

Abstract
Emerging evidence have shown that resting quiescent hematopoietic stem cells (HSCs) prefer to utilize anaerobic glycolysis rather than mitochondrial respiration for energy production. Compelling evidence has also revealed that altered metabolic energetics in HSC underlies the onset of certain blood diseases. However, the mechanisms responsible for energetic reprogramming remain elusive. We recently found that Fanconi anemia (FA) HSCs are more dependent on mitochondrial respiration relative to glycolysis in their resting state for energy metabolism. Here we have investigated the role of deficient glycolysis in FA HSC maintenance. We observed significantly reduced glucose consumption, lactate production and ATP production in HSCs but not less primitive multipotent progenitors or restricted hematopoietic progenitors of Fanca−/− and Fancc−/− mice compared to those of wild-type mice, which was associated with an over-activated p53-TIGAR metabolic axis. We have utilized Fanca−/− HSCs deficient for p53 to show that the p53-TIGAR axis suppressed glycolysis in FA HSCs, leading to enhanced pentose phosphate pathway and cellular antioxidant function, and consequently reduced DNA damage and attenuated HSC exhaustion. Furthermore, by using Fanca−/− HSCs carrying the separation-of-function mutant p53R172P transgene that selectively impairs the p53 function in apoptosis but not cell-cycle control, we demonstrated that the cell-cycle function of p53 was not required for glycolytic suppression in FA HSCs. Finally, ectopic expression of the glycolytic rate-limiting enzyme PFKFB3 specifically antagonized p53-TIGAR-mediated metabolic reprogramming in FA HSCs. Together, our results suggest that p53-TIGAR metabolic axis-mediated glycolytic suppression may play a compensatory role in attenuating DNA damage and proliferative exhaustion in FA HSCs.
Keywords: Hematopoietic stem cells (HSCs), Fanconi anemia, glycolysis, p53, TP53-inducible glycolysis, apoptosis regulator (TIGAR)
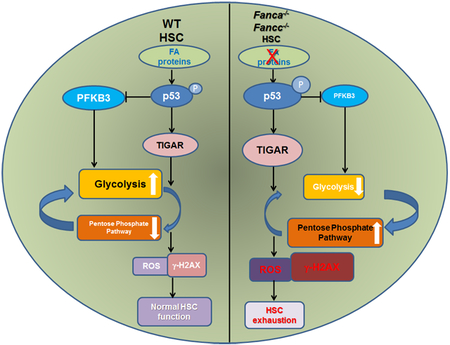
Graphical Abstract:
Introduction
Fanconi anemia (FA) is a rare inherited disease characterized by bone marrow (BM) failure and high risk of neoplasia including leukemia from dysfunctional hematopoietic stem cells (HSCs; 1, 2). FA is genetically heterogeneous with at least 22 genes (FANCA-W) identified thus far [1-11]. The hematologic complications of FA are nearly universal among patients with FA and defects in HSCs have been postulated as the root cause of BM failure and leukemia in FA [6, 12]. Emerging evidence suggests that dysregulated energy metabolism is one possible mechanism underlying FA HSC defect. For example, compared to wild-type (WT) HSCs, FA HSCs are more dependent on oxidative phosphorylation (OXPHOS) relative to glycolysis for energy metabolism and undergo glycolysis-to-OXPHOS switch in response to oxidative stress through a p53-dependent mechanism [12]. However, how p53 regulates energy metabolism in FA HSCs is not well defined.
The role of p53 in tumor suppression is well-known and loss of p53 function can lead to cancer [13, 14]. While p53 mutations are common in solid tumors, such mutations are found at lower frequencies in hematologic malignancies [15, 16]. Recent studies have shown that p53 plays a crucial role in hematopoiesis by regulating HSC self-renewal and quiescence [17-20]. p53 is also a key regulator of cellular energy metabolism [21-23]. For example, p53 can inhibit glycolysis through transcriptional activation of its target gene TP53-induced glycolysis regulator (TIGAR; an inhibitor of the fructose-2,6-bisphosphate) [24]. However, since HSCs predominantly utilize glucose for energy metabolism, how p53 functions in glycolytic regulation in the context of HSC homeostasis is not known.
TIGAR shares functional sequence similarities with the bisphosphatase domain (FBPase-2) of the bi-functional enzyme PFK-2/FBPase-2 (6-phosphofructo-e-kinase/fructose-2,6-bisphosphatase), which degrades fructose-2,6-bisphosphate (Fru-2,6-BP; 25). TIGAR causes a decline in Fru-2,6-BP levels in cells, resulting in glycolysis inhibition and an overall decrease in intracellular reactive oxygen species (ROS) and thus directing the pathway into the pentose phosphate shunt to produce NADPH [26, 27]. We propose that the p53-TIGAR axis may be responsible for the suppression of glycolysis in FA HSCs that we previously observed. In the present study, we have used the Fanca−/−, p53−/− and p53−/−Fanca−/− knockout mouse models to show that p53-TIGAR-mediated glycolytic suppression attenuates ROS production, reduces DNA damage, ameliorates chromosomal instability, and consequently prevents premature exhaustion of FA HSCs. Our findings reveal a novel function of the p53-TIGAR metabolic axis in the maintenance of FA HSCs.
Material and Method
Animals
Fanca+/−, Fancc+/− and p53R172P mice were generously provided by Dr. Madeleine Carreau (Laval University), Dr. Manuel Buchwald (Hospital for Sick Children, University of Toroto), and Dr. Guillermina Lozano (University of Texas M.D. Anderson Cancer Center), respectively [28-30]. p53−/−Fanca−/− and p53R172PFanca−/− mice were generated by interbreeding the heterozygous Fanca+/− with p53+/− mice (C57BL/6: B6, CD45.2+; Jackson Laboratories; 31) or heterozygous Fanca+/− with p53R172P mice. 8-12 week-old animals were used for all the experiments. All the animals including BoyJ (C57BL/6: B6, CD45.1+) recipient mice were maintained in the animal facility at West Virginia University (WVU) Health Science Center. All experimental procedures conducted in this study were approved by the Institutional Animal Care and Use Committee of WVU.
