

Abstract
Sindbis virus (SINV), the prototype alphavirus, contains a macro domain in the highly conserved N-terminal region of nonstructural protein 3 (nsP3). However, the biological role of the macro domain is unclear. Mutations of amino acids 10 and 24 from asparagine to alanine in the ADP-ribose binding region of the macro domain impaired SINV replication and viral RNA synthesis particularly in neurons, but did not alter binding of poly(ADP-ribose). Mutation at position 10 had the greatest effect and caused nsP3 instability in neurons, decreased SINV-induced death of mature, but not immature neurons, and attenuated virulence in 2 week-old, but not 5 day-old mice. A compensatory mutation at amino acid 31 in the macro domain of nsP3, as well as reversion of mutated amino acid 10, occurred during replication of double mutant SINV in vitro and in vivo. The nsP3 macro domain is important for SINV replication and age-dependent susceptibility to encephalomyelitis.
Keywords: alphavirus, encephalitis, mice, neurons, poly(ADP-ribose)
Introduction
In humans, alphaviruses can cause encephalitis, arthritis, or rash (Calisher, 1994;Laine, Luukkainen et al., 2004). Sindbis virus (SINV), the prototype alphavirus, causes encephalomyelitis in mice and is a model system for study of the pathogenesis of diseases caused by alphavirus infections. In the nervous system, SINV infection targets neurons, and neuronotropism is determined in part by the glycoproteins E1 and E2 on the virion surface (Lustig, Jackson et al., 1988). The outcome of infection depends on SINV neurovirulence and on host factors, including age and genetic background (Thach, Kimura et al., 2000;Johnson, McFarland et al., 1972). Age-dependent susceptibility is a characteristic of alphavirus encephalitis and is due to intrinsic properties of neurons, not differences in the immune response (Fazakerley, Pathak et al., 1993;Griffin, 1976). Newborn mice develop fatal disease, while adult mice survive infection. Likewise, undifferentiated neurons replicate SINV to higher titer in vitro and are more susceptible to SINV-induced cell death than differentiated neurons (Vernon & Griffin, 2005;Burdeinick-Kerr & Griffin, 2005).
SINV is enveloped and has a single positive-strand RNA genome of 11.7kb. The 3′-terminal one-third of the genome encodes structural proteins, and the 5′-terminal two-thirds of the genome encodes nonstructural proteins (nsPs) as polyproteins (P123, P1234). The nonstructural polyproteins are processed into four nsPs that form replication complexes in which the polyproteins and individual nsPs play different roles in virus replication. The replication complexes containing polyproteins synthesize negative-strand RNA and after the polyproteins are cleaved into individual nsPs, synthesize positive-strand RNA (Strauss & Strauss, 1994). nsP1 has guanine-7-methyltransferase and guanyltransferase activities and plays a role in capping viral RNAs (Mi, Durbin et al., 1989;Scheidel, Durbin et al., 1987). nsP2 has protease activity and cleaves nonstructural polyproteins (Ding & Schlesinger, 1989;Hardy & Strauss, 1989), and also exhibits RNA helicase and 5′ triphosphatase activities (Gomez, Ehsani et al., 1999;Vasiljeva, Merits et al., 2000). nsP4 is the RNA-dependent RNA polymerase (Kamer & Argos, 1984).
The function of nsP3 is least understood. nsP3 mutants generated from random insertion and chemical mutagenesis show reduced minus-strand and subgenomic RNA synthesis (LaStarza, Lemm et al., 1994;De, Fata-Hartley et al., 2003). nsP3 also interacts with cellular proteins, which are presumed to participate in the formation and/or function of viral replication complexes (Cristea, Carroll et al., 2006;Frolova, Gorchakov et al., 2006;Gorchakov, Garmashova et al., 2008). However, data supporting an essential role for nsP3 is limited and it has little homology with other nsPs of positive-strand RNA viruses (Ahlquist, Strauss et al., 1985;Haseloff, Goelet et al., 1984;LaStarza, Grakoui et al., 1994). The C-terminal domain is highly variable even in alphaviruses and is heavily phosphorylated at serine/threonine (Li, La Starza et al., 1990). The phosphorylated C-terminal region is not essential for SINV replication, but nsP3 phosphorylation may play a host cell-dependent role in SINV minus-strand RNA synthesis (De, Fata-Hartley et al., 2003;LaStarza, Grakoui et al., 1994).
Unlike the C-terminus, the N-terminal region of nsP3 is well conserved among alphaviruses. The N-terminal 150 amino acids of nsP3 was first recognized as a domain of unknown function (X domain) (Gorbalenya, Koonin et al., 1991;Koonin, Gorbalenya et al., 1992) and later identified as a macro domain based on homology to the non-histone region of histone macroH2A (Pehrson & Fried, 1992;Pehrson & Fuji, 1998). Macro domains are ancient and widely distributed throughout all eukaryotic organisms, bacteria, and archaea, indicating a ubiquitous and important basic biological function. Macro domains are also found in nsPs of several positive-strand RNA viruses, including hepatitis E virus, rubella virus and coronaviruses, as well as alphaviruses (Gorbalenya, Koonin et al., 1991;Koonin, Gorbalenya et al., 1992;Liang, Yao et al., 2000).
Despite the wide distribution and evolutionary conservation of macro domains, the physiological role is not known. MacroH2A, the protein with the best studied macro domain, is enriched in heterochromatin and is involved in regional chromatin silencing and cell-type specific regulation of transcription (Changolkar, Singh et al., 2008;Ladurner, 2003;Nusinow, Hernandez-Munoz et al., 2007;Agelopoulos & Thanos, 2006). These properties are associated with the nonhistone macro domain which interacts with histone deacetylases and poly(ADP-ribose) polymerase (PARP) to inhibit histone acetylation and nucleosome remodeling and repress initiation of transcription (Chakravarthy, Gundimella et al., 2005;Agelopoulos & Thanos, 2006). In addition, macro domains of yeast, severe acute respiratory syndrome (SARS) coronavirus and hepatitis E virus proteins exhibit ADP-ribose-1″ phosphatase activity in vitro (Martzen, McCraith et al., 1999;Saikatendu, Joseph et al., 2005). This enzymatic activity is dispensable for coronavirus replication, but appears to play a role in the pathogenesis of mouse hepatitis virus infection (Eriksson, Cervantes-Barragan et al., 2008;Putics, Filipowicz et al., 2005). The role of macro domains in other proteins is less clear (Chakravarthy, Gundimella et al., 2005).
Many macro domains can bind various forms of ADP-ribose, such as mono ADP-ribose, poly(ADP-ribose) (PAR), poly(A) and the SirT1 metabolite O-acetyl-ADP ribose (OAADPR) (Comstock & Denu, 2007;Egloff, Malet et al., 2006;Karras, Kustatscher et al., 2005;Kustatscher, Hothorn et al., 2005). However, individual eukaryotic and viral protein macro domains vary in the forms of ADP-ribose bound and in the affinity of binding (Neuvonen & Ahola, 2009). The macro domain of macroH2A binds OAADPR, but not PAR (Kustatscher, Hothorn et al., 2005;Comstock & Denu, 2007). The macro domain of Semliki Forest virus (SFV) binds PAR well, but monomeric ADP-ribose only poorly (Neuvonen & Ahola, 2009). The significance of these different interactions remains unknown.
To improve understanding of the function and importance of the nsP3 macro domain in alphavirus encephalitis, we have generated mutations in the putative ADP-ribose binding site. We characterized phenotypes of the mutants and revertants that appeared during replication. We also assessed the effect of mutations on SINV replication in neurons, synthesis of viral RNAs, and expression of viral proteins. Finally, we determined the effect of mutations on SINV virulence in mice at different maturation stages as well as on replication in undifferentiated and differentiated neuronal cell lines.
