluorescence stopped-flow spectroscopy, targeted molecular dynamics simulations revealed multiple pathways for chymotrypsin activation [24]. Banerjee and Pal studied the correlation of the conformational dynamics at the active site of α-chymotrypsin with enzyme activity. Interestingly, they found that the highest catalytic efficiency α-chymotrypsin is at 37 °C, the typical body temperature of homeo- thermal animals. Site selective fluorescence circular dichroism (FDCD) studies reveal that the conformational flexibility of the enzyme affects the structural perturbation at the active site. Consistently, the hydrodynamic diameter of the α-chymotrypsin decreases considerably with increasing temperature, indicating that the enzyme is more compact at higher temperatures [25].
Amide H/D exchange kinetics indicated that glycosylation can stabilize α-chymotrypsin and reduce its structural dynamics [26]. Enzyme catalysis kinetics indicated that glycosylation does not affect substrate binding (Ks) but decreases the rate of the catalytic steps. Therefore, for α-chymotrypsin, it is clear that the higher structural fluctuations decrease the catalytic rate [27]. With a dynamic- activity correlation, one would expect that macromolecular crowding should decrease the structural dynamics and the activity of α-chymotrypsin. However, it has been observed that crowding could either increase or decrease the catalytic rate of α-chymotrypsin, depending on the crowding molecules that are added to the solution. The addition of poly(ethylene glycol) (PEG) as crowding molecules increases the affinity of the enzyme to its substrate, however, followed by a decrease in the turnover number (kcat) [28]. Chemical linkage of PEG to α-chymotrypsin may represent an ‘extreme’ of a crowded environment. PEG conjugation has been shown to increase the α-chymotrypsin thermostability and to decrease the protein structural dynamics [29]. While the dehydration/hydration processes may also play a role, solvation is energetically coupled to the conformational changes of α-chymotrypsin [28]. Adding dextrans of various molecular weights to the reaction solutions was also tested. An increase in the dextran concentration decreases vmax and increases Km. While the increase in Km can be attributed to the slower protein diffusion rate due to crowding [30], the overall change in the catalytic rate is consistent with the rate using the PEG as the crowding molecules, indicating protein stabilization by dextran crowding. It was also found that the exclusion volume, not the size of the dextran, changes the catalytic rate of α-chymotrypsin. However, the different behavior was observed using much larger nanoparticles to represent the crowded environment. Tetraethylene glycol (TEG) functionalized gold nanoparticles with 2 nm core diameters (AuTEG) enhance the α-chymotrypsin (ChT) enzyme activity in a substrate-selective fashion [31]. While the hydrolysis of three substrates (N-succinyl-L-phenylalanine-p-nitroanilide (SPNA), N- glutaryl-phenylalanine-p-nitroanilide (GPNA), and N-benzyoyl- tyrosine-p-nitroanilide (BTNA)) was not affected by the crowding, a marked increase in activity (Kcat/Km) with the most hydrophobic substrate N-succinyl-alanine-alanine-proline-phenylalanine-p-nitroanilide (TP) was observed. Interestingly, high molecular weight PEG was shown to have similar effects as the functionalized gold nanoparticles, in contrast to the lower molecular weight PEG [31].
3. Allosteric control and crowding
The change in protein dynamics in a crowded environment reflects an intrinsic property of dynamic proteins: allostery [32]. Direct kinetic assays using isothermal calorimetry have shown that molecular crowding and allosteric activators affect pyruvate kinase kinetics in similar ways [33]. Allosteric regulation of enzyme activity is always accompanied by changes in protein dynamics [34]. Figure 1 shows two examples of allosteric enzymes. The first (Figure 1A) is a 145 residue mini intein, which catalyzes protein splicing [35]. Inteins are phylogenetically diverse self-splicing proteins that are of great functional, evolutionary, biotechnological, and medical interest [36]. It has been shown that intein activity can be regulated by a loop (connecting two β-strands from the N- and C-terminal intein subdomains of the mini-intein) and by the V67L mutation [35]. Active intein has a longer loop (VR96DVE99TGE102--L404). While reducing the loop length to VR96DVE99L404 (with the 100- through-403 region deleted) still preserves the activity, further shortening to VR96L404 inactivates the enzyme. The V67L mutation similarly illustrates the dynamic nature of the allosteric regulation of this intein, which globally enhances splicing and related cleavage reactions. While this mutation in the mini intein causes little change in crystal structures, it significantly slows the hydrogen-exchange rates globally, indicating a shift to more stable conformations and narrowing the ensemble distribution [35, 37]. The enhanced activity, together with the reduced dynamics of intein is similar to the crowding effects on α-chymotrypsin discussed in the last section.
Fig. 1.