Flow analysis and cell sorting
Femurs and tibias were flushed to dissociate the BM fraction. Cells were resuspended in 5mL PBS/0.5% BSA and filtered through a 70-μm filter (BD Biosciences, Cat # 3523350, San Jose, CA). The mononuclear cells were isolated by Ficoll-Paque (GE Healthcare, Cat # 95040-394, Pittsburgh, PA) gradient centrifugation. The following antibodies were used for flow cytometry analyses: APC-Cy7-anti-c-Kit (BioLegend, Cat # 105825, San Diego, CA), PE-Cy7-anti-Sca-1 (BD Biosciences, Cat # 105825, San Jose, CA), APC-anti-CD150 (BioLegend, Cat # 115909, San Diego, CA), Pacific Blue-anti-CD48 (BioLegend, Cat # 103417, San Diego, CA), PE-anti-CD45.1, APC-anti-CD45.2 (Both from BD Biosciences, Cat # 553776 and 558702, San Jose, CA). For LSK (Lineage−Sca-1+c-Kit+) staining, cells were incubated by the biotin conjugated anti-lineage antibody cocktail (BioLegend, Cat # 133307, San Diego, CA) followed by staining with a secondary PerCP-Cy5.5-anti Streptavidin antibody (BioLegend, Cat # 405214, San Diego, CA), PE-Cy7-anti-Sca-1 antibody (BD Biosciences, San Jose, CA), and APC-Cy7-anti-c-Kit antibody (BD Biosciences, San Jose, CA). To measure long-term HSC subpopulation, cells were stained with LSK antibodies in addition to CD150-APC, CD48-Pacific blue (Both from BioLegend, Cat # 115909 and 103417, San Diego, CA). Flow cytometry was performed on LSRFortessa (BD Biosciences, San Jose, CA) and analysis was done with FCS Express 6 software (De Novo Software, Los Angeles, CA). For the cell sorting, lineage negative cells were enriched using lineage depletion reagents (Miltenyi Biotec, Cat # 130-090-858, Auburn, CA) according to the manufacturer’s instruction. The Lin+ or LSK (Lin−c-kit+Sca-1+) fractions were acquired by using the FACSAria II sorter (BD Biosciences, San Jose, CA).
To detect reactive oxygen species (ROS) level, surface marker stained cells were washed with PBS twice followed by incubation with H2DCFDA working solution (10 μM H2DCFDA in pre-warmed PBS; Molecular Probes, Cat # D399, Eugene, OR) at 37˚C for 15 min. H2DCFDA intensity was analyzed by Flow cytometry.
For cell cycle analysis, surface marker stained cells were fixed and permeabilized using Cytofix/Cytoperm buffer (BD PharMingen, Cat # 554722, San Jose, CA) followed by intensive wash using Perm/Wash Buffer (BD PharMingen, Cat # 554723, San Jose, CA). Cells were then labeled with Pyronin Y staining buffer (150ng/ml Pyronin Y in Perm/Wash buffer; Sigma-Aldrich, Cat # 92-32-0, St Louis, MO) at 37˚C for 1 hour followed by Flow cytometry analysis on CD45.2+ SLAM (Lin−Sca1+c-kit+CD150+CD48−) gated population.
For γ-H2AX staining, surface marker stained cells were fixed and permeabilized using Cytofix/Cytoperm buffer (BD PharMingen, Cat # 554722, San Jose, CA) followed by intensive wash using Perm/Wash Buffer (BD PharMingen, Cat # 554723, San Jose, CA). Cells were then blocked with 2% BSA/PBS for 20 min at room temperature in dark followed by incubation with primary anti-γ-H2AX antibody (biotin-conjugated, Millipore, Cat #: 16-193, Billerica, MA). After washing, cells were then incubated with secondary FITC-anti-streptavidin antibody (BD Pharmingen, Cat # 554060, San Jose, CA) and analyzed by Flow Cytometry analysis.
For lineage differentiation, single cell suspensions were incubated with various combination of the following cell-surface marker antibodies: CD45.2-FITC, Gr1-APC, Mac1-PE-Cy7, CD45.2-APC, CD3e (All from BD PharMingen, Cat # 553772, 553129, 552850, 558702, 553062) and B220-PE (eBioscience, Cat # 12-0452-85). Immunolabeled cells were analyzed by flow cytometry.
Measurements of glucose consumption, lactate production, ATP and PFK1 activity
Glucose consumption, lactate production and phosphofructokinase (PFK) activity were measured using the Glucose Assay Kit, the Lactate Assay Kit and the Phosphofructokinase Activity Colorimetric Assay Kit (All from Biovision, Cat # K686, K607, and K776, Milpitas, CA), according to the manufacturer’s instructions, respectively. Intracellular ATP levels and Fructose-6-phosphate (F6P) levels were determined in cell lysates using the ATP Bioluminescent Somatic Cell Assay Kit and Fructose-6-Phosphate Assay Kit (Sigma-Aldrich, Cat # FLASC and MAK020, St Louis, MO), following the manufacturer’s instructions.
Measurements of Fru-2, 6-P2 Level, NADPH/NADP+ ratio and Intracellular GSH level
PBS washed cells were lysed in NaOH/Tris acetate by heating at 80˚C for 5 min. Lysates were neutralized to PH 7.2 by adding ice-cold acetic acid and HEPES (Both from Sigma-Aldrich, Cat # A6283 and H3375, St Louis, MO). Fru-2,6-P2 concentration was determined using the previously described method [32, 33]. NADPH/NADP+ ratios were determined by using an enzyme-based colorimetric reaction kit (NADP+/NADPH Quantification Kit, BioVision, Cat # K347-100, Milpitas, CA).
Intracellular reduced glutathione (GSH) levels were measured enzymatically with glutathione reductase [34] using the BIOXYTECH GSH-400 kit (Oxis Research, Cat # 21011, Abingdon, Oxfordshire, UK), according to the manufacturers’ instructions. Briefly, cells from the indicated mice were washed, and lysed in 0.9 ml of 100 mM Tris-Cl by freeze-thaw cycling. Supernatants from the cells suspension after centrifuge were transferred to Eppendorf tubes containing 0.1 ml of 30% 5-sulfosalicyclic acid and incubated on ice for 15 min. Cell extracts were then centrifuged for 2 min at 12,000 rpm to get the supernatants. Protein concentration was assessed through the BioRad protein assay (Bio-Rad, Cat # 5000002, Hercules, CA). GSH levels per 100 μl sample were then measured spectrophotometrically by conversion of 5,5′-dithio-bis (2-nitrobenzoic acid) to its colored product upon reduction by GSH-dependent glutathione reductase, and expressed as nmol GSH/mg protein.
Western blotting
30,000 of LSK cells from indicated mice were washed with ice-old PBS, and lysed in an ice-cold lysis buffer containing 50mM Tris HCL (pH7.4), 0.1% NP40, and 1M NaCl supplemented with protease and phosphatase inhibitors (10 μg/ml of aprotinin, 25 μg/ml of leupeptin, 10 μg/ml of pepstatin A, 2mM PMSF, 0.1M NaP2O4, 25mM NaF, and 2mM sodium orthovanadate) for 30 minutes on ice. Cell debris was removed from the lysate by centrifugation. Protein lysate was resolved on SDS-PAGE and transferred onto nitrocellulose membranes. Immunoblots were then probed with primary antibodies for TIGAR (Clone ab189164; Abcam, Cambridge, MA), total p53 (Clone pAb240; Calbiochem), p53-S18 (Clone ab1431; Abcam, Cambridge, MA), γ-H2AX (NB100-384, Novus, Littleton, CO) and β-actin (Sigma-Aldrich, Cat # A5316, St Louis, MO) for 12-16 hours at 4°C. Signals were visualized by incubation with anti-mouse secondary antibody, followed by ECL chemiluminescence (Amersham Biosciences, Cat # RPN2106, Pittsburgh, PA).
