

Abstract
A method for the annulation of amines and carboxylic acids to form pharmaceutically-relevant azaheterocycles via organophosphorus PIII/PV redox catalysis is reported. The method employs a phosphetane catalyst together with a mild bromenium oxidant and terminal hydrosilane reductant to drive successive C -N and C -C bond forming dehydration events via the serial action of a catalytic bromophosphonium intermediate. These results demonstrate the capacity of PIII/PV redox catalysis to enable iterative redox-neutral transformations in complement to the common reductive driving force of PIII/PV couple.
Graphical Abstract
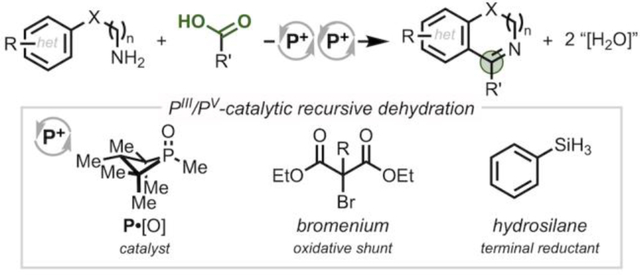
Progress over the past decade has established the viability of the PIII/PV=O redox couple for catalysis.1–3 In contrast to prior notions about the kinetic inertness of the P=O bond, the incorporation of P into a ring structure can lead to swift deoxygenation by mild reagents such as hydrosilanes.4,5 By virtue of the reducing potential of the pIII/ pV couple, m any of these transformations are reductive in nature (Figure 1A, left).6–8 In this vein, we have shown that four-membered ring organophosphorus catalysts effect reductive conversion of nitro9 and sulfonyl10 substrates (cf. Figure 1B) in which the ability to recursively renew a reactive PIII species under conditions of pIII/pV catalysis enables a single catalyst to perform successive deoxygenative operations on a substrate (i.e. auto-tandem catalysis11).
Figure 1.

(A) Complementary reductive and redox-neutral modes of PIII/pV catalysis. (B) Catalytic heterocyclization of nitrobiaryl by recursive deoxygenation. (C) Catalytic annulation of amines and carboxylic acids by recursive dehydration.
In addition to reductive chemistry, the versatile PIII/PV driving force can be adapted to achieve net redox-neutral transformations when paired with an appropriate oxidant as evoked in Mukaiyama’s conceptualization of an “oxidation-reduction condensation.”12 Within a PIII/PV-catalytic context,13 the introduction of a mild chemoselective halenium oxidant, for instance, can shunt the reductive manifold into a net redox-neutral mode where the key reactive catalytic intermediate is not a phosphine but rather a halophosphonium cation14,15 (Figure 1A, right). Indeed, halophosphonium intermediates have been invoked by Rutjes and van Delft5 and Mecinović16 in the context of PIII/PV-catalyzed Appel halogenation and N-acylation reactions, respectively.17 In view of the fact that phosphonium reagents have been described as having “virtually ideal properties as selective oxygen extractors for net dehydration reactions,”18,19 the potential to achieve recursive dehydrations in an auto-tandem catalytic manner via a net redox-neutral mode of pIII/pV catalysis could be expected to present new opportunities for serial bond formation.
We show here an annulation of amines and carboxylic acids via recursive dehydration driven by a redox-active organophosphorus catalyst cycling in the PIII/PV couple (Figure 1C). This auto-tandem catalytic system enables the elaboration of simple, commercially available starting materials into pharmaceutically- relevant azaheterocycles through a condensation/cyclodehydration sequence in a one-pot catalytic protocol. The success of the approach relies on the mutual compatibility and functional interplay of the reducing and oxidizing reagents with the organophosphorus catalyst in order to orchestrate a sequence of distinct C -N and C -C bond-forming events. The ability of PIII/PV redox catalysis to encompass such recursive dehydration stands as a complement to existing deoxygenation methods, thus broadening the scope of transformations accessible to this catalytic mode.