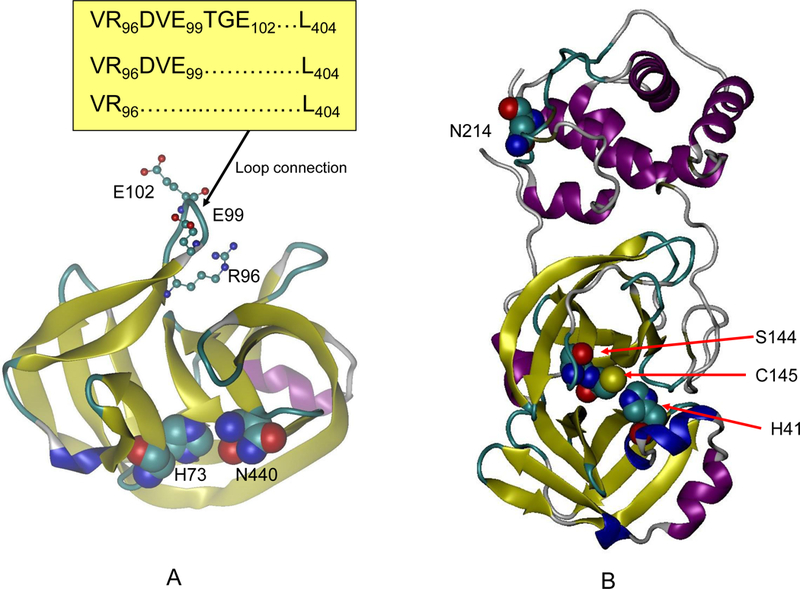
Allosteric proteins are sensitive to crowding effects. (A) The mini intein activity can be allosterically modulated by a loop distant from the active site. Two residues in the active site (H73 and N440) are represented by large balls; and residues in the loop are represented by small balls and sticks. Three loops with different sizes were tested in reference [35]. Active intein has a longer loop (VR96DVE99TGE102L404). While reducing the loop length to VR96DVE99... L404 still preserved the activity, further shortening to VR96L404 inactivates the enzyme. (B) The structure of allosteric protein SARS-Co 3CL peptidase, for which the catalytic activity increases in a crowded environment. Three active site residues (H41, S144, and C145) are shown as large balls in one domain. The mutant position N214 is shown as large balls in another domain.
Figure 1B shows the structure of another allosteric protein SARS-Co 3CL (3CLpro) peptidase, which is structurally and functionally similar to a-chymotrypsin. The 3CLpro peptidase activities increase in crowded environments [38]. One possible reason is that the active form of 3CLpro is a dimer [38]. Alternatively, dynamics change, rather than dimer formation, could underlie the crowding effects of 3CLpro’s activity.
Crowding could shift the monomer-dimer equilibrium to favor the dimer formation [38, 39]. This is the case for RNase A, which forms 3D domain-swapped oligomers with novel enzymatic and biological activities [40]. However, crowding effects do not necessarily favor the association of proteins to form an active oligomer state. A counter-example can be found in the muscle glycogen phosphorylase b (Phb). The Phb is a classical-like allosteric protein with T - R transition, with the active R state being the smaller dimer and the inactive T state the tetramer. Adding osmolytes as crowding molecules favors the inactive unassociated T state [41, 42]. The reason could be that the dimer form of Phb is more compact. It is interesting to see that under the same crowding conditions, the association of phosphorylase kinase (PhK) and its interaction with glycogen and the heat shock protein Hsp27 can be highly stimulated [43, 44].
On its own, the dimer form does not guarantee the activity of the 3CLpro. An inactive mutant with the N214A mutation in the extracellular domain of the 3CLpro still adopts a dimer structure which is almost identical to that of the wild-type [45]. The mutation site is distant from the active site. Molecular dynamics (MD) simulations revealed that the N214A mutant has much higher conformational flexibility than the WT enzyme [45]. Combining the crowding effects and the N214A mutational effects, one can see that a crowded environment restricts the conformational flexibility of the 3CLpro dimer; and thus increases its activity.
Isochorismate synthase (EntC) is another example which illustrates that macromolecular crowding increases the intrinsic activity of an enzyme by inducing conformational changes in the enzyme rather than through macromolecular association [46]. The EntC is a monomeric enzyme which catalyzes the reversible conversion between chorismate and isochorismate in Escherichia coli. Ficoll addition leads to two fold increase of Kcat/Km. In comparison to the EntC-catalyzed reaction, other reactions catalyzed by two homologous trimer and tetramer enzymes (EntB and LDH) are similarly stimulated by the same crowding conditions, indicating that protein-protein association is not a controlling factor for these enzymes. Changes of CD spectrum and Trp fluorescence revealed that conformational changes are responsible for the increase of the intrinsic activities of these enzymes [46].
Proteins can accommodate a crowded environment by different conformational changes. While red shift in Trp fluorescence was observed for EntC in crowded environments [46], blue shift was observed for the allosteric ADP-glucose pyrophosphorylase [47]. Escherichia coli ADP-sugar pyrophosphatase (AspP) is an ultrasensitive hydrolase that catalyzes the hydrolytic breakdown of ADP-glucose linked to glycogen biosynthesis. The AspP activity is strongly enhanced by macromolecular crowding [48]. Molecular crowding renders AGPase more sensitive to the interplay between the allosteric regulators and consequently enhances the ultrasensitive response. Fourth-derivative spectroscopy and size-exclusion chromatography indicated that the ultrasensitive behavior is correlated with intramolecular conformational changes induced in the tertiary structure of the homo-tetrameric enzyme [47].