Results
Mutations in the predicted ADP ribose-binding site in the nsP3 macro domain do not affect PAR binding, but do affect SINV replication
Modeling of nsP3 against a macro domain protein from Archaeoglobus fulgidus (Fig. 1A) predicted that asparagines at amino acids 10 and 24 of nsP3 are at corresponding positions to the two amino acids in close contact with ADP-ribose in Archaeoglobus fulgidus (Fig. 1B) (10). These asparagines were changed to alanines to generate SINV with a double mutation (SINV DM) or with single mutations at amino acid 10 (SINV SM10) or 24 (SINV SM24) (Fig. 1C). To assess the effect of these mutations on PAR binding, his-tagged WT and mutant proteins were tested for interaction with PAR at different concentrations (Fig. 1D). The mutations had no effect on binding of PAR.
Fig. 1.
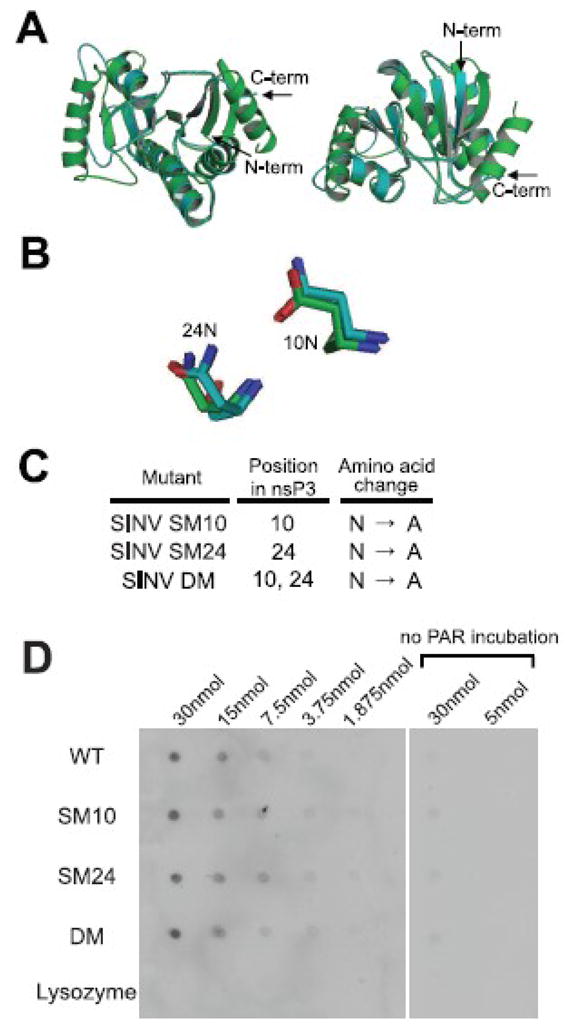
Mutation of the nsP3 macro domain does not affect binding of PAR. Homology modeling of the SINV nsP3 macro domain using Af1521 (a macro domain protein from Archaeoglobus fulgidus) as a template (A,B). The predicted structure of the nsP3 macro domain (blue) and the published structure of Af1521 (green) are superimposed, and overall front and rear view images (A) and close view of mutated amino acids (B) are presented. (C) Mutagenesis of the nsP3 macro domain. Amino acids for mutagenesis were chosen based on identification of conserved residues and homology modeling with the Archaeoglobus fulgidus macro domain. One amino acid at position 10 or 24 in nsP3 was mutated from asparagine (N) to alanine (A) for SINV single mutant (SM10 and SM24, respectively), and amino acids at positions 10 and 24 were mutated from N to A for SINV double mutant (DM). (D) PAR binding assay. The nsP3 macro domain WT, SM10, SM24 and DM, and lysozyme (negative control) were dot-blotted onto nitrocellulose membrane, and the membrane was incubated with or without PAR, followed by incubation with antibody to PAR for detection.
To assess the potential importance of the amino acids at these positions for SINV replication, undifferentiated (Fig. 2A) and differentiated (Fig. 2B) CSM14.1 cells were infected with SINV WT, SM10, SM24, or DM. In differentiated CSM14.1 cells, the replication of SINV SM10 and DM was impaired at early times after infection (P < 0.01 before 24 h). After 24 h, replication of SINV WT plateaued, while replication of SINV SM10 and DM continued to increase to approximately the same level as SINV WT. Replication of SINV SM24 was less impaired, suggesting that the amino acid at position 10 of nsP3 is more important for SINV replication in neurons than the amino acid at position 24. The results in undifferentiated CSM14.1 cells were similar. In BHK21 cells, however, replication of SINV WT and DM was not different (Fig. 2C), suggesting that the effect of mutations on SINV replication is greatest in neurons.
Fig 2.
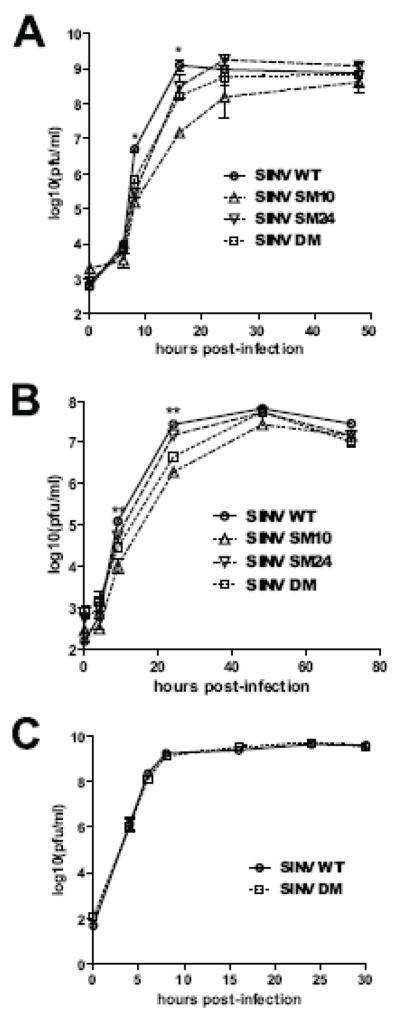
SINV replication in neurons is affected by mutations in the nsP3 macro domain. Undifferentiated CSM14.1 (A), 3 week-differentiated CSM14.1 (B) and BHK21 (C) cells were infected with SINV WT, SM10, SM24, or DM (MOI = 10). Virus released into cell culture media was assessed by plaque assay. Average data of two independent experiments are presented. Error bars indicate standard error of the mean. * P < 0.05; ** P < 0.01; *** P < 0.001;Student’s t-test comparing SINV WT and DM.
Revertants arise during growth of nsP3 macro domain mutants in neurons
SINV DM grown in BHK21 cells showed a small plaque phenotype compared to the large plaques produced by SINV WT (Fig. 3A). Viruses produced by SINV WT-infected differentiated CSM14.1 cells also formed large plaques (average diameter of 3 mm). In contrast, viruses produced by SINV DM-infected differentiated (Fig. 3A) and undifferentiated (data not shown) CSM14.1 cells were a mixture of small plaques (average diameter of 1.5 mm) and a few large plaques (Fig. 3A, arrow). To determine whether these large plaques were revertants, plaques from SINV DM grown in undifferentiated or differentiated CSM14.1 cells, or BHK21 cells were isolated and sequenced. In BHK21 cells, all plaques were original SINV DM with no reversion. In undifferentiated and differentiated CSM14.1 cells, however, small plaque viruses retained the original mutations, while large plaque viruses had reverted alanine (GCT) at amino acid 10 of nsP3 to threonine (ACT) or aspartate (GAT) (Fig. 3B). These reversions required only a single nucleotide change and were detected as early as 8 h after infection of both undifferentiated and differentiated CSM14.1 cells. SINV DM grown in CSM14.1 cells was also sequenced directly without plaque isolation (Fig. 3C) and showed coexistence of the original SINV DM and variants with reversions at amino acid 10. No reversion was detected at amino acid 24, and SINV SM24 formed large plaques (data not shown).
Fig. 3.