Quantitative PCR
Total RNA was extracted using RNeasy kit (Qiagen, Cat # 74104, Valencia, CA) following the manufacturer’s procedure. Reverse transcription was carried out at 42 °C for 60 min and stopped at 95 °C for 5 min using random hexamers and Superscript II RT (Invitrogen, Cat # 18064014, Grand Island, NY). First-strand cDNA was used for real-time polymerase chain reaction (PCR) using primers listed in Table S1. Samples were normalized to the level of Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) mRNA.
Chromosomal breakage analysis
Chromosomal breakage analysis was performed on LSK (Lin−Sca1+c-kit+) cells from WT, Fanca−/−, p53−/− and p53−/−Fanca−/− mice as previously described [35]. In brief, cells were treated with 0.05 mg/ml Colcemid (Invitrogen, Cat # 15210-016, Grand Island, NY) for 1.5 hours, followed by 0.4% KCl hypotonic solution at 37 ˚C for 20 min. Cells were then fixed with methanol and acetic acid (Sigma-Aldrich, Cat # A6283, St Louis, MO) at 4 ˚C for 15 min and dropped onto microscope slides. The cells were then rinsed with isoton, stained with Giemsa for 5 min. The slides were then rinsed with Gurr Buffer (CTL Scientific, Cat # 50-300-398, Deer Park, NY) and Milli-Q-filtered deionized water. A total of 50 cells from each sample were scored for chromosome aberrations.
Lentiviral vector construction, virus production and transduction
To generate lentiviral expression vectors for TIGAR and PFKFB3, the TIGAR cDNA was amplified from Transomic plasmid OMu13565 and PFKFB3 cDNA was amplified from Transomic plasmid OMu19009 (Both were from GenScript, Piscataway Township, NJ), respectively, and cloned into the pLVX-IRES-mCherry vector (Clontech, Cat # 631237, Mountain View, CA).
Lentivirus was produced in 293 T cells after transfection of 20 μg plasmid, 15 μg pCMVΔ8.91 helper plasmid and 6 μg pMD.G [36] using standard calcium phosphate transfection procedures. Fresh medium change was performed 12 hours after transfection. Supernatants from the cell culture were collected 48 hours after transfection, filtered through 0.45 μm-pore-size filters, and concentrated at 25,000rpm for 2.5 hours at 4°C to harvest viral particles. Virus pellet was resuspended in sterile PBS and stored at −80°C.
For lentivirus transduction, the sorted LSK cells from WT, p53−/−, Fanca−/−, and p53−/−Fanca−/− were maintained in StemSpan™ Serum-Free Expansion Media (StemCell Technologies, Cat # 09600, Vancouver, BC, Canada) supplemented with 50 ng/ml SCF and 50 ng/ml TPO (Both from Peprotech, Cat # 500-P71 and 315-14, Rocky Hill, NJ) for 24 hours before transduction. A multiplicity of infection (MOI) of 5 were used for TIGAR or PFKFB3 overexpression. After overnight transduction, cells were cultured in the presence of 0.5 mg/ml polybrene and re-fed with fresh medium the next day. Transduced cells were used in further assays at least 3 to 4 days post transduction.
Bone Marrow Transplantation
For competitive repopulating units (CRUs), serial dilutions of LSK cells (CD45.2+) transduced with mCherry or mCherry-TIGAR lentivirus (25, 75, 150 and 250 mCherry+ cells) were mixed with 2X105 protector BM cells from congenic mice (BoyJ) and injected into lethally irradiated (11.75 Gy) BoyJ mice (CD45.1+). The CRU frequencies were then calculated from the proportions of negative mice (<1% donor engraft). Poisson statistics at 8 weeks and 16 weeks after transplantation was performed using L-Calc software (StemCell Technologies, Vancouver, BC, Canada).
For serial BM transplantation, 1,000 mCherry-sorted mCherry or mCherry-TIGAR lentiviral vector transduced LSK cells (CD45.2+), mixed with 200,000 c-kit-depleted protector cells, were transplanted into lethally irradiated BoyJ mice. 16 weeks after BM transplantation, the recipients were subjected to Flow cytometry analysis for donor-derived cells. For secondary BM transplantation, one million BM cells from the primary recipient mice were pooled and transplanted into sublethally irradiated (7.0 Gy) secondary CD45.1+ recipient mice. Four months later, donor blood chimera was determined by staining peripheral blood samples with APC-CD45.2 (BD Biosciences, Cat # 558702, San Jose, CA) and analyzed on a FACS canto instrument (BD Biosciences, San Jose, CA).
Statistics
Statistics was done using a standard t test (Prism v6.0). The level of the statistical significance stated in the text was based on the P values. P < 0.05 was considered statistically significant.
Results
FA HSCs exhibit reduced glycolysis
To examine the role of glycolysis in FA HSC maintenance, we first compared glycolytic flux in different hematopoietic cell compartments (Fig. 1A) of WT, Fanca−/− and Fancc−/− mice. We found that glucose consumption rate in FA HSCs (Lin−Sca1+c-kit+CD150+CD48−) was significantly lower than those in WT HSCs (Fig. 1B), while the glucose usage in less primitive multipotential progenitors (MPPs; Lin−Sca1+c-kit+CD150−CD48−), restricted hematopoietic progenitor cells (HPCs; Lin−Sca1+c-kit+CD150−CD48+ and Lin−Sca1+c-kit+CD150+CD48+) and Lin+ cells were comparable among the three genotypes (Supporting Information Fig. S1A). Similar reduction was observed in lactate production (Fig. 1C) and intracellular ATP level (Fig. 1D) in FA HSCs in comparison to WT HSCs but not in MPPs, HPCs or Lin+ cells from Fanca−/− or Fancc−/− mice (Supporting Information Figs. S1B, S1C).
Fig 1. Reduced glycolysis in FA HSCs.

(A) Gating strategy for FACS sorting. (B) Reduced glucose consumption in FA HSCs. SLAM (Lin−Sca-1+c-Kit+CD150+CD48−) cells from the bone marrow (BM) of Fanca−/−, Fancc−/− and wild-type (WT) mice were isolated by FACS sorting and subjected to analysis for glucose consumption analysis. (C) Reduced lactate production in FA HSCs. SLAM cells described in (B) were analyzed for lactate production (C). (D) Lower ATP levels in FA HSCs. Relative intracellular ATP concentrations were measured in cells described in (A). Results depicted in B-D are means ± SD of three independent experiments. (n=6-9 per group). (E) Schematic presentation of glycolytic pathway. (F) Expression of the genes encoded for the key enzymes of the glycolysis pathway in WT and FA HSCs. RNA were extracted from SLAM cells described in (A) followed by q-PCR analysis for the indicated genes using primers listed in Supplementary Table S1. Samples were normalized to the level of GAPDH mRNA. (n=6-9 per group). (G) Increased TIGAR protein in FA HSPCs. Whole cell lysates (WCLs) were extracted from 30,000 LSK (Lin−Sca1+c-kit+) cells isolated from WT, Fanca−/− and Fancc−/− mice followed by immunoblotting using antibodies against TIGAR or β-actin. *, p<0.05; **, p<0.01.
