

Abstract
Metastatic cancer involving spread to the peritoneal cavity is referred to as peritoneal carcinomatosis and has a very poor prognosis. Our previous studies demonstrated a toll-like receptor 4 (TLR4) and C-type lectin receptor (CLR; Mincle/MCL) agonist pairing of monophosphoryl lipid A (MPL) and trehalose-6,6’-dicorynomycolate (TDCM) effectively inhibits peritoneal tumor growth and ascites development through a mechanism dependent upon B-1a cell-produced natural IgM, complement, and phagocytes. In the current study, we investigated the requirement for TLR4 and Fc receptor common γ chain (FcRγ), required for Mincle/MCL signaling, in the MPL/TDCM-elicited response. MPL/TDCM significantly increased macrophages and Ly6Chi monocytes in the peritoneal cavity of both TLR4−/− and FcRγ−/− mice, suggesting redundancy in the signals required for monocyte/macrophage recruitment. However, B1 cell activation, antibody secreting cell (ASC) differentiation, and tumor-reactive IgM production was defective in TLR4−/−, but not FcRγ−/− mice. TRIF was required for production of IgM reactive against tumor- and mucin-related antigens, but not phosphorylcholine, whereas TLR4 was required for production of both types of reactivities. Consistent with this, B1 cells lacking TLR4 or TRIF did not proliferate or differentiate into tumor-reactive IgM-producing cells in vitro and did not reconstitute MPL/TDCM-dependent protection against peritoneal carcinomatosis in CD19−/− mice. Our results indicate a TLR4/TRIF-dependent pathway is required by B1 cells for MPL/TDCM-elicited production of protective tumor-reactive natural IgM. The dependency on TRIF signaling for tumor-reactive, but not phosphorylcholine-reactive, IgM production reveals unexpected heterogeneity in TLR4-dependent regulation of natural IgM production, thereby highlighting important differences to consider when designing vaccines/therapies targeting these specificities.
Keywords: B-1 cells, peritoneal carcinomatosis, Toll-like receptor 4, TRIF
Precis:
A combinatorial treatment of monophosphoryl lipid A and trehalose-6,6’-dicorynomycolate potently inhibits peritoneal carcinomatosis by activating B1 cells to secrete protective tumor-reactive IgM via a pathway dependent on TLR4/TRIF signaling.
Introduction
Metastatic disease is a major cause of morbidity and mortality in cancer patients. In particular, the spread of malignant cells to the peritoneal cavity carries a grave prognosis, especially when associated with ascites development [1]. The peritoneal cavity provides extensive surface area and volume for tumor growth and supports growth of malignant epithelial (carcinomatosis), mesenchymal (sarcomatosis) and more rarely, lymphoid (lymphomatosis) cells [2]. Treatment options are limited and often palliative rather than curative, although cytoreductive surgery and hyperthermic intraperitoneal chemotherapy and emerging treatments show promise in some patients [3]. Immunotherapeutic approaches to treat peritoneal malignancies have been limited, although results obtained from some mouse models offer hope for future treatments [3].
Understanding the distinctive environment of the peritoneal cavity is key to devising optimal immunotherapies for peritoneal metastasis. The peritoneal space represents a unique immune environment [4]. Monocytes and macrophages comprise the majority of leukocytes in the cavity under normal conditions. Innate-like B-1 cells, composed of CD5+ B-1a cells and CD5− B-1b cell populations, are the second most numerous [4, 5]. These B cells have been studied most in mice, but have been identified in non-human primate peritoneal and omental tissue [5, 6] as well as in human blood [7]. B-1 cells produce natural IgM and IgA as well as pathogen-specific antibodies (Abs), which are critical for host defense and clearance of apoptotic debris [8]. Although B-1 cells are known to have a critical role in protection against infectious diseases, their role in cancer is not well understood. Our recent work and that of others has highlighted that natural reactivities against tumor associated carbohydrate antigens (TACAs) are present in the B1 cell Ab repertoire [9–11]. TACAs found on aberrantly glycosylated glycoproteins and glycolipids selectively expressed on cancer cells include Tn, sialyl Tn, TF, LeY, GM2, GD2, and GD3 [12–14]. In mice and humans, Abs against TACAs largely belong to the IgM subclass [9, 10, 12] and in some cases exhibit oncolytic effects[12, 15]. Additional studies indicate the production of TACA-specific Abs are associated with increased survival rates in patients [16–19] and in mice [9, 10]. Under normal conditions, both peritoneal B-1 cells and macrophages inhibit T cell activation and peritoneal macrophages additionally inhibit B cell proliferation and Ab production [20–24]. Overcoming immune suppression in the peritoneal cavity may provide opportunities for effective treatment of peritoneal carcinomatosis.