One of the common features of allosteric proteins is that their dynamics is designed to facilitate conformational change [49]. At the same time, from the functional standpoint, high sensitivity to crowding can present problems. The LRP protein is an example which illustrates that crowding effects could be coupled with function. The LRP transcriptional regulators are widely distributed among prokaryotes, bacteria and archaea. The architecture of LRP proteins includes two distinct domains that harbor the regulatory (effector-binding) site and the active (DNA-binding) site, and the two domains are connected by a flexible hinge region. This structural feature and experiments suggest an allosteric switch for the Lrp-like regulators [50–52]. LRP binding with DNA is stimulated under macromolecular crowding conditions [53]. In comparison, another DNA binding protein fis has no noticeable response to the same crowding conditions, indicating synergism of allostery and crowding effects for LRP DNA binding [53].
DNA binding proteins are similarly allosteric [54], and experimental data present mixed results with regard to the crowding effects. The hydrolysis of a 29-mer double-stranded DNA by DNase I and S1 nuclease was substantially enhanced by molecular crowding using PEG; however, molecular crowding had little effect on hydrolysis by exo III and exo I exonucleases, suggesting differential crowding effects on the catalytic activities under these conditions [55].
DNA replication is under high specificity pressure to ensure fidelity and retain efficiency under crowding conditions [56–60]. Crowding can help regulated stress response [61]. Under crowded conditions, dramatic improvements in all parameters of RT-PCR were observed, including 8- to 10-fold greater sensitivity, enhanced polymerase processivity, higher specific amplicon yield, greater primer annealing and specificity, and enhanced DNA polymerase thermal stability [62]. Similarly, polymerase activity assays under various crowding conditions demonstrated that the activities of T7 and Taq DNA polymerases depend on the molecular weight and concentration of the crowding agent [63].
Inter- and intra-domain motions in well-defined regions of non-ribosomal peptide synthetases (NRPS) are important for its activity [64]. One of the NPRS, enterobactin synthetase, experiences significant conformational change in crowded solutions mimicking the intracellular environment. The structural change correlates well with the extent of the crowding-induced side product suppression in the nonribosomal enterobactin synthesis [65].
4. Enzyme dynamics and energy landscapes with artificial uniform and in vivo structured crowding
The free energy landscapes of protein folding and binding provide insights into protein structure and function which relate to allosteric regulation and enzyme catalysis [1, 2, 32, 66–68]. Macro- molecular crowding changes the landscape, as evidenced by computational [69–71] and experimental studies [72, 73]. However, there are differences between the crowding effects caused by the artificial crowding under random conditions and the in vivo cellular environment. For example, there are differences (1) between proteins and artificial crowding agents; and (2) between many copies of few proteins and the proteome. The corresponding free energy landscapes could differ substantially. Here we consider two kinds of crowding effects on the protein free energy landscape which represent two extreme cases. The first is uniform crowding and the second is structured crowding (Figure 2). The question is how the free energy landscape, here represented by two conformations (α and β, figure 2A), could be affected.
Fig. 2.
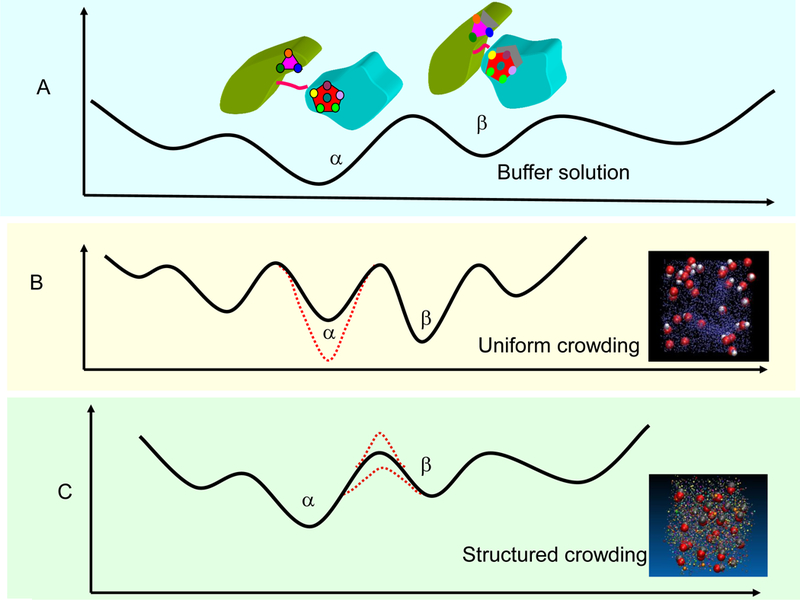
Changes of the protein free energy landscape under uniform crowding and structured crowding environment. (A) The original energy landscape is represented by two conformational states α and β in a buffer solution. (B) Uniform crowding considers the effects caused by hard sphere crowding agents, which have uniform size and repulsive interactions with protein solute. This represents the strong confinement of proteins. In an environment of the uniform crowding, the protein energy landscape may experience strong compression which may reflect protein folding-unfolding transition. Depending on the molecular size of the crowding agents, the relative stabilities of different conformers could increase or decrease in a uniform crowded environment. (C) Structured crowding refers to a highly coordinated cellular environment, where the overall proteome and other macromolecules pre-organized into structured clusters. In a structured crowded environment, the protein free energy landscape may be more similar to that in buffer solution. However, protein dynamics could be more sensitive than thermal stability.