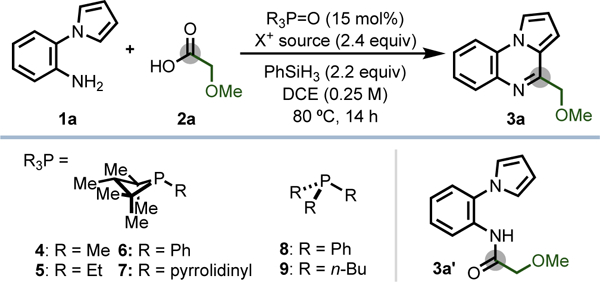
To evaluate the possibility of recursive dehydration driven by PIII/PV redox cycling, the tandem amidation/cyclodehydration of amine 1a and carboxylic acid 2a to generate pyrroloquinoxaline 3a was evaluated (Table 1). Optim al conditions using 1,2,2,3,4,4- hexamethylphosphetane P-oxide 4• [O]20 as catalyst, diethyl bromomalonate (DEBM ) as oxidant, and phenylsilane as terminal reductant yielded the desired product in 94% yield, isolable on 0.4 mmol scale in 84% yield (Table 1, entry 1). A mild, weakly oxidizing bromenium reagent was found to be essential for the redox compatibility of the system , as demonstrated by Rutjes and van Delft;5 the related diethyl (methyl)bromomalonate (DEMBM) was similarly competent in the transformation (entry 2), but the more strongly-oxidizing N -bromosuccinimide resulted in poor conversion to product (entry 3). Chlorenium oxidants such as diethyl chloromalonate (DECM) and carbon tetrachloride gave no dehydrative heterocyclization (entries 4 and 5); instead, am ide 3a’ was obtained in 70% yield, indicating chlorophosphonium ion com petency in C -N forming amidation but not C -C forming cyclodehydration.21
Table 1.
Discovery and Optimization of Phosphacatalytic Iterative Condensation/Annulation of Amine and Carboxylic Acid
| Entry | R3P=O | X+ source | Yield (%)a |
|---|---|---|---|
| 1 | 4•[O] | DEBM | 94 (84)b |
| 2 | 4•[O] | DEMBM | 90 |
| 3 | 4•[O] | NBS | 10 |
| 4 | 4•[O] | DECM | 0 |
| 5 | 4•[O] | CCl4 | 0 |
| 6 | 5•[O] | DEBM | 26 |
| 7 | 6•[O] | DEBM | 46 |
| 8 | 7•[O] | DEBM | 50 |
| 9 | 8•[O] or 9•[O] | DEBM | 0 |
| 10 | none | DEBM | 0 |
| 11 | 4•[O] | none | 0 |
| 12 | 4•[O], no PhSiH3 | DEBM | 0 |
| 13 | 4 | DEBM | 90 |
| 14 | [4•Br]Br | DEBM | 87 |
Yields determined through 1H NMR analysis with the aid of an internal standard. See Supporting Information for full synthetic details and yields of intermediate amide 3a’.
Isolated yield on 0.4 mmol scale. DCE = 1,2-dichloroethane; DEBM = diethyl bromomalonate; DEMBM = diethyl (methyl)bromomalonate; NBS = N-bromosuccinimide; DECM = diethyl chloromalonate.
With respect to catalyst, variation of the phosphetane exocyclic moiety to ethyl (5•[O], entry 6), phenyl (6• [O], entry 7), or pyrrolidino (7• [O], entry 8) all resulted in substantial decrease in the efficiency of the reaction, while the use of acylic phosphine oxides (8•[O] or 9• [O], entry 9) fail to promote the cyclocondensation (see SI). Control experiments confirm that no azaheterocycle product is observed in the absence of any of 4•[O], phenylsilane, or DEBM (entries 10–12).22 Furthermore, employing PIII species 4 or pregenerated bromophosphonium [4•Br]Br23 in place of phosphine oxide 4 •[O] resulted in com parable efficiency, consistent with the notion of PIII/PV=O redox cycling (entries 13–14).
The optimized phosphacatalytic protocol provides direct access to complex azaheterocycles from simple and readily available amine and carboxylic acid starting materials (Figure 2). A variety of carboxylic acids are efficiently incorporated into pyr-roloquinoxalines, including those possessing olefinic and aryl functionalities (3c-3i, 54–89% yields). This protocol was also readily translated to larger scale reactions, as a 5 mmol scale reaction of 1-(2-am inophenyl)pyrrole and butyric acid provided 1.04 g of com pound 3b in 99% yield with 8 mol% loading of organophosphorus catalyst 4 •[O]. As demonstrated by products 3d- 3i, the reaction efficiency is relatively independent of substitution on benzoic acid coupling partners, including changing steric profile (54–89% yields). Critically, acids containing polar functionalities, including alkyl ethers, thioethers, sulfonamides, and alkyl halides, undergo efficient iterative dehydration to provide heterocycles in good to excellent yields (3a, 3j-3p, 41–90% yields). Of particular note are the amino acid-incorporating products 3k and 3p, which originate from protected Ts-Gly-OH and Ts-Phe-OH. Further, both primary and secondary haloalkane functionalities are conserved under this PIII/PV-catalytic m anifold (3l and 3m, 93% and 98% yields, respectively). Substitution on the aniline ring was well- tolerated; both ortho- and meta-substituted pyrroloanilines were readily incorporated into heterocyclic scaffolds (3n and 3o, 86% and 95% yields, respectively). Chiral carboxylic acids, such as ibuprofen and naproxen, are incorporated with good yield and high stereochemical fidelity (3q, 67 %, 95:5 e.r.; 3r, 68%, 92.5:7.5 e.r.) under modified recursive dehydration conditions (precatalyst [4•Br]Br, MeCN, 50 °C; see Supporting Information for full synthetic details).