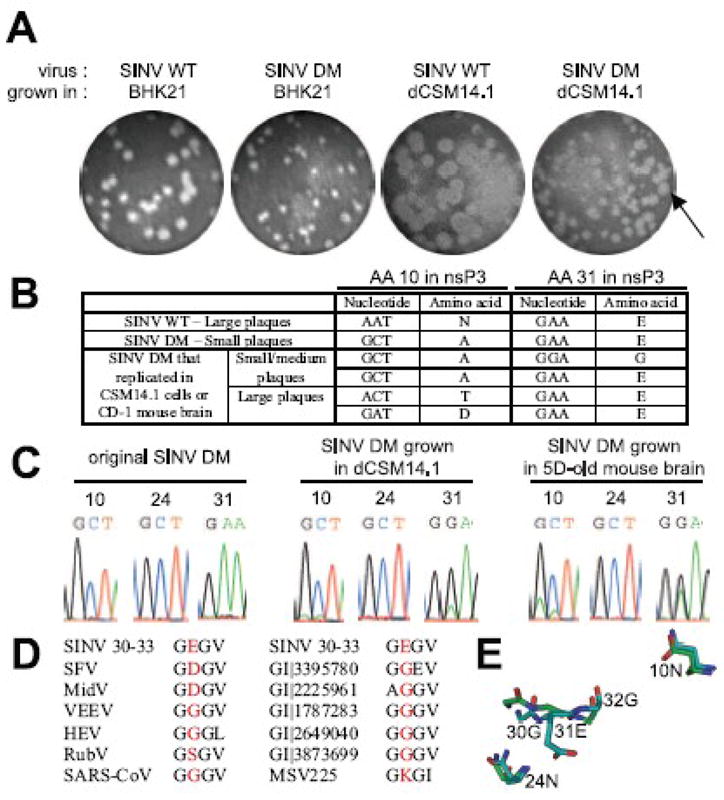
Reversions occur in the nsP3 macro domain of SINV DM during SINV replication. (A) The effect of mutations on plaque phenotype. P0 stocks of SINV WT and DM were produced in BHK21 cells, and the plaque phenotype observed (2 pictures on the left). 3 week-differentiated CSM14.1 (dCSM14.1) cells were infected with SINV WT or SINV DM, and viruses released into culture media were collected at 8 h post-infection. Plaque assay was performed in BHK21 cells to observe plaque phenotype (2 pictures on the right). (B) Analysis of plaque phenotype and reversions. Plaques from (A) were isolated, grown in BHK21 cells, and sequenced. (C)Assessment of reversions in SINV DM-infected CSM14.1 cells and the brains of CD-1 mice. P0 stock of SINV DM produced in BHK cells (left), virus released from SINV DM-infected differentiated CSM14.1 cells at 48 h post-infection (middle), and virus collected from the brains of SINV DM-infected 5 day-old CD-1 mice at 1 day post-infection (right) were sequenced. Electropherogram at amino acids 10, 24, and 31 of nsP3 is shown. (D) Alignment of SINV nsP3 amino acids 30–33 with other macro domain containing proteins. SFV, Semliki Forest virus; MidV, Middleburg virus; VEEV, Venezuelan equine encephalomyelitis virus; HEV, hepatitis E virus; RubV, rubella virus; SARS-CoV, severe acute respiratory syndrome coronavirus; GI|3395780, histone macroH2A1.1 from Gallus gallus; GI|2225961, Mycobacterium tuberculosis; GI|1787283, Escherichia coli; GI| 2649040, Archaeoglobus fulgidus; GI|3873699, Caenorhabditis elegans; MSV225, Melanoplus sanguinipes entomopoxvirus. (E) Homology modeling of the SINV nsP3 macro domain using Af1521 (a macro domain protein from Archaeoglobus fulgidus) as a template. The predicted structure of the nsP3 macro domain (blue) and the published structure of Af1521 (green) are superimposed, and close view image of nsP3 amino acids 10, 24, and 31 is presented.
In addition to the reversion at amino acid 10, a second-site mutation was detected at amino acid 31. A change from glutamate (GAA) (31E) to glycine (GGA) (31G) was present in plaques from SINV DM without the reversion at amino acid 10 (Fig. 3B). This second-site mutation was detected as early as 8 h after infection of CSM14.1 cells and represented more than 50 % of the total virus population (Fig. 3C). However, this change at amino acid 31 did not increase the plaque size (Fig. 3B). Although most alphaviruses contain glutamate or aspartate at the position corresponding to nsP3 31E, most other macro domain proteins contain glycine (Fig. 3D). When Ca and side chain positions of nsP3 31E are compared to those of corresponding residues in the Af1521 macro domain protein from Archaeoglobus fulgidus (Karras, Kustatscher et al., 2005), 31E has a bulky side chain that the template glycine residue does not have, and Cα positions of 31E and nearby residues (30G and 32G) are not similar to those of corresponding residues in the template (Fig. 3E). In contrast, Ca and side chain positions of nsP3 amino acids 10N and 24N are highly similar to those of corresponding residues in Af1521.
Mutations in the nsP3 macro domain affect formation of SINV replication complexes
To understand how mutations in the nsP3 macro domain affect SINV replication, we examined the levels of nsPs that form SINV replication complexes by immunoblotting (Fig. 4). In undifferentiated (Fig. 4A) and differentiated (Fig. 4B) CSM14.1 cells infected with SINV DM or SM10, less nsP3 was present at all times examined. In differentiated CSM cells, levels of nsP2 were also slightly lower, although nsP2 itself is not mutated. SINV SM24 showed minimal defects in the level of nsPs consistent with the less pronounced defects in viral replication. To examine the synthesis and stability of nsPs, differentiated CSM14.1 cells were labeled with tran35S at 20 h post-infection and chased for 45, 90, and 120 min, and levels of nsP2, nsP3, and nsP23 were analyzed by immunoprecipitation (Fig. 4C). Similar amounts of nsP3 were synthesized in SINV WT- and DM-infected cells, but nsP3 was rapidly degraded in SINV DM-infected cells. Instability of mutant nsP3 was not a result of proteasome-mediated degradation because treatment with a proteasome inhibitor MG132 decreased, rather than increased, the amount of detectable nsP3 (data not shown). Furthermore, mutations in the nsP3 macro domain did not affect nsP3 stability in BHK21 cells (Fig. 4D), indicating that the folding of mutant nsP3 is intact. The synthesis and stability of nsP2 and nsP23 were not significantly affected, suggesting that the mutations did not affect cleavage of P23 (Fig. 4C).
Fig. 4.
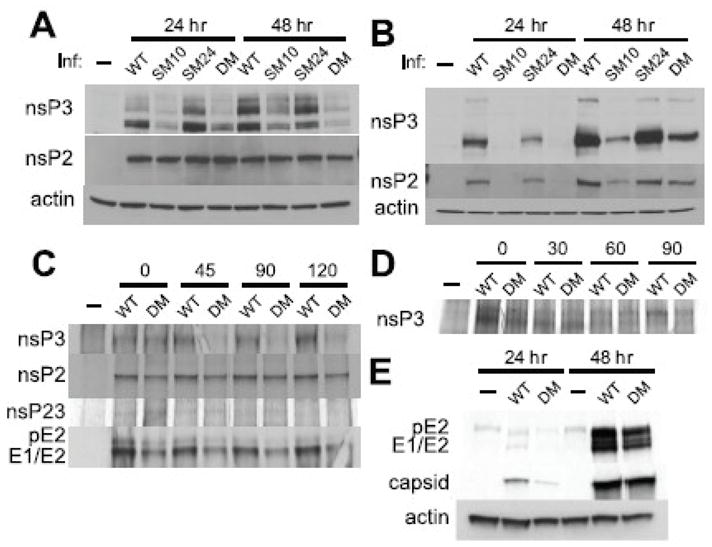
SINV replication complexes are diminished by mutations in the nsP3 macro domain. The effect of mutations on nsP expression in undifferentiated (A) and 3 week-differentiated (B) CSM14.1 cells. Cells were infected with SINV WT, SM10, SM24, or DM (MOI = 10), or mock infected. Cell lysates were analyzed by immunoblotting using antibodies to nsP2, nsP3 and actin. (C) The effect of mutations on the stability of nonstructural and structural proteins in CSM14.1 cells. 3 week-differentiated CSM14.1 cells were infected with SINV WT or DM (MOI = 10), or mock infected. At 20 h after infection, newly synthesized proteins were labeled with tran35S for 2 h and chased for 120 min. Cell lysates were incubated with antibody to nsP3, nsP2, or structural proteins, and then incubated with protein A/G. Immunoprecipitated proteins were separated by PAGE and visualized by autoradiography.(D) The effect of mutations on the stability of nsP3 in BHK21 cells. Cells were infected with SINV WT or DM (MOI = 10), or mock infected. At 6 h after infection, newly synthesized proteins were labeled with tran35S for 1 h and chased for 90 min followed by immunoprecipitation. (E) The effect of mutations on structural proteins in CSM14.1 cells. 3 week-differentiated CSM14.1 cells were examined for the expression of structural proteins by immunoblotting using antibody to structural proteins.