Uniform crowding (Figure 2B) considers effects caused by hard sphere crowding agents, which have uniform size and repulsive interactions with the protein solute. This scenario represents strong protein confinement. Crowding agents with sizes smaller than the protein solute could have larger effects. When the size of the crowding agent increases, there is an upper limit for the crowding effects, which has been shown by Milklos et al [74]. Milklos et al studied the stability and ps-ns internal dynamics of a small globular protein (radius ~2 nm) crowded by a large synthetic microgel particle (radii of ~300 nm). They found no change in protein rotational or ps-ns backbone dynamics; only mild stabilization at a volume occupancy of 70%, which approaches the occupancy of closely packed spheres [74]. However, it is difficult to predict the crowding effect when the size of the crowding agent is uniformly distributed. For example, proteins with aspherical shape may experience large conformational change in the presence of synthetic Ficoll70 [75]. Similarly using Ficoll70 as crowding agent, a recent study with combined experiment and computer simulation demonstrated that macromolecular crowding dramatically affects the structure, function, and folding landscape of phosphoglycerate kinase (PGK) [73]. Overall, the relative stabilities among different conformers could either increase or decrease by uniform crowding (Figure 2B).
Structured crowding refers to the highly coordinated cellular environment, where the proteome and other macromolecules are preorganized. Proteins often cluster and pre-organize to perform functions, for example, by forming transcriptional factories [76], which increases the effective local concentration. Pre-organization goes beyond clustering, because it further reduces the diffusion-and- collision times: it positions the proteins in an orientation which is closer to the native functional one. Signal transduction occurs in an organized microenvironment [7, 13] where elements of a signaling pathway are connected functionally and spatially [77, 78]. The organization of proteins with different charges and sizes can be important for fast cellular response in fluctuating environments. There are hundreds of proteins in the cytoplasm, with different sizes and shapes. Our study of 206 kinds of proteins in a theoretical minimal proteome model indicated that proteins with different sizes and charges are well organized into clusters (Xu, Y., Nussinov, R, and Ma, B. To be published). Therefore, while the particle diffusion can be severally compromised in a highly crowded environment, in a crowded structured environment, the overall protein energy landscape may be less perturbed (Figure 2C). This scenario also arises from the study of a cytoplasmic model that includes 50 of the most abundant types of macromolecules [79]. McGuffee and Elcock found that the overall perturbation of protein thermal stability is small, because when considering the excluded volume, the crowding effect may stabilize the folded state; however, the effect can be counterbalanced by the favorable energetic interactions which take place in unfolded conformations [79]. Proteins are better crowding agents than synthetic particles. For example, using two globular proteins (bovine serum albumin and hen-egg-white lysozyme) as crowding agents, chymotrypsin inhibitor 2 (CI2) was found to be only slightly destabilized [72]. Moving closer to a real crowded environment in the cell, hen egg white has been used to study the dynamics and stability of several proteins [80]. It was found that while the dynamic parameters of the studied protein are clearly affected by the crowded medium, the thermal stability of the protein is similar to that in buffer [80]. In this sense, in the structured crowded environment, the protein energy landscape may be more similar to that in buffer solution. However, protein dynamics are more sensitive than thermal stability, and the dynamics of globular protein may be more sensitive than the dynamics of intrinsically disordered protein [81, 82].
The classification into uniform and structured crowding may be simplified; and realistic crowding effects could be between these two ends of the spectrum. Protein dynamics may contribute more to the change in enzyme catalysis in crowded environments than thermal stability. This is well illustrated by the effect of a crowded solution on the reaction rates of the decarboxylating enzymes urease, pyruvate decarboxylase and glutamate decarboxylase, with the crowding agent being proteins and synthetic polymers [83]. It was found that increasing the concentration of globular proteins up to 30% crowding concentration caused a dramatic rise in enzyme activity, however, then the activity decreased; on the other hand, polymers caused a concentration-dependent decrease in activity [83].
5. Overview and Conclusions
Cells are crowded; and as John Ellis pointed out over a decade ago, while these effects obvious, they are (often) underappreciated [84] . For the E. coli, it was estimated that the cytosol contains about 300–400 (mg/ml) of macromolecules [85]. The bacterium contains up to 4,288 different types of proteins, with ~1,000 of these types produced at sufficiently high levels to be easily detected, DNA and RNA molecules [86]. Crowding reduces the volume of solvent available for other molecules in solution, thus increasing their effective concentrations. In eukaryotes, the interior of the cell is even more crowded: cells also contain protein filaments that make up the cytoskeleton. Pielak has highlighted crowding effects [87], and emphasized that experiments which are carried out on dilute samples (under10g/liter) are extremely different than those in the living cell. This high crowding led Allen Minton to ask in his 2006 Commentary [88], “How can biochemical reactions within cells differ from those in test tubes?” Minton argued that nonspecific interactions in the interior of the cell can greatly influence the equilibriums and rates of reactions. Minton classified the consequences of nonspecific interactions in the cellular interior into three types of phenomena: macromolecular crowding, confinement and adsorption. He further pointed out that these nonspecific ‘background’ interactions may be repulsive, which would lead to exclusion, thus increase the effective concentration and chemical activity; or attractive, which would lead to nonspecific associations or adsorption. The predominantly repulsive background interactions may alter the dissociation constants by enhancing the rate and extent of macromolecular associations in solution [89]. Crowding may also affect enzyme reactions involving small molecules if there is a large change in the shape of the enzyme [90]. Reaction dynamics under crowded conditions are affected by a high concentration of reactants [91].