Figure 2.

Examples of auto-tandem phosphacatalytic annulation of amines and carboxylic acids. All yields isolated on 0.4 mmol scale unless indicated otherwise. See Supporting Information for full synthetic details. aIsolated yield on 5.0 mmol scale, with 8 mol% of 4•[O]. bReaction conducted with Ph2SiH2 (4.4 equiv) in MeCN. cReaction conducted at 60 °C for 20 h. dReaction conducted on 0.2 mmol scale using 15 mol% of [4•Br]Br in MeCN at 50°C for 20–40 h. eYield determined by 1H NMR with internal standard. fReaction conducted with DEMBM. DEBM = diethyl bromomalonate; DEMBM = diethyl(methyl)bromomalonate; DCE = 1,2-dichloroethane; Bn = benzyl; Ts = tosyl; iBu = iso-butyl.
When N-alkyl amine substrates were initially employed under standard conditions, an undesired byproduct arising from N-alkylation by DEBM was identified by GCMS. However, replacing DEBM with its methyl-substituted analogue, DEMBM, abated this deleterious pathway, restoring the high degree of redox compatibility necessary for the catalytic system. Consequently, o-pyrrolobenzylamine could be efficiently coupled with carboxylic acids via iterative dehydration to provide the corresponding pyrrolo-benzodiazepines, a prevalent bioactive scaffold,24 in good yields (3s-3u, 66–86% yields).25 Heterocycles 3t and 3u, compounds investigated by Janssen for their antifungal activity,26 could be prepared in a single synthetic operation in 86% and 73% yields, respectively. Further, tryptamines and phenethylamines could be transformed into dihydro-β-carboline and dihydroisoquinoline products in good yields (3v-3x, 70–77% yields).27 Notably, both of these scaffolds are found in bioactive pharmaceutical agents and natural products,28 as demonstrated by the assembly in a single step of dihydroisoquinoline natural product 3,4- dihydropapaverine from commercial starting materials with good efficiency (3x, 70% yield).
Concerning the catalytic mechanism, in situ1H NMR spectroscopy revealed a rapid initial conversion of reactants 1a and 2a into am ide intermediate 3a’, followed by comparatively slow formation of heterocycle 3a (see SI, Sect. VI), establishing a stepwise reaction sequence for the auto-tandem catalytic process in which C -C bond-forming cyclodehydration is kinetically limiting. Despite the observation that 4, 4•[O], or 4•Br+ are each competent precatalysts (vide supra), in situ31P NMR and DART- MS analyses show that none of these com pounds represent the catalytic resting state. Rather, experiments are most consistent with resting state B1, an adduct of the phosphacyclic catalyst and am ide A1 (Figure 3A). Indeed, independent reaction of [4•Br]Br with A1 gives rise to spectroscopic signals indistinguishable from those observed under the catalytic steady state, and this species was shown to lead to C -C bond-forming cyclodehydration.29 Furthermore, spectroscopically indistinguishable species are observed by31P NMR spectroscopy when either N-methylacetamide and N,N-dimethylacetamide are introduced in lieu of reactive am ides to a mixture containing catalytic components (i.e. 4•[O], PhSiH3, DEBM).
Figure 3.

(A) 31P NMR studies. (B) Proposed mechanism of auto-tandem catalytic dehydrative annulation of amines and carboxylic acids. Methyl groups excluded from 4 for clarity. DEBM = diethyl bromomalonate.
Figure 3B depicts a plausible auto-tandem catalytic reaction mechanism consistent with the foregoing experimental data. From phosphine oxide 4•[O] as precatalyst, entry to the C -N bond-forming cycle (Figure 3B) is initiated by kinetically-facile phenylsilane-mediated reduction to phosphine 4, followed by rapid halophilic reaction30 with DEBM leading to bromophosphonium ion 4•Br+. Bromophosphonium cation 4•Br+ effects intermolecular amidation between acid 1 and amine 2, presumably via intermediate C in analogy to established precedent for amine N-acylation by activated acyloxyphosphoniums, thereby returning phosphine oxide 4•[O]. In C -C bond-forming second phase, phosphonium ion 4•Br+ is again generated by a reduction- oxidation sequence with PhSiH3 and DEBM, respectively. Exchange of bromide for the am ide substrate A then leads to activated species B, which is assigned as the catalytic resting state. Cyclization ensues to provide the product 3, liberating phosphine oxide 4•[O] and closing the catalytic cycle.31 The two noteworthy conclusions emerging from this mechanistic picture are: (1) Turnover of phosphine oxide 4•[O] to phosphine 4 is not kinetically limiting; and (2) The concentration of reducing phosphine 4 remains negligibly low during catalysis as a function of the efficient reaction with the oxidative halenium shunt.