It is known that glycolysis is tightly regulated by a series of enzymes and that crucial adjustments in glycolytic flux are set by the intracellular levels of fructose-2,6-bisphosphate (Fru-2,6-BP), which is controlled by a family of bifunctional enzymes, the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFB), including PFKFB3 and TIGAR [37, 38]. PFKFB3 has a much higher kinase activity than bisphosphatase activity, thus favoring the production of Fru-2,6-BP; whereas TIGAR functions as a fructose-2,6-bisphosphatase, thus decreasing the intracellular levels of Fru-2,6-BP (Fig. 1E). We thus measured the expression of the genes encoded for several key enzymes of the glycolysis pathway in WT and FA HSCs. Although there was no significant changes in the expression of HK1, GPI1, PFKFB3, PFK1 and PKM1, we observed a significantly (>2 fold) increased expression of TIGAR in Fanca−/− and Fancc−/− HSCs compared to that in WT HSCs (Fig. 1F). In addition, TIGAR protein levels in FA HSCs were also found markedly increased in LSK cells from Fanca−/− and Fancc−/− mice (Fig. 1G). Accordingly, fructose-6-phosphate (F6P) was increased in FA HSPCs (Supporting Information Fig. S2). These results indicate a correlation between upregulated TIGAR and reduced glycolysis in FA HSCs.
p53-dependent suppression of glycolysis in FA HSCs
Since p53 regulates glycolysis and since TIGAR is a p53 target, we then attempted to delete the p53 gene in Fanca−/− mice and determined the role of p53-TIGAR axis in glycolytic suppression and HSC maintenance in FA. We first determined p53 levels in BM LSK cells from mice of four genotypes (WT, Fanca−/−, p53−/− and Fanca−/−p53−/− DKO) produced from the Fanca+/− × p53+/− cross. The level of p53 protein in Fanca−/− LSK cells was substantially higher than that in WT cells, and no p53 was detected in p53−/− and Fanca−/−p53−/− double knockout (DKO) HSPCs (Fig. 2A). Consistently, Fanca−/− HSPCs showed increased TIGAR compared to WT cells, which was not expressed in p53−/− and Fanca−/−p53−/− DKO HSPCs (Fig. 2A), suggesting that the expression of TIGAR is dependent on p53.
Fig 2. p53-dependent suppression of glycolysis in FA HSCs.

(A) Increased p53 and TIGAR proteins in FA HSPCs. Whole cell lysates (WCL) were extracted from 30,000 LSK isolated from WT, Fanca−/−, p53−/− or p53−/−Fanca−/− mice followed by immunoblotting using antibodies against p53, TIGAR, or β-actin. (B) Deletion of p53 increases F2,6BP level in Fanca−/− HSCs. SLAM cells isolated from WT, Fanca−/−, p53−/− or p53−/−Fanca−/− mice were subjected to MicroAssay for F26BP. (C, D) Deletion of p53 increases glucose consumption and lactate production in FA HSCs. SLAM cells isolated from WT, Fanca−/−, p53−/− or p53−/−Fanca−/− mice were subjected to glucose uptake (C) and lactate production analysis (D). Results depicted in B-D are means ± SD of three independent experiments. (n=6-9 per group). *, p<0.05; **, p<0.01.
Consistent with increased TIGAR expression, the levels of F-2,6-BP were significantly lower in Fanca−/− HSCs but higher in p53−/− HSCs compared to that in WT HSCs (Fig. 2B). Deletion of p53 in Fanca−/− HSCs also increased the levels of F-2,6-BP (Fig. 2B). Furthermore, loss of p53 not only abolished the inhibitory effect of Fanca deficiency but also further boosted glucose uptake (Fig. 2C) and lactate production (Fig. 2D). These results suggest a novel metabolic reprogramming by which FA deficiency collaborates with p53 in suppressing glycolysis.
p53-TIGAR attenuates ROS, DNA damage and chromosomal instability in FA HSPCs
To understand the role of p53-TIGAR-mediated glycolytic suppression in FA HSCs, we first determined p53 activation in Fanca−/− HSPCs. We observed increased phosphorylation of p53 at Serine 18 (p53Ser18), an established marker for p53 activation [39, 40], in Fanca−/− BM LSK cells compared to WT cells (Fig. 3A).
Fig 3. p53-TIGAR-mediated glycolytic suppression attenuates ROS, DNA damage and chromosomal instability in FA HSPCs.

(A) Overactivation of p53 in FA HSPCs. WCLs were extracted from LSK cells isolated from WT, Fanca−/−, p53−/− or p53−/−Fanca−/− mice followed by western blotting using antibodies against total p53, p53ser18 and β-actin. (B) Lentiviral transduction and FACS sorting. LSK cells isolated from WT, Fanca−/−, p53−/− or p53−/−Fanca−/− mice were transduced with lentiviral vector expressing mCherry or mCherry-TIGAR. Flow plots after FACS sorting for mCherry are shown. The sorted mCherry+ cells were then subjected to ex vivo expansion for 72 h followed by the analyses described below. (C) Expression of TIGAR in transduced HSCs. RNA were extracted from mCherry+ SLAM cells described in (B) were subjected to q-PCR analysis for TIGAR expression. Samples were normalized to the level of GAPDH mRNA. (D) Overexpression of TIGAR reduces ROS level in HSCs. Cells described in (B) were gated for mCherry+ SLAM population and subjected to flow cytometry analysis for ROS. Representative plots (Left) and quantification (Right) are shown. (E) Overexpression of TIGAR reduces DNA damage in FA HSPCs. WCLs were extracted from mCherry+ cells described in (B) followed by western blotting using antibodies against γ-H2AX and β-actin. (F) Overexpression of TIGAR reduces spontaneous chromosomal breakage in FA HSPCs. mCherry+ cells described in (B) were subjected to chromosomal breakage assay. Representative images (Left) and quantification of 50 cells in random fields (Right) are shown. Arrowheads point to chromosome breaks or radial structures. (G, H) Overexpression of TIGAR enhances the pentose phosphate pathway (PPP) and cellular antioxidant function in FA HSCs. Cells described in (B) were gated for mCherry+ population and subjected to assays for NADPH/NADP+ ratio (F) or intracellular GSH analysis. Results depicted in C-H are means ± SD of three independent experiments. *, p<0.05; **, p<0.01;***, p<0.001.
To specifically explore the function of TIGAR in glycolytic suppression in the context of FA cellular deficiency, we ectopically expressed mCherry or mCherry-TIGAR in WT, Fanca−/−, p53−/− and Fanca−/−p53−/− DKO LSK cells (Fig. 3B). Quantitative PCR (q-PCR) analysis shows that the sorted mCherry+ cells overexpressed TIGAR (Fig. 3C). Since TIGAR suppresses glycolysis and promotes the pentose phosphate pathway (PPP), leading to increase in NADPH production and reduction of reactive oxygen species (ROS) [41], we first measured intracellular ROS level. Flow cytometry analysis showed that ROS levels in p53−/− and p53−/−Fanca−/− DKO HSCs were remarkably higher than those in WT and Fanca−/− controls (Fig. 3D). Forced expression of TIGAR markedly reduced ROS production in p53−/− and p53−/−Fanca−/− DKO HSCs (Fig. 3D). TIGAR expression also reduced ROS in WT and Fanca−/− cells, albeit not statistically significant when compared to their mCherry controls (Fig. 3D). Consequently, ectopic expression of TIGAR markedly reduced DNA damage in p53−/− and p53−/−Fanca−/− DKO HSPCs and, to a less extend in Fanca−/− cells, as determined by immunoblotting for γ-H2AX, a robust marker of DNA double-strand breaks (42; Fig. 3E). Furthermore, overexpression of TIGAR significantly reduced spontaneous chromosomal breakage in Fanca−/−, p53−/− and p53−/−Fanca−/− DKO LSK cells (Fig. 3F).