The effect of mutations in the nsP3 macro domain on the synthesis and stability of structural proteins (E1, E2, and capsid) was also assessed in differentiated CSM14.1 cells (Fig. 4E). Lower amounts of both capsid and E1/E2 were present at 24 h after infection with SINV DM, but comparable amounts of structural proteins were detected by 48 h. In general, the differences in the amounts of structural proteins between SINV DM and WT were less apparent than the differences in the amounts of nonstructural proteins, suggesting that the impairment in the replication of SINV DM results from defects in nsPs. In addition, the stability of the structural proteins was not affected (Fig. 4C).
Mutations in the nsP3 macro domain affect the synthesis of SINV RNAs
To understand which step of virus replication is affected by mutations in the nsP3 macro domain, we compared the synthesis of viral genomic and subgenomic RNAs as well as plus and minus strand RNAs (Fig. 5). In differentiated CSM14.1 cells infected with SINV WT or DM, newly synthesized viral RNAs were labeled with 3H-uridine for 2 h, and genomic and subgenomic RNAs were analyzed on an agarose gel. Initial RNA synthesis was similar between SINV WT and DM, but the later amplification of RNA synthesis observed in SINV WT infection did not occur in SINV DM infection (Fig. 5A). The ratios of genomic to subgenomic RNAs were comparable, and similar to those observed in previous studies of SINV in differentiated CSM14.1 cells (Burdeinick-Kerr & Griffin, 2005).
Fig. 5.

SINV RNA synthesis is affected by mutations in the nsP3 macro domain. (A) The effect of mutations on the synthesis of viral genomic and subgenomic RNAs. 3 week-differentiated CSM14.1 cells were infected with SINV WT or SINV DM, or mock infected. Cells were treated with Dactinomycin to inhibit cellular RNA synthesis, and newly synthesized RNAs were labeled with 3H-uridine for 2 h. Purified RNAs were separated by agarose gel electrophoresis and visualized by fluorography. (B, C) The effect of mutations on the synthesis of viral plus (B) and minus (C) strand RNAs. 3 week-differentiated CSM14.1 cells were infected with SINV WT or SINV DM (MOI = 10), or mock infected. RNAs were purified from cells, and SINV plus or minus strand RNA was quantitated by real time RT-PCR. Representative data from two independent experiments are presented. No viral RNA was detected in mock infected cells. Error bars indicate SEM. * P < 0.05; ** P < 0.01; *** P < 0.001 (Student’s t-test comparing SINV WT and DM); AU, artificial unit.
To compare the synthesis of plus and minus strand viral RNAs, RNAs were purified from differentiated CSM14.1 cells infected with SINV WT or DM, and real time RT-PCR was performed using primers specifically recognizing plus or minus strand SINV RNA. Synthesis of both plus (Fig. 5B) and minus (Fig. 5C) strand RNAs was affected by mutations in the nsP3 macro domain at 16, 24, and 30 h post-infection.
Mutations in the nsP3 macro domain reduce SINV-induced death of mature neurons
Next, we assessed the effect of mutations on SINV-induced neuronal death. In undifferentiated CSM14.1 cells, no differences in SINV-induced death were observed between SINV WT and mutants (Fig. 6A). In differentiated CSM14.1 cells, however, SINV SM10 and DM caused less death than SINV WT (p < 0.01, 3 days after infection; p < 0.05, 4 days after infection) (Fig. 6B). Survival of SINV SM24-infected neurons was modestly improved from that of SINV WT-infected neurons.
Fig. 6.
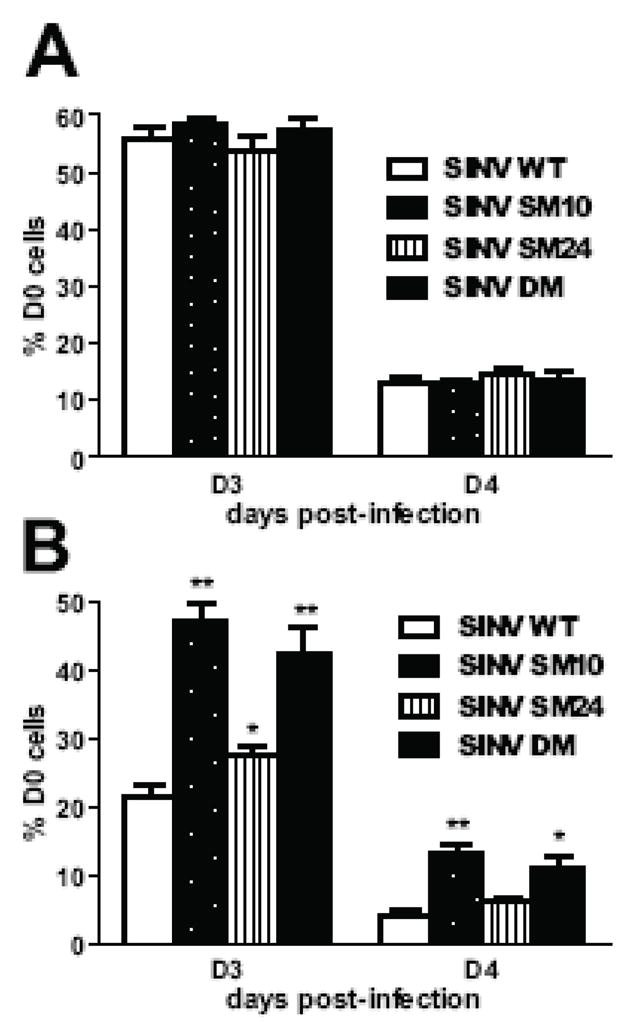
SINV virulence is affected by mutations in the nsP3 macro domain in mature neurons. The effect of mutations on SINV virulence in undifferentiated (A) and 3 week-differentiated (B) CSM14.1 cells. Cells were infected with SINV WT, SINV SM10, SINV SM24, or SINV DM (MOI = 10), or mock infected. Live cells were counted at indicated days post-infection by trypan blue exclusion assay, and percentage of D0 cells was calculated. Representative data from 3 independent experiments are presented. In each experiment, triplicate samples were assessed. Error bars indicate SEM. * P < 0.05; **P < 0.01; *** P < 0.001; Student’s t-test comparing SINV WT and mutants.