The net redox neutral character of the recursive dehydration (and the absence of appreciable concentrations of phosphine 4) enables chemoselective annulation in preference to established PIII/PV-catalyzed reductive transformations. This orthogonality is illustrated in the context of p-nitrohydrocinnam ic acid (10), possessing both carboxylic acid and nitroarene moieties (Figure 4). Under the recursive dehydration conditions described herein, catalyst 4•[O] drives the selective annulation of carboxylic acid 10 to yield pyrroloquinoxaline 11 in 97% yield. With the same catalyst (4•[O]) but omission of the DEBM shunt, the nitro group is then reductively addressed to effect intermolecular C -N cross coupling9c and yield fully deoxygenated species 12 in 72% yield. This sequence, in which a single organophosphorus catalyst executes four distinct oxygen excisions, demonstrates the complementarity of the reductive and redox-neutral PIII/PV-catalytic manifolds owing to the disparate reactivity of the phosphetane in its different oxidation states as a function of the addition or exclusion of an exogenous oxidant.
Figure 4.

Selective functionalization of carboxylic acid- and nitro-containing substrate via sequential redox-neutral recursive dehydration, then reductive recursive deoxygenation, using a single catalyst 4•[O]. Reaction conditions: (a) 1a (1.0 equiv), 10 (1.05 equiv), DEBM (2.4 equiv), PhSiH (2.2 equiv), 4•[O] (15 mol%), DCE, 80 °C; (b) 11 (1.0 equiv), 4-MeO-C6H4 -B(OH)2 (1.1 equiv), PhSiH3 (2.0 equiv), 4•[O] (15 mol%), mxylene, 120 °C.
In conclusion, we have demonstrated that a small-ring phosphetane catalyst can induce iterative dehydrative C -N and C -C bond-forming reactions, enabling direct azaheterocycle synthesis from carboxylic acids and amines via recursive dehydration. Through the synergistic use of mild hydrosilane reductant and bromenium oxidant, the elements of water can be catalytically removed in the form of an O-atom and two protons with complete redox compatibility. We anticipate that this phosphacatalytic dehydration manifold will prove generally enabling for the redox-neutral functionalization of oxygenated organic functionalities to accomplish C -C and C-heteroatom bond forming events via condensation, especially in a recursive fashion.
Supplementary Material
ACKNOWLEDGMENT
Financial support was provided by NIH NIGMS (GM 114547). M.L. thanks the Belgian American Educational Foundation (BAEF) for a postdoctoral fellowship. J.M .L. thanks the Camille and Henry Dreyfus Foundation for a postdoctoral fellow ship in Environmental Chemistry. S.-H. K.-L. thanks Prof. Pablo Mauleon, Universidad Autónoma de Madrid for a mobility grant and Ministerio de Educación, Cultura y Deporte (M ECD) for a FPU predoctoral fellowship. The authors acknowledge the Buchwald laboratory (MIT) for access to HPLC equipment, and J. Connor Gilhula and Hye Won Moon for assistance in acquiring HRMS data.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Additional optimization results, mechanistic studies, and synthetic procedures.1H,13C, and 31P NMR spectra
The authors declare no competing financial interest.