It is well established that TIGAR has fructose-2,6-bisphosphatase activity, which depletes Fru-2,6-BP, resulting in inhibition of glycolysis and thus directing the pathway into the pentose phosphate shunt to produce NADPH and reduce ROS [26, 27]. Indeed, overexpression of TIGAR enhanced the pentose phosphate pathway (PPP) and cellular antioxidant function in Fanca−/−, p53−/− and p53−/−Fanca−/− DKO HSPCs evidenced by increased NADPH/NADP+ ratio and intracellular GSH level in TIGAR-expressing cells compared to the cells expressing mCherry only (Figs. 3G, 3H). Together, these findings indicate that p53-TIGAR-mediated glycolytic suppression attenuates DNA damage and chromosomal instability in FA HSPCs through promoting the PPP pathway and enhancing cellular antioxidant function.
p53-TIGAR ameliorates exhaustion of FA HSCs
To determine the role of p53-TIGAR in FA HSC function, we first performed a well-established competitive repopulating unit (CRU) assays using limiting dilutions of LSK cells expressing mCherry or mCherry-TIGAR from WT, Fanca−/−, p53−/− and Fanca−/−p53−/− DKO mice. Increasing doses of transduced LSK cells (25, 75, 150 and 250 mCherry+ cells) along with protector BM cells were transplanted into lethally irradiated BoyJ mice. Peripheral blood (PB) samples were collected at 8 and 16 weeks post-transplantation and analyzed for donor engraftment. Consistent with previous report, in which the reduced long-term repopulation of p53−/− SLAM was shown by a competitive repopulating unit assays [43], we constantly observed the decreased donor-derived cells in both primary and secondary recipients injected with p53−/− LSK cells 4 months post-BMT, which can be arguably considered long-term repopulation. Poisson statistical analysis at 8 weeks post-transplant showed >5-fold reduction in CRUs in DKO mice than in WT mice (CRUs: 1 in 28 for WT-mCherry compared to 1 in 142.9 for DKO-mCherry; Fig. 4A). This reduction in CRUs in DKO mice was found further increased to >30-fold when estimated at 16 week post-transplantation (CRUs: 1 in 14.2 for WT-mCherry compared to1 in 442 for DKO-mCherry; Table 1). Forced expression of TIGAR significantly increased CRUs in Fanca−/−, p53−/− and Fanca−/−p53−/− DKO mice compared to WT controls at both 8-week and 16-week post-transplant (Table 1; Fig. 4A). These results indicate that the p53-TIGAR metabolic axis plays an important role in ameliorating exhaustion of FA HSCs, a hallmark of FA BM failure [6, 43].
Fig 4. p53-TIGAR ameliorates exhaustion of FA HSCs.

(A) Limited dilution assay. Various doses of sorted transduced LSK cells (25, 75, 150 and 250 mCherry+ cells) expressing mCherry or mCherry-TIGAR were mixed with 2X105 protector BM cells (CD45.1+) and transplanted into irradiated congenic recipients. (n=8-10 per group). Plotted are the percentages of recipient mice containing less than 1% CD45.2+ blood nucleated cells (negative) at 8 weeks after transplantation. Frequency of functional HSCs was calculated according to Poisson statistics. (B) Overexpression of TIGAR improves hematopoiesis reconstitution capacity of FA HSPCs. 1,000 sorted mCherry or mCherry-TIGAR lentiviral vector transduced LSK cells were transplanted into lethally irradiated BoyJ mice. The recipients were subjected to Flow cytometry analysis for donor-derived cells 16 weeks after BM transplantation. Representative flow plots (Left) and quantification (Right) are shown. (n=9 per group). (C) Overexpression of TIGAR improves long-term repopulating activity of FA HSCs. One million BM cells from the primary recipient mice described in (B) were transplanted into sublethally irradiated secondary CD45.1+ recipient mice, and donor-derived chimera in secondary recipients were determined by flow cytometry 16 weeks later. (n=9 per group). (D) Overexpression of TIGAR reduces DNA damage in donor cells. CD45.2+ LSK cells from the recipients described in (B) were subjected to Flow cytometry analysis for γ-H2AX. Representative plots (Upper) and quantification (Lower) are shown. (E) Overexpression of TIGAR increases donor HSC quiescence. Percentage of Pyronin Y+ cells in donor HSC compartment (CD45.2+ SLAM) were analyzed by Flow cytometry. Representative flow plots (Left) and quantification (Right) are shown. (n=6 per group). p<0.05; **, p<0.01;***, p<0.001.
Table 1.
Competitive repopulating units (CRUs)
| Genotype | WT | Fanca−/− | p53−/− | Fanca−/−;p53−/− | ||||
|---|---|---|---|---|---|---|---|---|
| mCherry | TIGAR | mCherry | TIGAR | mCherry | TIGAR | mCherry | TIGAR | |
| CRU frequency | 1/14.2 | 1/13.74 | 1/76.4 | 1/33.4 | 1/54.2 | 1/32.6 | 1/442 | 1/53.43 |
n=8-10 per group
We next performed serial BM transplantation experiments by transplanting 1,000 mCherry+ LSK cells into lethally irradiated syngeneic recipient mice. We found that overexpression of TIGAR significantly improved the repopulating capacities of Fanca−/−, p53−/− and Fanca−/−p53−/− DKO HSCs in primary recipient mice at 16 weeks post-transplantation (Fig. 4B). Noticeably, alteration of p53-TIGAR axis did not affect lineage differentiation in the recipient mice (Supporting Information Fig. S3). Similar results were observed in secondary transplantation, in which TIGAR enhanced the repopulating capacity of the donor-derived Fanca−/−, p53−/− and Fanca−/−p53−/− DKO HSCs, as compared to mCherry controls (Fig. 4C). Significantly, overexpression of TIGAR reduced the levels of γH2AX in donor-derived Fanca−/−, p53−/− and Fanca−/−p53−/− DKO HSCs (Fig. 4D). Furthermore, cell cycle analysis showed that TIGAR-expressing Fanca−/−, p53−/− or p53−/−Fanca−/− HSCs were more quiescent than those expressing mCherry only (Fig. 4E). Therefore, these data suggest that p53-TIGAR attenuates FA HSC exhaustion by reducing DNA damage and maintaining HSC quiescence.