However, cells are not only crowded; they are also structured; and this holds not only for the membrane environment but for the cytoplasm as well [92, 93]. Hence, it is not only the membrane- bound compartmentalization and biochemical association with subcellular organelles; it also relates to the spatial regulation inside the cell. The microtubule-dependent organization of non-membranous components helps direct cellular function. The sequestration of GEF-H1 provides a clear example [94]. Thus, in contrast to the view of the cytoplasm as a free and fluid environment, the microtubule network provides a platform for a ‘structured cytoplasm’. Binding to microtubules can be either directly, or indirectly, through other microtubule-binding scaffolding molecules; all dynamic and allosteric. This can cause partial sequestration of proteins by the microtubule network. The extent of sequestration can depend on binding affinities, and microtubule density. The cytoskeleton mesh also effectively divides the cytosol into a network of narrow pores. Further, while highly dynamic, the genome is also pre-organized and highly structured. Structuring provides docking surfaces, and is critical for function. The so-called ‘matrix’ or scaffolding proteins which organize the spatial localization of protein assemblies, have also been demonstrated to be hubs for controlling function as in the case of Ste5 in the MAPK signaling pathway [95, 96], and cullin [97]. Matrix proteins are also dynamic and allosterically regulate function [34, 98]. Thus, in vivo, proteins do not diffuse freely, and do not travel over large distances. Spatially organized crowded environment is a more realistic description; it also implies that allosteric effects can be transmitted via crowders. Protein crowders should not be viewed as necessarily inert media or of specific uniform shapes. During (specific or nonspecific) binding both partners can change their shapes or dynamics, which has been viewed as a ‘molecular dance’ [99]. Further, residence times and molecular motility of the players involved are also important in the organization of the intracellular space. Overall, both cooperative protein-protein interactions and membrane properties play key roles in cell signaling and vesicle trafficking and thus in diffusion in the crowded cell. They allow switching between different protein states, either soluble in the cytoplasm or bound to the membrane, for example, by modulating the chemical composition of the membrane [100]; by changing its physical properties, such as curvature or fluidity; or by lipophilic post- translational modification, such as myristoylation, palmitoylation, prenylation, etc which are often allosteric. For the Escherichia coli Min system, in vitro studies clarified the order of events in space and time and the interplay between cooperative binding of MinD to the membrane and positive feedback during protein detachment [101]. These in vivo considerations limit the space, distance to be travelled and thus diffusion time in the spatially pre-organized and structured cell and may buffer conformational dynamic consequences.
To conclude, here, we focused on macromolecular crowding effects on enzyme conformations, dynamics, and catalysis. Computational and experimental studies revealed that enzyme conformations, dynamics and catalytic rates differ under crowding conditions. Most of the experimental studies use synthetic crowding agents to probe crowding effects, which may not be realistic and may have only limited relevance to in vivo conditions [102, 103]; however, some in-vivo-like conditions have also been published [79, 80, 82, 83]. Nevertheless, no matter what kinds of crowding agents were used, all provided valuable insights into the dynamic nature of proteins and protein functional adaptation to crowded environments. We suggest that crowded environments can be classified into uniform and structured crowding. Uniform crowding represents crowded conditions created by synthetic particles with narrow size distribution. Such conditions may elicit large protein structure and dynamic perturbations, and facilitate experimental investigation. Structured crowding refers to the highly coordinated cellular environment, where protein and other macromolecules are clustered and organized. Structured crowded environments may perturb less the protein thermal stability, and allow dynamic modulation of protein function. Allosteric proteins with flat free energy landscape may be more sensitive to the crowded cellular environment. Proteins with rougher landscapes may be more robust. Dynamic regulation of allosteric enzymes may result in efficiency and specificity under crowding conditions.
While most studies focus on the crowded cytoplasm environment, it behooves us to note that cell membranes are also crowded. For example, the enzymatic activity of a proteolytic enzyme, the Subtilisin Carlsberg (SC) in anionic sodium dodecyl sulfate (SDS) micellar medium, has been explored and found to be retarded compared to that in bulk buffer [104]. Escherichia coli dihydrofolate reductase (DHFR) is also a well studied allosteric enzyme [105–107], which also experiences large magnitudes of crowding effects on the vesicle surface [108]. Finally, similar to protein, RNA enzymes are also subjected to crowding effects. For example, the hammerhead ri- bozyme activity can be increased by a factor of 2–6 by PEG [109]. In particular, membrane environments are well known to be preorganized. Thus, the underlying principle of coupling of enzyme dynamics to activity in structured crowded environments is expected to be general.