REFERENCES
- 1. (a).O’Brien CJ; Tellez JL; Nixon ZS; Kang LJ; Carter AL; Kunkel SR; Przeworski KC; Chass GA Recycling the Waste: The Development of a Catalytic Wittig Reaction. Angew. Chem. Int. Ed 2009, 48, 6836–6839. [DOI] [PubMed] [Google Scholar]; (b) O’Brien CJ Catalytic Wittig and Mitsunobu Reactions. U.S. Patent 8,901,365, December 2, 2014. [Google Scholar]
- 2. (a).van Kalkeren HA; van Delft FL; Rutjes FPJT Organo-phosphorus Catalysis to Bypass Phosphine Oxide Waste. Chem. Sus. Chem 2013, 6, 1615–1624. [DOI] [PubMed] [Google Scholar]; (b) Guo H; Fan YC; Sun Z; Wu Y; Kwon O Phosphine Organocatalysis. Chem. Rev 2018, 118, 10049–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For reviews discussing PV=O catalysis, see:; (a) Marsden SP Catalytic Variants of Phosphine Oxide-Mediated Organic Transformations In Sustainable Catalysis; Dunn PJ, Hii KK, Krische MJ, Williams MT, Eds.; John Wiley & Sons, Inc.: New York, 2013; pp 339–361. [Google Scholar]; (b) Denmark SE; Stavenger RA Asymmetric Catalysis of Aldol Reactions with Chiral Lewis Bases. Acc. Chem. Res 2000, 33, 432–440. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE; Beutner GL Lewis Base Catalysis in Organic Synthesis. Angew. Chem. Int. Ed 2008, 47, 1560–1638. [DOI] [PubMed] [Google Scholar]; (d) Benaglia M; Rossi S Chiral Phosphine Oxides in Present-Day Organocatalysis. Org. Biomol. Chem 2010, 8, 3824–3830. [DOI] [PubMed] [Google Scholar]
- 4.Marsi KL Phenylsilane Reduction of Phosphine Oxides with Complete Stereospecificity. J. Org. Chem 1974, 39, 265–267. [Google Scholar]
- 5. (a).van Kalkeren HA; Leenders SHAM; Hommersom CRA; Rutjes FPJT; van Delft FL In Situ Phosphine Oxide Reduction: A Catalytic Appel Reaction. Chem. Eur. J 2011, 17, 11290–11295. [DOI] [PubMed] [Google Scholar]; (b) van Kalkeren HA; van Delft FL; Rutjes FPJT Catalytic Appel Reactions. Pure Appl. Chem 2013, 85, 817–828. [Google Scholar]
- 6.Zhao W; Yan PK; Radosevich AT A Phosphetane Catalyzes Deoxygenative Condensation of α-Keto Esters and Carboxylic Acids via PIII/PV=O Redox Cycling. J. Am. Chem. Soc 2015, 137, 616–619. [DOI] [PubMed] [Google Scholar]
- 7.For phosphacatalytic Staudinger and related reactions:; (a) van Kalkeren HA; Bruins JJ; Rutjes FPJT; van Delft FL Organophosphorus-Catalysed Staudinger Reduction. Adv. Synth. Catal 2012, 354, 1417–1421. [Google Scholar]; (b) van Kalkeren HA; te Grotenhuis C;Haasjes FS; Hommersom CRA; Rutjes FPJT; van Delft FL Catalytic Staudinger/Aza-Wittig Sequence by In Situ Phosphane Oxide Reduction. Eur. J. Org. Chem 2013, 7059–7066. [Google Scholar]; (c) Cai L; Zhang K; Chen S; Lepage RJ; Houk KN; Krenske EH; Kwon O Catalytic Asymmetric Staudinger-Aza-Wittig Reaction for the Synthesis of Heterocyclic Amines. J. Am. Chem. Soc 2019, 141, 9537–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kosal AD; Wilson EE; Ashfeld BL Phosphine-Based Redox Catalysis in the Direct Traceless Staudinger Ligation of Carboxylic Acids and Azides. Angew. Chem. Int. Ed 2012, 51, 12036–12040. [DOI] [PubMed] [Google Scholar]
- 8.For phosphacatalytic Wittig and related reactions:; (a) O’Brien CJ; Lavigne F; Coyle EE; Holohan AJ; Doonan BJ Breaking the Ring Through a Room Temperature Catalytic Wittig Reaction. Chem. Eur. J 2013, 19, 5854–5858. [DOI] [PubMed] [Google Scholar]; (b) O’Brien CJ; Nixon ZS; Holohan AJ; Kunkel SR; Tellez JL; Doonan BJ; Coyle EE; Lavigne F; Kang LJ; Przeworski KC Part I: The Development of the Catalytic Wittig Reaction. Chem. Eur. J 2013, 19, 15281–15289. [DOI] [PubMed] [Google Scholar]; (c) Coyle EE; Doonan BJ; Holohan AJ; Walsh KA; Lavigne F; Krenske EH; O’Brien CJ Catalytic Wittig Reactions of Semi- and Nonstabilized Ylides Enabled by Ylide Tuning. Angew. Chem. Int. Ed 2014, 53, 12907–12911. [DOI] [PubMed] [Google Scholar]; (d) Lee C; Chang T; Yu J; Reddy GM; Hsiao M; Lin W Synthesis of Functionalized Furans via Chemoselective Reduction/Wittig Reaction Using Catalytic Triethyla-mine and Phosphine. Org. Lett 2016, 18, 3758–3761. [DOI] [PubMed] [Google Scholar]; (e) Saleh N; Voituriez A Synthesis of 9H-Pyrrolo[1,2-a]indole and 3H-Pyrrolizine Derivatives via a Phosphine-Catalyzed Umpolung Addition/Intramolecular Wittig Reaction. J. Org. Chem 