p53-dependent cell-cycle control is not required for TIGAR-mediated metabolic reprogramming
Because deletion of p53 reduced the quiescence of Fanca−/− HSCs (Fig. 4E), we wondered if the effect of TIGAR was specifically dependent on the cell-cycle function of p53. We recently demonstrated a cell-cycle-specific function of p53 in FA HSPC proliferation [43], using a mutant p53 mouse strain harboring a separation-of-function mutation in p53, p53R172P, in which its apoptotic function is disturbed but its cell-cycle checkpoint activities remain intact [29]. We thus crossed the p53R172P strain to Fanca−/− mice and transduced LSK cells from the mice of the resulting four genotypes (WT, Fanca−/−, p53R172P and Fanca−/−p53R172P) with lentivirus expressing mCherry-TIGAR or mCherry alone. We found that like p53-null mutation, p53R172P mutation significantly increased glucose consumption in Fanca−/− HSCs, and that forced expression of TIGAR reduced glucose consumption to near WT levels (Fig. 5A). Similar results were obtained with the measurements of phosphofructokinase-1 ( PFK1) activity (Fig. 5B), intracellular ROS (Fig. 5C) and DNA damage (Fig. 5D). These data indicate that the cell-cycle function of p53 is not required for TIGAR-mediated metabolic reprogramming in HSCs.
Fig 5. p53-dependent cell-cycle control is not required for TIGAR-mediated metabolic reprograming.

(A) Overexpression of TIGAR reduces glucose consumption in p53R172P HSCs. BM LSK cells from WT, Fanca−/−, p53R172P and Fanca−/−p53R172P mice were transduced with lentivirus expressing mCherry-TIGAR or mCherry alone. The sorted mCherry+ cells were expanded for 72 hours. The transduced cells were gated for mCherry+ SLAM population and assayed for glucose consumption. (B) Overexpression of TIGAR reduces PFK1 activity in p53R172P HSPCs. Cells described in (A) were gated for mCherry+ population and assayed for PFK1 activity. (C) Overexpression of TIGAR reduces ROS production in p53R172P HSPCs. Cells described in (A) were gated for mCherry+ SLAM population and analyzed for intracellular ROS by flow cytometry. (n=6 per group). (D) Overexpression of TIGAR reduces DNA damage in p53R172P HSPCs. WCLs were extracted from mCherry+ cells described in (A) followed by western blotting using antibodies against γ-H2AX and β-actin. Results depicted in A-B are means ± SD of three independent experiments. p<0.05; **, p<0.01.
PFKFB3 specifically antagonizes p53-TIGAR metabolic function in FA HSCs
Both TIGAR and another rate-limiting enzyme, PFKFB3, play critical roles in controlling glycolytic flux, which is adjusted by the level of Fructose 2,6-bisphosphate (F2,6BP). F2,6BP activates PFK1 and is by itself synthesized and degraded by a family of biofunctional enzymes, the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFB; 45). Of all PFKFB isoenzymes, PFKFB3 exhibits a much higher (700-fold) kinase activity than bisphosphatase activity, thus favoring F2,6BP production and controlling its abundance [38, 44]. On the other hand, TIGAR functions as a fructose-2,6-bisphosphatase and therefore lowering the activity of PFK1 and flux through the main glycolytic pathway, leading to increase in Fructose-6-phosphate [16, 17, 41]. Given its closely relative role in regulating glycolytic flux, we wondered whether increased expression of PFKFB3 could counteract p53-TIGAR function in FA HSCs. Ectopic expression of PFKFB3 in WT and Fanca−/−, p53−/− and DKO LSK cells was achieved by lentiviral transduction (Figs. 6A, 6B). We found that overexpression of PFKFB3 increased F2,6BP levels in WT and Fanca−/− SLAM cells but had no further effect on F2,6BP in p53−/− or DKO HSCs (Fig. 6C). Consistently, PFK1 activity was increased in PFKFB3-overexpressing WT or Fanca−/− HSPCs but not in p53−/− or DKO cells (Fig. 6D). Furthermore, overexpression of PFKFB3 decreased intracellular levels of GSH in WT and Fanca−/− mCherry+ cells but not in HSPCs deleted for p53 (p53−/− and DKO; Fig. 6E). Consequently, intracellular ROS were significantly increased in PFKFB3-overexpressing WT or Fanca−/− HSCs but not in p53-deficient cells (Fig. 6F). These results indicate that PFKFB3 specifically antagonizes p53-TIGAR metabolic function in HSCs.
Fig 6. PFKFB3 specifically antagonizes p53-TIGAR metabolic function in FA HSCs.

(A) Lentiviral mCherry-PFKFB3 transduction and FACS sorting. LSK cells isolated from WT, Fanca−/−, p53−/− or p53−/−Fanca−/− mice were transduced with lentiviral vector expressing mCherry or mCherry-PFKFB3. Flow plots after FACS sorting for mCherry are shown. The sorted mCherry+ cells were then subjected to ex vivo expansion for 72 h followed by the analyses described below. (B) Expression of PFKFB3 in transduced HSCs. Cells described in (A) were gated for mCherry+ SLAM population, and RNA were extracted for q-PCR analysis for PFKFB3 expression. Samples were normalized to the level of GAPDH mRNA. (n=6 per group). (C) Overexpression of PFKFB3 increases Fru-2,6-BP production. Cells described in (A) were gated for mCherry+ SLAM population, and analyzed for F26BP by MicroAssay. (D) Overexpression of PFKFB3 increases PFK1 activity. Cells described in (A) were gated for mCherry+ population and subjected to analysis for PFK1 activity. (E) Overexpression of PFKFB3 increases intracellular GSH. Cells described in (A) were gated for mCherry+ population and subjected to GSH assay. (F) Overexpression of PFKFB3 increases ROS. Cells described in (A) were gated for mCherry+ population and subjected to analysis for ROS by flow cytometry. Representative plots (Left) and quantification (Right) are shown. (n=6 per group). p<0.05; **, p<0.01.
Discussion
The present study provides several pieces of evidence that the p53-TIGAR metabolic axis suppresses glycolysis, which serves to enhance the competitive fitness of FA HSCs. Specifically, we show that 1) Compared to WT HSCs, FA HSCs exhibit a p53-TIGAR-dependent glycolytic suppression; 2) the p53-TIGAR metabolic axis functions to ameliorate FA HSC exhaustion; 3) p53-TIGAR-dependent glycolytic suppression attenuates ROS, DNA damage and chromosomal instability in FA HSCs; 4) Although the p53-TIGAR metabolic axis is essential for maintaining FA HSC quiescence, p53-dependent cell-cycle control is not required for metabolic reprogramming in FA HSCs; 5) the rate-limiting glycolytic enzyme PFKFB3 specifically antagonists p53-TIGAR metabolic function in FA HSCs.