Acknowledgments
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, NCI, Center for Cancer Research.
References:
- 1.Ma B, Nussinov R (2010) Curr Opin Chem Biol 14: 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma B, Kumar S, Tsai CJ, Hu Z, Nussinov R (2000) J Theor Biol 203: 383. [DOI] [PubMed] [Google Scholar]
- 3.Ansari A, Berendzen J, Bowne SF, Frauenfelder H, Iben IET, Sauke TB, Shyamsunder E, Young RD (1985) Proceedings of the National Academy of Sciences of the United States of America 82: 5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller DW, Dill KA (1997) Protein Sci 6: 2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dill KA, Chan HS (1997) Nature Structural Biology 4: 10. [DOI] [PubMed] [Google Scholar]
- 6.Ohmine I, Tanaka H, Wolynes PG (1988) Journal of Chemical Physics 89: 5852 [Google Scholar]
- 7.Ma B, Nussinov R (2009) Proc Natl Acad Sci U S A 106: 6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de la Cruz L, Nguyen TH, Ozawa K, Shin J, Graham B, Huber T, Otting G (2011) J Am Chem Soc [DOI] [PubMed] [Google Scholar]
- 9.Ekonomiuk D, Caflisch A (2009) Protein Sci 18: 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith AJ, Muller R, Toscano MD, Kast P, Hellinga HW, Hilvert D, Houk KN (2008) J Am Chem Soc 130: 15361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osmulski PA, Hochstrasser M, Gaczynska M (2009) Structure 17: 1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hindie V, Stroba A, Zhang H, Lopez-Garcia LA, Idrissova L, Zeuzem S, Hirschberg D, Schaeffer F, Jorgensen TJ, Engel M, Alzari PM, Biondi RM (2009) Nat Chem Biol 5: 758. [DOI] [PubMed] [Google Scholar]
- 13.Nussinov R (2011) Mol Biosyst [Google Scholar]
- 14.Konopka MC, Shkel IA, Cayley S, Record MT, Weisshaar JC (2006) J Bacteriol 188: 6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konopka MC, Sochacki KA, Bratton BP, Shkel IA, Record MT, Weisshaar JC (2009) J Bacteriol 191: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olsen SN, Ramlov H, Westh P (2007) Comp Biochem Physiol A Mol Integr Physiol 148: 339. [DOI] [PubMed] [Google Scholar]
- 17.Belluzo S, Boeris V, Farruggia B, Pico G (2011) Int J Biol Macromol 49: 936. [DOI] [PubMed] [Google Scholar]
- 18.Pozdnyakova I, Wittung-Stafshede P (2010) Biochim Biophys Acta 1804: 740. [DOI] [PubMed] [Google Scholar]
- 19.Pioselli B, Bettati S, Mozzarelli A (2005) FEBS Lett 579: 2197. [DOI] [PubMed] [Google Scholar]
- 20.Minh DD, Chang CE, Trylska J, Tozzini V, McCammon JA (2006) J Am Chem Soc 128: 6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin S, Minh DD, McCammon JA, Zhou HX (2010) J Phys Chem Lett 1: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Somalinga BR, Roy RP (2002) J Biol Chem 277: 43253. [DOI] [PubMed] [Google Scholar]
- 23.Matrai J, Verheyden G, Kruger P, Engelborghs Y (2004) Protein Sci 13: 3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matrai J, Jonckheer A, Joris E, Kruger P, Carpenter E, Tuszynski J, De Maeyer M, Engelborghs Y (2008) Eur Biophys J 38: 13. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee D, Pal SK (2008) Langmuir 24: 8163. [DOI] [PubMed] [Google Scholar]
- 26.Sola RJ, Griebenow K (2006) FEBS Lett 580: 1685. [DOI] [PubMed] [Google Scholar]
- 27.Sola RJ, Griebenow K (2006) FEBS J 273: 5303. [DOI] [PubMed] [Google Scholar]
- 28.Verma PK, Rakshit S, Mitra RK, Pal SK (2011) Biochimie 93: 1424. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Martinez JA, Sola RJ, Castillo B, Cintron-Colon HR, Rivera-Rivera I, Barletta G, Griebenow K (2008) Biotechnol Bioeng 101: 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pastor I, Vilaseca E, Madurga S, Garces JL, Cascante M, Mas F (2011) J Phys Chem B 115: 1115. [DOI] [PubMed] [Google Scholar]
- 31.Jordan BJ, Hong R, Han G, Rana S, Rotello VM (2009) Nanotechnology 20: 434004. [DOI] [PubMed] [Google Scholar]
- 32.Gunasekaran K, Ma B, Nussinov R (2004) Proteins 57: 433. [DOI] [PubMed] [Google Scholar]
- 33.Lonhienne TG, Winzor DJ (2002) Biochemistry 41: 6897. [DOI] [PubMed] [Google Scholar]
- 34.del Sol A, Tsai CJ, Ma B, Nussinov R (2009) Structure 17: 1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiraga K, Derbyshire V, Dansereau JT, Van Roey P, Belfort M (2005) J Mol Biol 354: 916. [DOI] [PubMed] [Google Scholar]