2016, 81, 4371–4377. [DOI] [PubMed] [Google Scholar]; (f) Saleh N; Blanchard F; Voituriez A Synthesis of Nitrogen Containing Heterocycles and Cyclopentenone Derivatives via Phosphine Catalyzed Michael Addition/Intramolecular Wittig Reaction. Adv. Synth. Catal 2017, 359, 2304–2315. [Google Scholar]; (g) Zhang K; Cai L; Yang Z; Houk KN; Kwon O Bridged [2.2.1] Bicyclic Phosphine Oxide Facilitates Catalytic y-Umpolung Addition-Wittig Olefination. Chem. Sci 2018, 9, 1867–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. (a).Nykaza TV; Harrison TS; Ghosh A; Putnik RA; Radosevich AT A Biphilic Phosphetane Catalyzes N-N Bond-Forming Cadogan Heterocyclization via PIII/PV=O Redox Cycling. J. Am. Chem. Soc 2017, 139, 6839–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nykaza TV; Ramirez A; Harrison TS; Luzung MR; Radosevich AT Biphilic Organophosphorus-Catalyzed Intramolecular Csp2-H Amination: Evidence for a Nitrenoid in Catalytic Cadogan Cyclizations. J. Am. Chem. Soc 2018, 140, 3103–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nykaza TV; Cooper JC; Li G; Mahieu N; Ramirez A; Luzung MR;Radosevich AT Intermolecular Reductive C-N Cross Coupling of Nitroarenes and Boronic Acids by PIII/PV=O Catalysis. J. Am. Chem. Soc 2018, 140, 15200–15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh A; Lecomte M; Kim-Lee S-H; Radosevich AT Organo-phosphorus-Catalyzed Deoxygenation of Sulfonyl Chlorides: Electrophilic (Fluoroalkyl)sulfenylation by PIII/PV=P Redox Cycling. Angew. Chem. Int. Ed 2019, 58, 2864–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fogg DE; dos Santos EN Tandem Catalysis: A Taxonomy and Illustrative Review. Coord. Chem. Rev 2004, 248, 2365–2379. [Google Scholar]
- 12.(a) Mukaiyama T Oxidation-Reduction Condensation. Angew. Chem. Int. Ed 1976, 15, 94–103. [Google Scholar]; (b) Mukaiyama T Explorations into New Reaction Chemistry. Angew. Chem. Int. Ed 2004, 43, 5590–5614. [DOI] [PubMed] [Google Scholar]
- 13.(a) Buonomo JA; Aldrich CC Mitsunobu Reactions Catalytic in Phosphine and a Fully Catalytic System. Angew. Chem. Int. Ed 2015, 54, 13041–13044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hirose D; Gazvoda M; Košmrlj J; Taniguchi T The “Fully Catalytic System” in Mitsunobu Reaction Has Not Been Realized Yet. Org. Lett 2016, 18, 4036–4039. [DOI] [PubMed] [Google Scholar]; (c) Beddoe RH; Sneddon HF; Denton RM The Catalytic Mitsunobu Reaction: A Critical Analysis of the Current State-of-the-Art. Org. Biomol. Chem 2018, 16, 7774–7781. [DOI] [PubMed] [Google Scholar]
- 14.Classical Appel conditions have been adapted for N-acylation reactions:; (a) Appel R Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P-N Linkage. Angew. Chem. Int. Ed 1975, 14, 801–811. [Google Scholar]; (b) Barstow LE; Hruby VJ A Simple Method for the Synthesis of Amides. J. Org. Chem 1971, 36, 1305–1306. [Google Scholar]; (c) Luo Q-L; Lv L; Li Y; Tan J-P; Nan W; Hui Q An Efficient Protocol for the Amidation of Carboxylic Acids Promoted by Trimethyl Phosphite and Iodine. Eur. J. Org. Chem 2011, 6916–6922. [Google Scholar]
- 15.For halophosphonium cations as Lewis acids, see:; (a) Caputo CB; Hounjet LJ; Dobrovetsky R; Stephan DW Lewis Acidity of Organofluorophosphonium Salts: Hydrodefluorination by a Saturated Acceptor. Science 2013, 341, 1374–1377. [DOI] [PubMed] [Google Scholar]; (b) Phosphorus Lewis Acids: Emerging Reactivity and Applications in Catalysis. Chem. Soc. Rev 2016, 45, 765–774. [DOI] [PubMed] [Google Scholar]
- 16.Lenstra DC; Rutjes FPJT; Mecinović J Triphenylphosphine-Catalysed Amide Bond Formation Between Carboxylic Acids and Amines. Chem. Commun 2014, 50, 5763–5766. [DOI] [PubMed] [Google Scholar]