Glycolysis is the preferential metabolic program utilized by HSCs to produce ATP, while mitochondrial oxidative phosphorylation (OXPHOS) is more active in hematopoietic progenitors [45]. One interesting observation of our study is that the reduced glycolysis is restricted in FA HSC compartment but not in the less primitive MPPs (Lin−Sca1+c-kit+CD150−CD48−), HPCs (Lin−Sca1+c-kit+CD150−CD48+ and Lin−Sca1+c-kit+CD150+CD48+) or Lin+ cells. This is consistent with our previously study that shows that FA HSCs are more dependent on OXPHOS relative to glycolysis in their resting state for energy metabolism [12]. Whether this abnormal metabolic reprogramming in FA HSCs contributes to clinical hematological symptoms in FA such as BM failure and leukemogenesis remains to be defined. Nevertheless, altered metabolic energetics has been demonstrated in HSCs at different stages of their life cycle and in certain blood disorders [46-48]. In this context, new insights into the metabolic differences between normal and diseased HSCs may prove valuable for developing better therapeutic strategies for hematologic diseases like BM failure and leukemia.
It is known that p53 regulates energy metabolism at both glycolytic and OXPHOS steps through transcriptional regulation of its target genes TIGAR and SCO2 (synthesis of cytochrome c oxidase; 23, 24). TIGAR shares functional sequence similarities with the bis-phosphatase domain (FBPase-2) of the bi-functional enzyme PFK-2/FBPase-2, which degrades fructose-2,6-bisphosphate (Fru-2,6-BP). Fru-2,6-BP can induce 6-phospho-1-kinase (PFK1), which is required for the conversion of fructose-6-phosphate to fructose-1,6-bisphosphate at the third step of the cellular glycolysis reaction [49]. TIGAR thus reduces the cellular levels of Fru-2,6-BP and thereby blocks glycolysis at this rate-limiting step. We observed an over-activated p53-TIGAR metabolic axis in HSCs from Fanca−/− and Fancc−/− mice (Figs. 1 and 2). This may explain why FA HSCs swift to OXPHOS for energy metabolism [12]. Ectopic expression of TIGAR directs the metabolism of cellular glucose through enhancing the pentose phosphate pathway (PPP) and cellular antioxidant function therefore reducing DNA damage and chromosomal breakages in FA HSCs (Fig. 3, 4). These results suggest that p53-TIGAR metabolic axis-mediated glycolytic suppression may play a compensatory role in attenuating DNA damage and genomic instability in FA HSCs. Indeed, inactivation of the p53-TIGAR metabolic axis by deleting p53 in Fanca−/− mice induced premature HSC exhaustion (Fig. 4). Thus, our study identifies a novel role of the p53-TIGAR metabolic axis in genomic maintenance.
PFKFB3 has been distinguished from the other PFKFB isoenzymes by its high kinase to bisphosphatase ratio. Overexpression of PFKFB3 in transformed cells has identified it as a powerful glycolytic activator [44, 49]. Our data suggest that PFKFB3 could counteract p53-TIGAR function in FA HSCs. PFKFB3 synthesizes F26BP, an allosteric stimulator of PFK-1 which is another potent activator of glycolysis [38]. Although we observed no difference in PFKFB3 mRNA levels between WT and FA HSCs (Fig. 1), the levels of F26BP and PFK1 enzyme activity were significantly lower in FA HSCs than in WT cells (Figs. 2, 6). Importantly, we showed that overexpression of PFKFB3 increased F2,6BP and PFK1 activity, decreased intracellular GSH and consequently increased ROS in WT and Fanca−/− HSCs but had no further effect on these glycolytic stimulators or DNA-damaging substances in HSCs deleted for p53 (p53−/− and DKO; Fig. 6). These results suggest that PFKFB3 specifically antagonizes p53-TIGAR metabolic reprogramming in FA HSCs.
Although further studies are required to reveal the underlying molecular mechanisms, we employed a separation-of-function mutant p53 mouse model to demonstrate that the cell cycle function of p53 is not required for its metabolic reprogramming in FA HSCs. This is especially intriguing, given that the p53-TIGAR metabolic axis acted to preserve FA HSC quiescence (Fig. 4). The role of p53 in both apoptosis and cell-cycle arrest have been well established. Recent study suggests that p53 downregulates the FA DNA repair pathway [50]. It has also been shown that BM failure in FA is triggered by an exacerbated p53/p21 DNA damage response that impairs HSPCs [51]. We recently demonstrated a cell-cycle specific function of p53 in FA HSPC proliferation and proposed that over-activated p53 might represent a compensatory checkpoint mechanism for FA HSC proliferation [43]. However, our present study shows that Fanca−/− HSCs carrying the p53R172P mutation exhibited increased glucose consumption and PFK1 activity (Fig. 5), exactly as observed in p53-null Fanca−/− HSCs (Fig. 2), suggesting that the cell-cycle activity of p53 is not required for TIGAR-mediated glycolytic suppression in FA HSCs.
Supplementary Material
Acknowledgments
We thank Dr. Madeleine Carreau (Laval University) for Fanca+/− mice, Dr. Guillermina Lozano (University of Texas M.D. Anderson Cancer Center) for p53R172P mice and Dr. Manuel Buchwald (University of Toronto) for Fancc+/− mice. W.D. is supported by NIH Tumor Microenvironment Center of Biomedical Excellence Award (P20GM121322), West Virginia University (WVU) Health Science Center (HSC) and School of Pharmacy (SOP) startup funds, a Leukemia Research Foundation (LRF) Award, and an American Cancer Society (ACS) Institutional Research Grant. This work was partially supported by the National Natural Science Foundation of China (NNSFC # 81470288, 81370608 and U1401221).