Mutations in the nsP3 macro domain reduce SINV virulence in mice
To assess the importance of the nsP3 macro domain for induction of encephalomyelitis in vivo, mice were infected with SINV WT or mutants, and morbidity, mortality and virus replication in brain were assessed (Fig. 7). Because mutations in the nsP3 macro domain affected SINV-induced neuronal death only in differentiated neurons (Fig. 6), we explored the possibility that the effect of mutations on SINV virulence in mice also depends on age. Two week-old or 5 day-old mice were infected with SINV WT, SM10, SM24, or DM, or mock infected with PBS. In 5 day-old mice, SINV WT infection resulted in severe paralysis and death 1.5 days earlier than SINV DM infection (median survival: WT, 4 days; DM, 5.5 days; P < 0.0001) (Fig. 7A and C). However, survival curves for WT- and DM-infected mice were not significantly different (P = 0.2059, Log-rank test), and all of the mice infected with WT or mutant viruses died by day 6 after infection. Viral replication in brain was not different between SINV WT and mutants (Fig. 7E). In contrast, in 2 week-old mice, SINV WT infection caused 90 % mortality, while DM-infected mice remained well (P < 0.0001, Log-rank test) (Fig. 7B and D). SINV SM10 and SM24 were less attenuated than SINV DM with 40% mortality (P = 0.0228, SM10; P = 0.0170, SM24). SINV WT, and all mutants replicated to high titers in the brains of 2 week-old mice, but only mutated viruses showed evidence of clearance 7 days after infection (Fig. 7F).
Fig. 7.

SINV virulence is affected by mutations in the nsP3 macro domain in 2 week-old mice. The effect of mutations on the survival of 5 day-old (A) or 2 week-old (B) CD-1 mice. Groups of 10 mice were infected with SINV WT, SM10, SM24 or DM (intracerebral injection of 1000 PFU virus), or mock infected with PBS, and monitored daily for survival. Percent survival was plotted using Kaplan-Meyer analysis and Log-rank test was used for statistics. (C,D) The effect of mutations on the clinical signs of 5 day-old (C) or 2 week-old (D) CD-1 mice. Mice were infected as in (A,B) and monitored daily for signs of illness. Disease was scored as: 0, no signs of illness; 1, weakness; 2, moderate paralysis in one hindlimb; 3, severe paralysis in both hind limbs; 4, death. (E,F) The effect of mutations on viral replication in the brains of 5 day-old (E) or 2 week-old (F) CD-1 mice. Mice were infected with SINV WT, SM10, SM24 or DM and brains collected from 3 mice in each group were homogenized and assayed for PFU using BHK21 cells. Error bars indicate SEM. * P < 0.05; ** P < 0.01; *** P < 0.001; Student’s t-test comparing SINV WT and mutants.
Virus produced from the brains of SINV-DM-infected mice was sequenced and the same reversions at nsP3 amino acids 10 and 31 that occurred during replication in CSM14.1 cells were detected (Fig. 3B and C).
Discussion
While the roles of nsP1, nsP2, and nsP4 are well characterized in SINV replication, the function of nsP3 has been elusive. Previous studies using SINV variants generated by random insertion or chemical mutagenesis of nsP3 have reported small reductions in virus replication and in minus strand or subgenomic RNA synthesis, but have not focused on the macro domain and have not examined the in vivo effects of these mutations (De, Fata-Hartley et al., 2003;LaStarza, Grakoui et al., 1994;LaStarza, Lemm et al., 1994). In this study we targeted the ADP-ribose binding site in the macro domain for mutational analysis and showed that the nsP3 macro domain plays an important role in SINV replication although the mutations did not affect PAR binding. Mutations at nsP3 amino acids 10 and 24 in the macro domain impaired virus replication and viral RNA amplification, and caused nsP3 instability in neurons. The greatest effects were detected when amino acid 10 was mutated. Mutated viruses formed small plaques in BHK21 cells, and virus variants with a reversion at amino acid 10 or with a second-site mutation at amino acid 31, also in the macro domain, were rapidly selected during replication of SINV in cultured neurons and mouse brains. This study also showed that the mutations in the nsP3 macro domain impaired virulence for 2 week-old mice and mature cultured neurons.
Previous studies support a role for the macro domain in SINV replication. SINV mutants generated by random insertion of linker sequences into the nsP3 macro domain show smaller plaque phenotypes, a decrease in plaque formation, and a defect in RNA synthesis (LaStarza, Lemm et al., 1994). The mutation present in SINV mutant ts138 (alanine to glycine at residue 68 of nsP3) is associated with decreased C-terminal nsP3 phosphorylation and decreased minus strand synthesis (De, Fata-Hartley et al., 2003).
The importance of the nsP3 macro domain is underscored by reversions that occurred during SINV DM replication in cultured neurons or the brains of infected mice. Virus variants with a reversion at nsP3 amino acid 10 to threonine or aspartate (the amino acid present in many macro domains at this position (Neuvonen & Ahola, 2009)) emerged shortly after infection, indicating selective pressure. Furthermore, viruses with a second-site change at nsP3 amino acid 31 (glutamate to glycine) were selected and thus became more homologous in amino acid sequence and structure to other macro domains, in which this is a highly conserved amino acid (Neuvonen & Ahola, 2009; Till & Ladurner, 2009). Viruses with reversions at amino acid 10 of nsP3 did not have the change at amino acid 31, and the population of viruses with amino acid 10 reversion increased as the infection progressed, while the population of viruses with amino acid 31 reversion decreased (data not shown), suggesting that virus variants with these amino acid changes are different populations or are selected at different times after infection. The reciprocal relationship between these amino acid reversions suggests cooperativity and that a change at residue 31 is not required once the mutation at amino acid 10 is reverted.
Several positive-strand RNA viruses have conserved macro domains in their nonstructural proteins, suggesting an evolutionarily preserved role (Koonin, Gorbalenya et al., 1992;Gorbalenya, Koonin et al., 1991). Viral macro domains are often closely associated with papain-like protease domains, but no functional link between the macro domain and protease activity has been reported (Liang, Yao et al., 2000;Putics, Filipowicz et al., 2005;Gorbalenya, Koonin et al., 1991). Some macro domains can hydrolyze ADP-ribose-1″ phosphate (ADPR-1″P), a product of tRNA splicing, to yield ADP-ribose. ADPR-1″P phosphatase activity is detected in the macro domain of hepatitis E virus and coronaviruses (Neuvonen & Ahola, 2009;Saikatendu, Joseph et al., 2005), but this enzymatic activity is dispensable for replication of human coronavirus 229E in vitro (Putics, Filipowicz et al., 2005). However, mutation of a residue important for this enzymatic activity in the coronavirus mouse hepatitis virus A59 results in altered liver pathology in infected mice associated with decreased induction of IL-6, decreased hepatic necrosis and improved virus clearance, but little change in peak virus replication.
The macro domains of alphaviruses have little ADPR-1″P phosphatase activity. Residue 24 of SINV nsP3 is in the APPR-1″P catalytic site, and mutation of this site in SFV, along with amino acid 21, eliminates the small amount of enzymatic activity present in the WT protein (Neuvonen & Ahola, 2009). SINV SM24 was least impaired of the mutated SINVs studied, suggesting that the ADPR-1″P phosphatase activity of nsP3 is not crucial for SINV replication. In differentiated neurons, SM24 nsP3 was stable, virus replication and induction of neuronal death were similar to that observed with WT virus, and revertants were not detected during replication. However, in 2 week-old mice virulence was attenuated and clearance was facilitated compared to WT virus, suggesting a potential role in the pathogenesis of encephalitis.
Many viral macro domains have the ability to interact with ADP-ribose in some form. The ability of macro domains to bind monomeric ADP-ribose appears unrelated to the ability to bind PAR. The macro domain of SFV binds PAR well, but does not efficiently bind monomeric ADP-ribose (Neuvonen & Ahola, 2009). Likewise, mutations at nsP3 amino acids 10 and 31 in the SFV macro domain that affect coronavirus binding of monomeric ADP-ribose (Karras, Kustatscher et al., 2005) did not affect binding of PAR (Neuvonen & Ahola, 2009). Our studies confirm the lack of effect of mutation at residues 10 and 24 for binding PAR and suggest that additional macro domain residues are important for this binding. Residues 10 and 24 may be involved in binding another NAD metabolite that has not yet been examined.