- 17.PV-catalyzed deoxychlorination and amidation reactions using phosphine oxide and oxalyl chloride have been developed:; (a) Denton RM; An J; Adeniran B Phosphine Oxide-Catalysed Chlorination Reactions of Alcohols Under Appel Conditions. Chem. Commun 2010, 46, 3025–3027. [DOI] [PubMed] [Google Scholar]; (b) Denton RM; Tang X; Przeslak A Catalysis of Phosphorus(V)-Mediated Transformations: Dichlorination Reactions of Epoxides Under Appel Conditions. Org. Lett 2010, 12, 4678–4681. [DOI] [PubMed] [Google Scholar]; (c) Denton RM; An J; Adeniran B; Blake AJ; Lewis W; Poulton AM Catalytic Phosphorus(V)-Mediated Nucleophilic Substitution Reactions: Development of a Catalytic Appel Reaction. J. Org. Chem 2011, 76, 6749–6767. [DOI] [PubMed] [Google Scholar]; (d) An J; Tang X; Moore J; Lewis W; Denton RM Phosphorus(V)-Catalyzed Deoxydichlorination Reactions of Aldehydes. Tetrahedron 2013, 69, 8769–8776. [Google Scholar]; (e) Yu T-Y; Wang Y; Xu P-F An Unusual Triphenylphosphine Oxide Catalyzed Stereoselective 1,3-Dichlorination of Unsaturated Ketoesters. Chem. Eur. J 2014, 20, 98–101. [DOI] [PubMed] [Google Scholar]; (f) Tang X; An J; Denton RM A Procedure for Appel Halogenations and Dehydrations Using a Polystyrene Supported Phosphine Oxide. Tetrahedron Lett. 2014, 55, 799–802. [Google Scholar]; (g) Jiang L; Yu J; Niu F; Zhang D; Sun X A High-Efficient Method for the Amidation of Carboxylic Acids Promoted by Triphenylphosphine Oxide and Oxalyl Chloride. Heteroat. Chem 2017, 28, e21364. [Google Scholar]
- 18.Hendrickson JB; Hussoin MS Seeking the Ideal Dehydrating Reagent. J. Org. Chem 1987, 52, 4137–4139. [Google Scholar]
- 19.Phosphonium salts are widely used as peptide-coupling reagents:; (a) El-Faham A; Albericio F Peptide Coupling Reagents, More than a Letter Soup. Chem. Rev 2011, 111, 6557–6602. [DOI] [PubMed] [Google Scholar]; (b) Berchel M; Jaffres P-A Recent Developments in Phosphonium Chemistry In Organ-ophosphorus Chemistry: From Molecules to Applications; Iaroshenko V, Ed.; Wiley-VCH: Weinheim, Germany, 2019; pp 78–84. [Google Scholar]
- 20.Strem item no. 15–8150.
- 21.Consistent with the notion of sequential C-N then C-C bond forming by serial dehydration events, the optimized protocol using DEBM can be applied to access a wide variety of azaheterocycles via catalytic Bischler-Napieralski-type cyclodehydration of preformed amides (see SI, Sect. V, Table S4, 18 examples).
- 22.Silanes have been shown to promote amidation, including in phosphine-catalyzed Staudinger amidation. However, low background conversion to amide was observed with PhSiH3 in the absence of either 4• [O] or DEBM (See SI, Sect II, Table S1). See:; (a) Ruan Z;Lawrence RM; Cooper CB Phenylsilane as an Active Amidation Reagent for the Preparation of Carboxamides and Peptides. Tetrahedron Lett. 2006, 47, 7649–7651. [Google Scholar]; (b) Sayes M; Charette AB Diphenylsilane as a Coupling Reagent for Amide Bond Formation. Green Chem. 2017, 19, 5060–5064. [Google Scholar]; (c) Andrews KG; Denton RM A More Critical Role for Silicon in the Catalytic Staudinger Amidation: Silanes as Non-Innocent Reductants. Chem. Com- mun 2017, 53, 7982–7985. [DOI] [PubMed] [Google Scholar]
- 23.For the synthesis of halophosphoniums from phosphine oxides, see:; (a) Nikitin K; Müller-Bunz H; Gilheany D Direct Evidence of a Multicentre Halogen Bond: Unexpected Contraction of the P-XXX-P Fragment in Triphenylphosphine Dihalides. Chem. Commun 2013, 49, 1434–1436. [DOI] [PubMed] [Google Scholar]; (b) Jennings EV; Nikitin K; Ortin Y; Gilheany DG Degenerate Nucleophilic Substitution in Phosphonium Salts. J. Am. Chem. Soc 2014, 136, 16217–16226. [DOI] [PubMed] [Google Scholar]; (c) Nikitin K; Jennings EV; Al Sulaimi S; Ortin Y; Gilheany DG Dynamic Cross-Exchange in Halophosphonium Species: Direct Observation of Stereochemical Inversion in the Course of an Sn 2 Process. Angew. Chem. Int. Ed 2018, 57, 1480–1484. [DOI] [PubMed] [Google Scholar]
- 24.(a) Gerratana B Biosynthesis, Synthesis, and Biological Activities of Pyrrolobenzodiazepines. Med. Res. Rev 2012, 32, 254–293. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hartley JA The Development of Pyrrolobenzodiazapines as Antitumor Agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [DOI] [PubMed] [Google Scholar]; (c) Dornisch E; Pletz J; Glabonjat RA; Martin F; Lembacher-Fadum C; Neger M; Högenauer C; Francesconi K; Kroutil W; Zangger K; Breinbauer R; Zechner EL Biosynthesis of the Enterotoxic Pyrrolobenzodiazepine Natural Product Tilivalline. Angew. Chem. Int. Ed 2017, 56, 14753–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.For comparison, 3s was formed in 31% yield using DEBM (yield by 1H NMR analysis with the aid of an internal standard).