- 36.Hiraga K, Soga I, Dansereau JT, Pereira B, Derbyshire V, Du Z, Wang C, Van Roey P, Belfort G, Belfort M (2009) J Mol Biol 393:1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du Z, Liu Y, Ban D, Lopez MM, Belfort M, Wang C (2010) J Mol Biol 400: 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okamoto DN, Oliveira LC, Kondo MY, Cezari MH, Szeltner Z, Juhasz T, Juliano MA, Polgar L, Juliano L, Gouvea IE 2010) Biol Chem 391: 1461. [DOI] [PubMed] [Google Scholar]
- 39.Wang W, Xu WX, Levy Y, Trizac E, Wolynes PG (2009) Proc Natl Acad Sci U S A 106: 5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ercole C, Lopez-Alonso JP, Font J, Ribo M, Vilanova M, Picone D, Laurents DV (2011) Arch Biochem Biophys 506: 123. [DOI] [PubMed] [Google Scholar]
- 41.Chebotareva NA, Harding SE, Winzor DJ (2001) Eur J Biochem 268: 506. [DOI] [PubMed] [Google Scholar]
- 42.Chebotareva NA, Kurganov BI, Harding SE, Winzor DJ (2005) Biophys Chem 113: 61. [DOI] [PubMed] [Google Scholar]
- 43.Chebotareva NA (2007) Biochemistry (Mosc) 72: 1478. [DOI] [PubMed] [Google Scholar]
- 44.Chebotareva NA, Makeeva VF, Bazhina SG, Eronina TB, Gusev NB, Kurganov BI (2010) Macromol Biosci 10: 783. [DOI] [PubMed] [Google Scholar]
- 45.Shi J, Han N, Lim L, Lua S, Sivaraman J, Wang L, Mu Y, Song J (2011) PLoS Comput Biol 7: e1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang M, Guo Z (2007) J Am Chem Soc 129: 730. [DOI] [PubMed] [Google Scholar]
- 47.Casati DF, Aon MA, Iglesias AA (2000) Biochem J 350 Pt 1: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moran-Zorzano MT, Viale AM, Munoz FJ, Alonso-Casajus N, Eydallin GG, Zugasti B, Baroja-Fernandez E, Pozueta- Romero J (2007) FEBS Lett 581: 1035. [DOI] [PubMed] [Google Scholar]
- 49.Li W, Wolynes PG, Takada S (2011) Proc Natl Acad Sci U S A 108:3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brinkman AB, Ettema TJ, de Vos WM, van der Oost J (2003) Mol Microbiol 48: 287. [DOI] [PubMed] [Google Scholar]
- 51.Ma B, Tsai CJ, Haliloglu T, Nussinov R (2011) Structure 19: 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen S, Iannolo M, Calvo JM (2005) J Mol Biol 345: 251. [DOI] [PubMed] [Google Scholar]
- 53.Pul U, Wurm R, Wagner R (2007) J Mol Biol 366: 900. [DOI] [PubMed] [Google Scholar]
- 54.Ma B, Tsai CJ, Pan Y, Nussinov R (2010) ACS Chem Biol 5: 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sasaki Y, Miyoshi D, Sugimoto N (2007) Nucleic Acids Res 35:4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santoso Y, Joyce CM, Potapova O, Le Reste L, Hohlbein J, Torella JP, Grindley ND, Kapanidis AN (2010) Proc Natl Acad Sci U S A 107: 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andrade P, Martin MJ, Juarez R, Lopez de Saro F, Blanco L 2009) Proc Natl Acad Sci U S A 106: 16203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eoff RL, Sanchez-Ponce R, Guengerich FP (2009) J Biol Chem 284: 21090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu C, Maxwell BA, Brown JA, Zhang L, Suo Z (2009) PLoS Biol 7: e1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rothwell PJ, Waksman G (2007) J Biol Chem 282: 28884. [DOI] [PubMed] [Google Scholar]
- 61.Simon SM, Sousa FJ, Mohana-Borges R, Walker GC (2008) Proc Natl Acad Sci U S A 105: 1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lareu RR, Harve KS, Raghunath M (2007) Biochem Biophys Res Commun 363: 171. [DOI] [PubMed] [Google Scholar]
- 63.Sasaki Y, Miyoshi D, Sugimoto N (2006) Biotechnol J 1: 440. [DOI] [PubMed] [Google Scholar]
- 64.Frueh DP, Arthanari H, Koglin A, Vosburg DA, Bennett AE, Walsh CT, Wagner G (2008) Nature 454: 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo ZF, Jiang M, Zheng S, Guo Z (2010) Bioorg Med Chem Lett 20: 3855. [DOI] [PubMed] [Google Scholar]
- 66.Ma B, Kumar S, Tsai CJ, Nussinov R (1999) Protein Eng 12: 713. [DOI] [PubMed] [Google Scholar]
- 67.Kumar S, Ma B, Tsai CJ, Sinha N, Nussinov R (2000) Protein Sci 9: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma B, Shatsky M, Wolfson HJ, Nussinov R (2002) Protein Sci 11: 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dong H, Qin S, Zhou HX (2010) PLoS Comput Biol 6: e1000833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheung MS, Klimov D, Thirumalai D (2005) Proc Natl Acad Sci U S A 102: 4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Friedel M, Shea JE (2004) J Chem Phys 120: 5809. [DOI] [PubMed] [Google Scholar]