The significance of the interaction between PAR and the nsP3 macro domain is unclear. It is possible that some cellular proteins modified with PAR are recruited by nsP3 into replication complexes. However, there is no evidence that a deficiency in PAR binding is important for the phenotypes of the mutants we have studied because all retained the ability to bind PAR.
In vivo studies of nsP3 macro domain mutants are few. Studies of SFV have shown that mutation of the opal termination codon between nsP3 and nsP4 to encode arginine increases neurovirulence (Tuittila & Hinkkanen, 2003;Tuittila, Santagati et al., 2000). In addition, mutations in the macro domain at nsP3 amino acid 11 (valine to isoleucine), 48 (alanine to glutamate) and 70 (glycine to alanine) collectively contribute to increased neurovirulence, but the mechanisms are not known (Tuittila & Hinkkanen, 2003). We have shown that mutations in the nsP3 macro domain significantly reduced SINV virulence in 2 week-old mice and impaired the ability of the virus to induce death in mature neurons. Although replication of SINV WT and mutants was similar in mice as indicated by titers at 24 h after infection, the mutations facilitated virus clearance from the nervous system, a process dependent on the responses of infected cells to immune effectors such as interferon-γ and antibody (Burdeinick-Kerr & Griffin, 2005;Levine, Hardwick et al., 1991). A similar effect on virus clearance was observed in the studies of MHV A59 with a single amino acid change in the macro domain (Eriksson, Cervantes-Barragan et al., 2008). These data further suggest that nsP3 interaction with host factors is affected by the macro domain amino acid sequence.
Materials and methods
Cell culture
The rat neuronal CSM14.1 cell line was derived from nigral-striatal neurons immortalized with a temperature-sensitive simian virus 40 T antigen and was a gift from Dale E. Bredesen (Buck Institute for Age Research) (Durand, Chugani et al., 1990;Zhong, Sarafian et al., 1993). CSM14.1 cells were grown at the permissive temperature (31°C) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), L-glutamine (2mM), penicillin (100U/mL), and streptomycin (100μg/ml) (Gibco BRL) in a 5% CO2 incubator. For differentiation, CSM14.1 cells were grown under permissive conditions until they were 95% confluent and then switched to the restrictive temperature (39°C) and DMEM-1% FBS supplemented as above. Baby hamster kidney 21 (BHK21) cells were grown at 37°C in DMEM-10% FBS supplemented as above. BHK21 cells were used to grow virus stocks and to quantitate virus by plaque assay.
Homology modeling
The full-length amino acid sequence of nsP3 was run with GENO3D (http://geno3d-pbil.ibcp.fr). A macro domain protein Af1521 (Protein Data Bank ID: 2bfq), which has an expectation value of 1.0e-32 when compared with amino acid 3-158 of SINV nsP3, was chosen for a template structure, and the structure of the nsP3 macro domain was predicted by homology modeling.
Viruses, mutagenesis and sequencing
TE, a recombinant strain of SINV, was previously constructed by replacing restriction fragments of the Toto1101 cDNA clone of SINV with the E2 fragment from neuroadapted SINV and the E1 fragment from the original SINV isolate AR339 (Lustig, Jackson et al., 1988). At amino acid positions 10 and 24 of nsP3, asparagines were changed to alanines in the TE plasmid using the Quick Change® Site-Directed Mutagenesis Kit (Stratagene) to create viruses with single mutations (SM10 and SM24) and double mutations (DM). Viral RNAs were transcribed using mMESSAGE mMACHINE® High Yield Capped RNA Transcription Kit (Ambion) and then transfected into BHK21 cells using Lipofectamine 2000 (Invitrogen). Virus titers were assessed by plaque formation in BHK21 cells. For sequencing, viral RNAs were purified from virus released into cell culture media using the QIAamp® Viral RNA Mini kit (Qiagen), or from mouse brains using RNease® Mini Kit (Qiagen), and were amplified by RT-PCR using primers TE 3958(+) (5′ GACCAGATTGTGTCTCAAGC 3′) and TE 4352(−) (5′ CAATTTCAAGGCTTCTGCTTC 3′). RT-PCR products were sequenced using the primer TE 3958(+).
For analysis of replication, virus was diluted in DMEM-1% FBS, and cells were infected at a multiplicity of infection (MOI) of 10 for 1 h at 31°C (undifferentiated CSM14.1 cells), 39°C (differentiated CSM14.1 cells), or 37°C (BHK21 cells). Cells were then washed with medium 3 times, and the medium was replaced.
To assess viability, cells were washed with PBS, trypsinized, resuspended, and incubated with 0.4% trypan blue. Unstained live cells were counted, and cell viability was calculated by dividing the number of live cells by the number of live cells at the time of infection.
Poly(ADP-ribose) (PAR) binding assay
The nsP3 macro domains from WT, SM10, SM24 and DM were amplified by PCR and inserted into pRSET (Invitrogen) to express hexahistidine (his)-tagged proteins. The macro domain proteins were purified over a nickel column using ProBond™ Purification System (Invitrogen) and were dot-blotted, along with lysozyme (Sigma), onto a nitrocellulose membrane. The membrane was blocked for 1 h at RT in TBS-T buffer (10 mM Tris, pH7.4, 150 mM NaCl, 0.04% Tween 20) containing 5% nonfat dry milk and incubated with PAR (1 μg/ml, Trevigen) in blocking buffer for 1h at RT. The membrane was then washed with TBS-T, blocked and incubated with antibody to PAR (1:1000, Trevigen) overnight at 4°C. Membrane was incubated with HRP-conjugated secondary antibody (Amersham), and ECL (Amersham) was used to visualize signals.
Morbidity, mortality and virus production in mice
Five day or 2 week-old CD-1 mice (Charles River Laboratories) were infected intracerebrally with 1,000 plaque forming units (PFU) of virus in 10 μl phosphate-buffered saline (PBS) under isoflurane anesthesia. To determine morbidity and mortality, groups of 10 infected animals were monitored daily and clinically scored. Survival was plotted and compared using Kaplan-Meyer survival curves. To assess virus replication, brains were collected and homogenized in PBS using FastPrep® (MP Biomedicals), and assayed for PFU on BHK21 cells.
Analysis of viral RNA synthesis
To assess viral genomic and subgenomic RNA synthesis, differentiated CSM14.1 cells were incubated with culture medium containing [5,6-3H] uridine (20μCi/ml) (PerkinElmer) and dactinomycin (1μg/ml) (Calbiochem) at 39°C for 2 h. Cells were then washed with PBS and lysed with RNA STAT-60 (Tel-Test). RNAs were purified by chloroform extraction and isopropanol precipitation, and were separated at 120 volt-hours on a 1% agarose gel. For fluorography, the gel was fixed with methanol at RT for 30 min, impregnated with 2.5% 2,5-diphenyloxazole in methanol overnight, and then washed with water. The gel was then dried with a vacuum dryer at 50°C for 1 h and exposed to Kodak BioMax MS film at −80°C until signals were detected.
To quantitate viral plus and minus strand RNA, SINV-infected differentiated CSM14.1 cells were lysed, and total RNAs were purified using the RNease® Mini Kit (Qiagen). Each cDNA was synthesized from RNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science) with a SINV-specific primer (SINV8456F 5′ CACGGCAATGTGTTTGCT 3′ for the cDNA synthesis of minus strand RNA; SINV9899R 5′ AGCATTGGCCGACCTAACGCAGCAC 3′ for the cDNA synthesis of plus strand RNA). Real time PCR was performed with the synthesized cDNA, primers SVE2F 5′ TGGGACGAAGCGGACGATAA 3′ and SVE2R 5′ CTGCTCCGCTTTGGTCGTAT 3′, and Taqman probe 5′ [FAM] CGCATACAGACTTCCGCCCAGT [TAMRA] 3′ (Applied Biosystems) using TaqMan™ PCR Core Reagent Kit (Applied Biosystems). Real time PCR was run and analyzed with the 7500 Real Time PCR System (Applied Biosystems).