- 26.(a) Meerpoel L; Van Gestel J; Van Gerven F; Woestenborghs F; Marichal P; Sipido V; Terence G; Nash R; Corens D; Richards RD Pyrrolo[1,2-a][1,4]benzodiazepine: A Novel Class of Non-Azole Anti-Dermatophyte Anti-Fungal Agents. Bioorg. Med. Chem. Lett 2005, 15, 3453–3458. [DOI] [PubMed] [Google Scholar]; (b) Paulussen C; de Wit K; Boulet G; Cos P; Meerpoel L; Maes L Pyrrolo[1,2-α][1,4]benzodiazepines Show Potent In Vitro Anti-fungal Activity and Significant In Vivo Efficacy in a Microsporum canis Dermatitis Model in Guinea Pigs. J. Antimicrob. Chemother 2014, 69, 1608–1610. [DOI] [PubMed] [Google Scholar]
- 27.3w is formed racemically under both standard and modified conditions (as used for 3p-3s). 3,4-Dihydroisoquinolines bearing α-stereogenic centers are known to undergo facile thermal racemization even at room temperature, see:; Movassaghi M; Hill MD A Versatile Cyclodehydration Reaction for the Synthesis of Isoquinoline and p-Carboline Derivatives. Org. Lett 2008, 10, 3485–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) McKenzie E; Nettleship L; Slaytor M New Natural Products from Peganum harmala. Phytochemistry 1975, 14, 273–275. [Google Scholar]; (b) Cao R; Peng W; Wang Z; Xu A p-Carboline Alkaloids: Biochemical and Pharmacological Functions. Curr. Med. Chem 2007, 14, 479–500. [DOI] [PubMed] [Google Scholar]; (c) Rommelspacher H; Susilo R Tetrahydroisoquinolines and β-Carbolines: Putative Natural Substances in Plants and Mammals In Progress in Drug Research; Jucker E, Ed.; Birkhauser Verlag: Basel, 1985; Vol. 29, pp 415–459. [DOI] [PubMed] [Google Scholar]
- 29.Whaley WM;Govindachari TR The Preparation of 3,4-Dihydroisoquinolines and Related Compounds by the Bischler-Napieralski Reaction. Org. React 1951, 6, 74–144. [Google Scholar]
- 30.(a) Hoffmann H; Diehr HJ Phosphonium Salt Formation of the Second Kind. Angew. Chem. Int. Ed 1964, 3, 737–746. [Google Scholar]; (b) Zefirov NS; Makhon’kov DI X-philic Reactions. Chem. Rev 1982, 82, 615–624. [Google Scholar]
- 31.The classical Bischler-Napieralski reaction is understood to proceed via elimination of oxyphosphonium to generate a nitrilium ion, as evidenced by the formation of side-product alkene deriving from retro-Ritter reaction. For no substrates described in this manuscript was alkene formation observed. The intermediacy of a nitrilium ion is not ruled out in this chemistry, and is logically consistent with the observed TOLS loss of proton. See:; (a) Fodor G; Nagubandi S Correlation of the von Braun, Ritter, Bischler-Napieralski, Beckmann and Schmidt Reactions via Nitrilium Salt Intermediates. Tetrahedron 1980, 36, 1279–1300. [Google Scholar]; (b) Nagubandi S; Fodor G The Mechanism of the Bischler-Napieralski Reaction. J. Heterocycl. Chem 1980, 17, 1457–1463. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.