Footnotes
Author Disclosure Statement
There is no conflict of interest to disclose on the part of any authors.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References:
- 1.Bagby GC Jr. Genetic basis of Fanconi anemia. Curr Opin Hematol 2003;10(1):68–76. [PubMed: 12483114] [DOI] [PubMed] [Google Scholar]
- 2.Tischkowitz MD, Hodgson SV. Fanconi anaemia. J Med Genet 2003;40(1):1–10. [PubMed: 12525534] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bluteau D, Masliah-Planchon J, Clairmont C et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest 2016;126:3580–3584. [PubMed: 27500492] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011;11(7):467–480. [PubMed: 21701511] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the human and mouse Fanconi anemia genes. Hum Genomics 2015;9:32. [PubMed: 26596371] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du W, Erden O, Pang Q. TNF-a signaling in Fanconi anemia. Blood Cells Mol Dis 2013;52(1):2–11. [PubMed: 23890415] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kennedy RD, D’Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev 2005;19(24):2925–2940. [PubMed: 16357213] [DOI] [PubMed] [Google Scholar]
- 8.Meyer S, Neitzel H, Tonnies H. Chromosomal aberrations associated with clonal evolution and leukemic transformation in Fanconi anemia: clinical and biological implications. Anemia 2012;2012:349837. [PubMed: 22675616] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JY, Virts EL, Jankowska A et al. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J Med Genet 2016;53:672–680. [PubMed: 27208205] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawyer SL, Tian L, Kähkönen M et al. Biallelic Mutations in BRCA1 Cause a New Fanconi Anemia Subtype. Cancer Discov 2015;5(2):135–142. [PubMed: 25472942] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knies K, Inano S, Ramírez MJ et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest 2017;127(8):3013–3027. [PubMed: 28691929] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du W, Amarachintha S, Wilson AF, Pang Q. SCO2 Mediates Oxidative Stress-Induced Glycolysis to Oxidative Phosphorylation Switch in Hematopoietic Stem Cells. Stem Cells 2016;34(4):960–971. [PubMed: 26676373] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancers 2009;9:749–758. [PubMed: 19776744] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol 2008;(9):702–712. [PubMed: 12855557] [DOI] [PubMed] [Google Scholar]
- 15.Krug U, Ganser A, Koeffler HP. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene 2002;21:3475–3495. [PubMed: 18719709] [DOI] [PubMed] [Google Scholar]
- 16.Prokocimer M, Rotter V. Structure and function of p53 in normal cells and their aberrations in cancer cells: projection on the hematologic cell lineages. Blood 1994;84:2391–2411. [PubMed: 7919359] [PubMed] [Google Scholar]
- 17.Liu Y, Elf SE, Miyata Y et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009;4:37–48. [PubMed: 19128791] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.TeKippe M, Harrison DE, Chen J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp Hematol 2003;31:521–527. [PubMed: 12829028] [DOI] [PubMed] [Google Scholar]
- 19.Asai T, Liu Y, Bae N, Nimer SD. The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J Cell Physiol. 2011;226:2215–2221. [PubMed: 21660944] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nii T, Marumoto T, Tani K. Roles of p53 in various biological aspects of hematopoietic stem cells. J Biomed Biotechnol 2012;2012:903435. [PubMed: 22778557] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev 2009;23:537–548. [PubMed: 19270154] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancers 2009;9:691–700. [PubMed: 19759539] [DOI] [PubMed] [Google Scholar]
- 23.Matoba S, Kang JG, Patino WD et al. p53 regulates mitochondrial respiration. Science, 2006;312:1650–1653. [PubMed: 16728594] [DOI] [PubMed] [Google Scholar]
- 24.Bensaad K, Tsuruta A, Selak MA et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006;126:107–120. [PubMed: 16839880] [DOI] [PubMed] [Google Scholar]
- 25.Dasgupta T, Croll DH, Owen JA et al. A fundamental trade-off in covalent switching and its circumvention by enzyme bifunctionality in glucose homeostasis. J Biol Chem 2014;289(19):13010–13025. [PubMed: 24634222] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green DR, Chipuk JE. p53 and metabolism: Inside the TIGAR. See comment in PubMed Commons belowCell 2006;126(1): 30–32. [PubMed: 16839873] [DOI] [PubMed] [Google Scholar]
- 27.Bensaad K, Tsuruta A, Selak MA et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006;126(1):107–120. [PubMed: 16839880] [DOI] [PubMed] [Google Scholar]
- 28.Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS. Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet 2003;12:2063–2076. [PubMed: 12913077] [DOI] [PubMed] [Google Scholar]
- 29.Liu G, Parant JM, Lang G et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 2004;36:63–68. [PubMed: 14702042] [DOI] [PubMed] [Google Scholar]
- 30.Chen M, Tomkins DJ, Auerbach W et al. Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. Nat Genet 1996;12(4):448–451. [PubMed: 8630504] [DOI] [PubMed] [Google Scholar]
- 31.Jacks T, Remington L, Williams BO et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994;4:1–7. [PubMed: 7922305] [DOI] [PubMed] [Google Scholar]
- 32.Van Schaftingen E, Lederer B, Bartrons R, Hers HG. A kinetic study of pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose 2,6-bisphosphate. Eur J Biochem 1982;129(1):191–195. [PubMed: 6297885] [DOI] [PubMed] [Google Scholar]
- 33.Imbert-Fernandez Y, Clem BF, O’Neal J et al. Estradiol stimulates glucose metabolism via 6-phosphofructo-2-kinase (PFKFB3). J Biol Chem 2014;289(13):9440–9448. [PubMed: 24515104] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davison K, Côté S, Mader S, Miller WH. Glutathione depletion overcomes resistance to arsenic trioxide in arsenic-resistant cell lines. Leukemia 2003;17:931–940. [PubMed: 12750708] [DOI] [PubMed] [Google Scholar]
- 35.Oostra AB, Nieuwint AW, Joenje H, de Winter JP. Diagnosis of Fanconi anemia: chromosomal breakage analysis. Anemia 2012;2012:238731. [PubMed: 22693659] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schambach A, Galla M, Modlich U et al. Lentiviral vectors pseudotyped with murine ecotropic envelope: increased biosafety and convenience in preclinical research. Exp Hematol 2006;34(5):588–592. [PubMed: 16647564] [DOI] [PubMed] [Google Scholar]
- 37.Moncada S, Higgs EA, Colombo SL. Fulfilling the metabolic requirements for cell proliferation. Biochem J 2012;446(1):1–7. [PubMed: 22835215] [DOI] [PubMed] [Google Scholar]
- 38.Yalcin A, Clem BF, Simmons A et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem 2009;284(36):24223–24232. [PubMed: 19473963] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chao C, Herr D, Chun J, Xu Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J 2006;25(11):2615–2622. [PubMed: 16757976] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carr MI, Roderick JE, Gannon HS, Kelliher MA, Jones SN. Mdm2 Phosphorylation Regulates Its Stability and Has Contrasting Effects on Oncogene and Radiation-Induced Tumorigenesis. Cell Rep 2016;16(10):2618–2629. [PubMed: 27568562] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinberg F, Hamanaka R, Wheaton WW et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 2010;107(19):8788–8793. [PubMed: 20421486] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Celeste A, Difilippantonio S, Difilippantonio MJ et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003;114(3):371–383. [PubMed: 12914701] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li X, Wilson AF, Du W, Pang Q. Cell-Cycle-Specific Function of p53 in Fanconi Anemia Hematopoietic Stem and Progenitor Cell Proliferation. Stem Cell Reports 2018;10(2):339–346. [PubMed: 29307578] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol 2009;86(3):174–179. [PubMed: 19454274] [DOI] [PubMed] [Google Scholar]
- 45.Snoeck HW. Mitochondrial regulation of hematopoietic stem cells. Curr Opin Cell Biol 2017;49:91–98. [PubMed: 29309987] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011;9(4):298–310. [PubMed: 21982230] [DOI] [PubMed] [Google Scholar]
- 47.Warr MR, Pietras EM, Passegué E. Mechanisms controlling hematopoietic stem cell functions during normal hematopoiesis and hematological malignancies. Wiley Interdiscip Rev Syst Biol Med 2011;3(6), 681–701. [PubMed: 21412991] [DOI] [PubMed] [Google Scholar]
- 48.Baumann K Stem cells: A metabolic switch. Nat Rev Mol Cell Biol 2013;14(2):64–65. [PubMed: 23340568] [DOI] [PubMed] [Google Scholar]
- 49.Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget 2011;2(12):948–957. [PubMed: 22248668] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaber S, Toufektchan E, Lejour V, Bardot B, Toledo F. (2016) P53 downregulates the Fanconi anemia DNA repair pathway. Nat Commun 2016;7:11091. [PubMed: 27033104] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freie B, Li X, Ciccone SL, Nawa K et al. Fanconi anemia type C and p53 cooperate in apoptosis and tumorigenesis. Blood 2003;102(12):4146–4152. [PubMed: 12855557] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.