Analysis of viral protein synthesis
For immunoblots, cells were lysed using radioimmune precipitation assay (RIPA) buffer (1% Nonidet P-40, 0.1% sodium dodecyl sulfate (SDS), 0.1% sodium deoxycholate, 10mM Tris-Cl, 150mM NaCl, 1mM EDTA, pH 7.5) containing a protease inhibitor cocktail (Sigma) and Halt™ phosphatase inhibitor cocktail (Pierce). Cell lysates were centrifuged at 13,000 rpm for 30 min to remove cell debris and used for immunoblot. Antibodies used for immunoblotting were against actin (Chemicon), nsP2 (polyclonal rabbit antibody raised against amino acid 113–130 of nsP2, Quality Controlled Biochemicals (QCB)), nsP3 (polyclonal rabbit antibody raised against amino acid 212–230 of nsP3, QCB), and polyclonal rabbit antibody to SINV structural proteins (Jackson, Moench et al., 1988).
To label newly synthesized proteins, differentiated CSM14.1 cells at 19 h post-infection or BHK21 cells at 6 h post-infection were incubated with Met/Cys-free DMEM (MP Biomedicals) for 1 h, followed by incubation with Met/Cys-free DMEM containing Tran35S (100μCi/ml) (MP Biomedicals) at 39°C for 2 h (CSM14.1 cells) or 1 h (BHK21 cells). To chase labeled proteins, cells were washed, incubated with cold media containing 15 mg/L of L-Met, and lysed with RIPA buffer. To inhibit proteasome activity, MG132 (1μM, Biomol) was added to the medium after infection. For immunoprecipitation, cell lysates were incubated with antibody to nsP3, nsP2, or SINV structural proteins (10μg) in RIPA buffer overnight with gentle rocking at 4°C. ImmunoPure® Immobilized Protein A/G (10μl of settled gel) (Pierce) was added to the mixture of cell lysate and antibody, followed by overnight incubation with gentle rocking at 4°C. The gel was washed with RIPA buffer 3 times, and bound proteins were eluted by boiling at 90°C with 2X SDS sample buffer for 5 min. Eluted proteins were analyzed by PAGE. Then, the gel was fixed in 50% methanol and 10% glacial acetic acid (in water) at RT for 30 min, incubated in 10% glycerol (in water) for 5 min to prevent cracking, dried using a vacuum dryer at 80°C for 1 h, and exposed to Kodak BioMax MS film at RT until signals were detected.
Statistical analysis
Statistical analyses were performed using Student’s unpaired t test or Kaplan Meyer survival analysis and log rank test using GraphPad QuickCalcs software.
Acknowledgments
This work was supported by research grant R01 NS18596 from the National Institutes of Health. We would like to thank Dr. Sean Prigge for helping with the homology modeling, Dr. Richard Kuhn for helpful discussions and Dr. Rebeca Burdeinick-Kerr for her helpful comments and support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Agelopoulos M, Thanos D. Epigenetic determination of a cell-specific gene expression program by ATF-2 and the histone variant macroH2A. EMBO J. 2006;25:4843–4853. doi: 10.1038/sj.emboj.7601364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlquist P, Strauss EG, Rice CM, Strauss JH, Haseloff J, Zimmern D. Sindbis virus proteins nsP1 and nsP2 contain homology to nonstructural proteins from several RNA plant viruses. J Virol. 1985;53:536–542. doi: 10.1128/jvi.53.2.536-542.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdeinick-Kerr R, Griffin DE. Gamma interferon-dependent, noncytolytic clearance of Sindbis virus infection from neurons in vitro. J Virol. 2005;79:5374–5385. doi: 10.1128/JVI.79.9.5374-5385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calisher CH. Medically important arboviruses of the United States and Canada. Clin Microbiol Rev. 1994;7:89–116. doi: 10.1128/cmr.7.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy S, Gundimella SK, Caron C, Perche PY, Pehrson JR, Khochbin S, Luger K. Structural characterization of the histone variant macroH2A. Mol Cell Biol. 2005;25:7616–7624. doi: 10.1128/MCB.25.17.7616-7624.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changolkar LN, Singh G, Pehrson JR. macroH2A1-dependent silencing of endogenous murine leukemia viruses. Mol Cell Biol. 2008;28:2059–2065. doi: 10.1128/MCB.01362-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock LR, Denu JM. Synthesis and biochemical evaluation of O-acetyl-ADP-ribose and N-acetyl analogs. Org Biomol Chem. 2007;5:3087–3091. doi: 10.1039/b710231c. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Carroll JW, Rout MP, Rice CM, Chait BT, MacDonald MR. Tracking and elucidating alphavirus-host protein interactions. J Biol Chem. 2006;281:30269–30278. doi: 10.1074/jbc.M603980200. [DOI] [PubMed] [Google Scholar]
- De I, Fata-Hartley C, Sawicki SG, Sawicki DL. Functional analysis of nsP3 phosphoprotein mutants of Sindbis virus. J Virol. 2003;77:13106–13116. doi: 10.1128/JVI.77.24.13106-13116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M, Schlesinger MJ. Evidence that Sindbis virus nsP2 is an autoprotease which processes the virus nonstructural polyprotein. Virology. 1989;171:280–284. doi: 10.1016/0042-6822(89)90539-4. [DOI] [PubMed] [Google Scholar]
- Durand MM, Chugani DC, Mahmoudi M, Phelps ME. Characterization of neuron-like cell line immortalized from primary rat mesencephalon cultures. Soc Neurosci. 1990;16:40. [Google Scholar]
- Egloff MP, Malet H, Putics A, Heinonen M, Dutartre H, Frangeul A, Gruez A, Campanacci V, Cambillau C, Ziebuhr J, Ahola T, Canard B. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J Virol. 2006;80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson KK, Cervantes-Barragan L, Ludewig B, Thiel V. Mouse hepatitis virus liver pathology is dependent on ADP-ribose-1″-phosphatase, a viral function conserved in the alpha-like supergroup. J Virol. 2008;82:12325–12334. doi: 10.1128/JVI.02082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazakerley JK, Pathak S, Scallan M, Amor S, Dyson H. Replication of the A7(74) strain of Semliki Forest virus is restricted in neurons. Virology. 1993;195:627–637. doi: 10.1006/viro.1993.1414. [DOI] [PubMed] [Google Scholar]
- Frolova E, Gorchakov R, Garmashova N, Atasheva S, Vergara LA, Frolov I. Formation of nsP3-specific protein complexes during Sindbis virus replication. J Virol. 2006;80:4122–4134. doi: 10.1128/JVI.80.8.4122-4134.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez DC, Ehsani N, Mikkola ML, Garcia JA, Kaariainen L. RNA helicase activity of Semliki Forest virus replicase protein NSP2. FEBS Lett. 1999;448:19–22. doi: 10.1016/s0014-5793(99)00321-x. [DOI] [PubMed] [Google Scholar]
- Gorbalenya AE, Koonin EV, Lai MM. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi-, alpha- and coronaviruses. FEBS Lett. 1991;288:201–205. doi: 10.1016/0014-5793(91)81034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov R, Garmashova N, Frolova E, Frolov I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J Virol. 2008;82:10088–10101. doi: 10.1128/JVI.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DE. Role of the immune response in age-dependent resistance of mice to encephalitis due to Sindbis virus. J Infect Dis. 1976;133:456–464. doi: 10.1093/infdis/133.4.456. [DOI] [PubMed] [Google Scholar]