- 72.Miklos AC, Sarkar M, Wang Y, Pielak GJ (2011) J Am Chem Soc 133: 7116. [DOI] [PubMed] [Google Scholar]
- 73.Dhar A, Samiotakis A, Ebbinghaus S, Nienhaus L, Homouz D, Gruebele M, Cheung MS (2010) Proc Natl Acad Sci U S A 107:17586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miklos AC, Li C, Sorrell CD, Lyon LA, Pielak GJ (2011) BMC Biophys 4: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Homouz D, Perham M, Samiotakis A, Cheung MS, Wittung- Stafshede P (2008) Proc Natl Acad Sci U S A 105: 11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bartlett J, Blagojevic J, Carter D, Eskiw C, Fromaget M, Job C, Shamsher M, Trindade IF, Xu M, Cook PR (2006) Biochem Soc Symp: 67 [DOI] [PubMed] [Google Scholar]
- 77.Reth M, Wienands J (1997) Annu Rev Immunol 15: 453. [DOI] [PubMed] [Google Scholar]
- 78.Maudsley S, Martin B, Luttrell LM (2005) J Pharmacol Exp Ther 314: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McGuffee SR, Elcock AH (2010) PLoS Comput Biol 6: e1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martorell G, Adrover M, Kelly G, Temussi PA, Pastore A 2011) Proteins 79: 1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Szasz CS, Alexa A, Toth K, Rakacs M, Langowski J, Tompa P (2011) Biochemistry 50: 5834. [DOI] [PubMed] [Google Scholar]
- 82.Li C, Charlton LM, Lakkavaram A, Seagle C, Wang G, Young GB, Macdonald JM, Pielak GJ (2008) J Am Chem Soc 130:6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Derham BK, Harding JJ (2006) Biochim Biophys Acta 1764: 1000. [DOI] [PubMed] [Google Scholar]
- 84.Ellis RJ (2001) Trends Biochem Sci 26: 597. [DOI] [PubMed] [Google Scholar]
- 85.Zimmerman SB, Trach SO (1991) J Mol Biol 222: 599. [DOI] [PubMed] [Google Scholar]
- 86.Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y (1997) Science 277: 1453. [DOI] [PubMed] [Google Scholar]
- 87.Pielak GJ (2005) Proc Natl Acad Sci U S A 102: 5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Minton AP (2006) J Cell Sci 119: 2863. [DOI] [PubMed] [Google Scholar]
- 89.Zhou HX, Rivas G, Minton AP (2008) Annu Rev Biophys 37: 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Minton AP (2001) J Biol Chem 276: 10577. [DOI] [PubMed] [Google Scholar]
- 91.Dorsaz N, De Michele C, Piazza F, De Los Rios P, Foffi G 2010) Phys Rev Lett 105: 120601. [DOI] [PubMed] [Google Scholar]
- 92.Zheng Y (2010) Nat Rev Mol Cell Biol 11: 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.St Johnston D (2005) Nat Rev Mol Cell Biol 6: 363. [DOI] [PubMed] [Google Scholar]
- 94.Birkenfeld J, Nalbant P, Yoon SH, Bokoch GM (2008) Trends Cell Biol 18: 210. [DOI] [PubMed] [Google Scholar]
- 95.Good M, Tang G, Singleton J, Remenyi A, Lim WA (2009) Cell 136: 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seeliger MA, Kuriyan J (2009) Cell 136: 994. [DOI] [PubMed] [Google Scholar]
- 97.Liu J, Nussinov R (2011) J Biol Chem 286: 40934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Good MC, Zalatan JG, Lim WA (2011) Science 332: 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Csermely P, Palotai R, Nussinov R (2010) Trends Biochem Sci 35: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Di Paolo G, De Camilli P (2006) Nature 443: 651. [DOI] [PubMed] [Google Scholar]
- 101.Loose M, Kruse K, Schwille P (2011) Annu Rev Biophys 40: 315. [DOI] [PubMed] [Google Scholar]
- 102.Elcock AH (2010) Curr Opin Struct Biol 20: 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pielak GJ, Miklos AC (2010) Proc Natl Acad Sci U S A 107: 17457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shaw AK, Pal SK (2007) J Photochem Photobiol B 86: 199. [DOI] [PubMed] [Google Scholar]
- 105.Hu Z, Bowen D, Southerland WM, del Sol A, Pan Y, Nussinov R, Ma B (2007) PLoS Comput Biol 3: e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bhabha G, Lee J, Ekiert DC, Gam J, Wilson IA, Dyson HJ, Benkovic SJ, Wright PE (2011) Science 332: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S (2002) Proc Natl Acad Sci U S A 99: 2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leventis R, Silvius JR (2010) Biophys J 99: 2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nakano S, Karimata HT, Kitagawa Y, Sugimoto N (2009) J Am Chem Soc 131: 16881. [DOI] [PubMed] [Google Scholar]