- Hardy WR, Strauss JH. Processing the nonstructural polyproteins of Sindbis virus: Nonstructural proteinase is in the C-terminal half of nsP2 and functions both in cis and in trans. J Virol. 1989;63:4653–4664. doi: 10.1128/jvi.63.11.4653-4664.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseloff J, Goelet P, Zimmern D, Ahlquist P, Dasqupta R, Kaesberg P. Striking similarities in amino acid sequence among nonstructural proteins encoded by RNA viruses that have dissimilar genomic organization. Proc Natl Acad Sci USA. 1984;81:4358–4362. doi: 10.1073/pnas.81.14.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Moench TR, Trapp BD, Griffin DE. Basis of neurovirulence in Sindbis virus encephalomyelitis of mice. Lab Invest. 1988;58:503–509. [PubMed] [Google Scholar]
- Johnson RT, McFarland HF, Levy SE. Age-dependent resistance to viral encephalitis: Studies of infections due to Sindbis virus in mice. J Infect Dis. 1972;125:257–262. doi: 10.1093/infdis/125.3.257. [DOI] [PubMed] [Google Scholar]
- Kamer G, Argos P. Primary structural comparison of RNA-dependent polymerases from plant, animal and bacterial viruses. Nucl Acids Res. 1984;12:7269–7282. doi: 10.1093/nar/12.18.7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, Bycroft M, Ladurner AG. The macro domain is an ADP-ribose binding module. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Gorbalenya AE, Purdy MA, Rozanov MN, Reyes GR, Bradley DW. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: delineation of an additional group of positive-strand RNA plant and animal viruses. Proc Natl Acad Sci USA. 1992;89:8259–8263. doi: 10.1073/pnas.89.17.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG. Splicing regulates NAD metabolite binding to histone macroH2A. Nat Struct Mol Biol. 2005;12:624–625. doi: 10.1038/nsmb956. [DOI] [PubMed] [Google Scholar]
- Ladurner AG. Inactivating chromosomes: a macro domain that minimizes transcription. Mol Cell. 2003;12:1–3. doi: 10.1016/s1097-2765(03)00284-3. [DOI] [PubMed] [Google Scholar]
- Laine M, Luukkainen R, Toivanen A. Sindbis viruses and other alphaviruses as cause of human arthritic disease. J Intern Med. 2004;256:457–471. doi: 10.1111/j.1365-2796.2004.01413.x. [DOI] [PubMed] [Google Scholar]
- LaStarza MW, Grakoui A, Rice CM. Deletion and duplication mutations in the C-terminal nonconserved region of Sindbis virus nsP3: effects on phosphorylation and on virus replication in vertebrate and invertebrate cells. Virology. 1994;202:224–232. doi: 10.1006/viro.1994.1338. [DOI] [PubMed] [Google Scholar]
- LaStarza MW, Lemm JA, Rice CM. Genetic analysis of the nsP3 region of Sindbis virus: evidence for roles in minus-strand and subgenomic RNA synthesis. J Virol. 1994;68:5781–5791. doi: 10.1128/jvi.68.9.5781-5791.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Hardwick JM, Trapp BD, Crawford TO, Bollinger RC, Griffin DE. Antibody-mediated clearance of alphavirus infection from neurons. Science. 1991;254:856–860. doi: 10.1126/science.1658936. [DOI] [PubMed] [Google Scholar]
- Li G, La Starza MW, Hardy WR, Strauss JH, Rice CM. Phosphorylation of Sindbis virus nsP3 in vivo and in vitro. Virology. 1990;179:416–427. doi: 10.1016/0042-6822(90)90310-n. [DOI] [PubMed] [Google Scholar]
- Liang Y, Yao J, Gillam S. Rubella virus nonstructural protein protease domains involved in trans- and cis-cleavage activities. J Virol. 2000;74:5412–5423. doi: 10.1128/jvi.74.12.5412-5423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig S, Jackson AC, Hahn CS, Griffin DE, Strauss EG, Strauss JH. The molecular basis of Sindbis virus neurovirulence in mice. J Virol. 1988;62:2329–2336. doi: 10.1128/jvi.62.7.2329-2336.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martzen MR, McCraith SM, Spinelli SL, Torres FM, Fields S, Grayhack EJ, Phizicky EM. A biochemical genomics approach for identifying genes by the activity of their products. Science. 1999;286:1153–1155. doi: 10.1126/science.286.5442.1153. [DOI] [PubMed] [Google Scholar]
- Mi S, Durbin R, Huang HV, Rice CM, Stollar V. Association of the Sindbis virus RNA methytransferase activity with the nonstructural protein nsP1. Virology. 1989;170:385–391. doi: 10.1016/0042-6822(89)90429-7. [DOI] [PubMed] [Google Scholar]
- Neuvonen M, Ahola T. Differential activities of cellular and viral macro domain proteins in binding of ADP-ribose metabolites. J Mol Biol. 2009;385:212–225. doi: 10.1016/j.jmb.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow DA, Hernandez-Munoz I, Fazzio TG, Shah GM, Kraus WL, Panning B. Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, MacroH2A, and contributes to silencing of the inactive X chromosome. J Biol Chem. 2007;282:12851–12859. doi: 10.1074/jbc.M610502200. [DOI] [PubMed] [Google Scholar]
- Pehrson JR, Fried VA. MacroH2A, a core histone containing a large nonhistone region. Science. 1992;257:1398–1400. doi: 10.1126/science.1529340. [DOI] [PubMed] [Google Scholar]
- Pehrson JR, Fuji RN. Evolutionary conservation of histone macroH2A subtypes and domains. Nucleic Acids Res. 1998;26:2837–2842. doi: 10.1093/nar/26.12.2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putics A, Filipowicz W, Hall J, Gorbalenya AE, Ziebuhr J. ADP-ribose-1″-monophosphatase: a conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J Virol. 2005;79:12721–12731. doi: 10.1128/JVI.79.20.12721-12731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikatendu KS, Joseph JS, Subramanian V, Clayton T, Griffith M, Moy K, Velasquez J, Neuman BW, Buchmeier MJ, Stevens RC, Kuhn P. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1″-phosphate dephosphorylation by a conserved domain of nsP3. Structure. 2005;13:1665–1675. doi: 10.1016/j.str.2005.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidel LM, Durbin RK, Stollar V. Sindbis virus mutants resistant to mycophenolic acid and ribavirin. Virology. 1987;158:1–7. doi: 10.1016/0042-6822(87)90230-3. [DOI] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. The alphaviruses: Gene expression, replication and evolution. Microbiol Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thach DC, Kimura T, Griffin DE. Differences between C57BL/6 and BALB/cBy mice in mortality and virus replication after intranasal infection with neuroadapted Sindbis virus. J Virol. 2000;74:6156–6161. doi: 10.1128/jvi.74.13.6156-6161.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till S, Ladurner AG. Sensing NAD metabolites through macro domains. Front Biosci. 2009;14:3246–3258. doi: 10.2741/3448. [DOI] [PubMed] [Google Scholar]
- Tuittila M, Hinkkanen AE. Amino acid mutations in the replicase protein nsP3 of Semliki Forest virus cumulatively affect neurovirulence. J Gen Virol. 2003;84:1525–1533. doi: 10.1099/vir.0.18936-0. [DOI] [PubMed] [Google Scholar]
- Tuittila MT, Santagati MG, Roytta M, Maatta JA, Hinkkanen AE. Replicase complex genes of Semliki Forest virus confer lethal neurovirulence. J Virol. 2000;74:4579–4589. doi: 10.1128/jvi.74.10.4579-4589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljeva L, Merits A, Auvinen P, Kaariainen L. Identification of a novel function of the alphavirus capping apparatus. RNA 5′-triphosphatase activity of nsP2. J Biol Chem. 2000;275:17281–17287. doi: 10.1074/jbc.M910340199. [DOI] [PubMed] [Google Scholar]
- Vernon PS, Griffin DE. Characterization of an in vitro model of alphavirus infection of immature and mature neurons. J Virol. 2005;79:3438–3447. doi: 10.1128/JVI.79.6.3438-3447.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong LT, Sarafian T, Kane DJ, Charles AC, Mah S, Edwards RH, Bredesen DE. Bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci USA. 1993;90:4533–4537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]