We recently found that a combination of pathogen-associated molecular pattern molecules (PAMPs), consisting of a toll-like receptor 4 (TLR4) and C-type lectin receptor (CLR; Mincle/MCL) agonist pairing of monophosphoryl lipid A (MPL) and trehalose-6,6’-dicorynomycolate (TDCM), effectively inhibits peritoneal tumor growth and ascites development[9]. Protection is elicited through a mechanism dependent upon B-1a cell-produced natural IgM reactive against TACAs and complement. Phagocytes also play a supportive role in the therapeutic effects of MPL/TDCM treatment through production of cytotoxic intermediates and likely engaging in mechanisms of complement-dependent cellular cytotoxicity[25]. MPL signals through the TLR4/MD2 complex[26] whereas TDCM relies on the CLRs, Mincle and MCL, which require the Fc receptor common γ chain (FcRγ) for signaling [27]. Understanding how these PAMPs activate the effector functions of key populations required for the therapeutic effects of MPL/TDCM is critical for devising effective therapies to treat patients suffering from peritoneal carcinomatosis. In the current study, we examined the role of TLR4 and FcRγ in eliciting monocyte/macrophage recruitment and B1 cell activation and tumor-reactive IgM production in response to MPL/TDCM treatment. A primary role for TLR4, along with its activation of the TRIF signaling pathway, was determined to be essential for B1 cell-elicited protection against peritoneal carcinomatosis. These findings will help to guide future strategies aimed at harnessing the anti-tumor potential of TACA-reactive B cells in humans.
Materials and Methods
Mice
Wild-type (WT), TLR4−/−, MyD88−/−, TRIF−/−, FcRγ−/− mice were from Jackson Laboratories and on a C57BL/6 background. Animal experiments were approved by Wake Forest Animal Care and Use Committee. Mice were housed in a specific pathogen-free animal facility and fed standard chow and housed in autoclaved cages. MyD88−/− mice were provided water containing 4 mg/ml sulfamethoxadole and 0.8 mg/ml trimethoprim where indicated. Experiments were conducted using 8- to 12-week old age-matched mice.
Tumor challenges and treatments
TA3-Ha cells were obtained from Dr. Richard Lo-Man (Pasteur Institute, Paris, France) in 2010, as previously described[9, 10]. Stocks were tested for rodent pathogens (IMPACT IV testing, IDEXX-RADIL). TA3-Ha cells were expanded in culture for 3 days prior to injection. Mice were given 2.5 × 104 TA3-Ha cells in 200 μl PBS intraperitoneally (i.p.). Mice developing ascites with signs of distress were humanely euthanized.
Mice were injected i.p. with Sigma adjuvant system containing 10 μg monophosphoryl lipid A (MPL) and 10 μg synthetic trehalose dicorynomycolate (TDCM) mixed in 0.4% squalene (Sigma) in a 200 μl volume as previously described[9]. Adoptive transfers of 2 ×106 B1 cells into CD19−/−mice were performed as previously described[9] one day prior to tumor challenge, using the B1 cell purification strategy described below.
Flow cytometry
Peritoneal cells were harvested using 10 ml of DPBS to lavage the peritoneal cavity. Splenocyte preparations were lysed, filtered, pelleted, and resuspended in staining buffer (DPBS + 2% newborn calf serum). Visceral adipose tissue (VAT) was diced with scissors and incubated for one hour in collagenase IV (1 mg/ml in RPMI) with gentle stirring, filtered (70 μm), and centrifuged at 300xg for 7 minutes to separate the floating adipocytes from pelleted cells. Cell pellets were washed twice with cold RPMI-1640 and resuspended in staining buffer. Cells were pre-incubated with 0.5 μg/ml FcBlock (eBioscience) and stained with mAbs conjugated to fluorochromes (Biolegend, eBioscience, and BD Biosciences): CD5 (53–7.3), Ly6G (Gr-1), CD86 (GL-1), CD19 (1D3), CD11b (M1/70), F4/80 (BM8), CD11c (N418), Ly6C (HK1.4), MHC II IA-IE (M5/114.15.2), and CD138 (281–2). BLIMP1 and Ki-67 staining was performed using eBioscience’s Foxp3/transcription factor staining set. Small and large peritoneal macrophages were distinguished as previously described by Cain et al [28]. Serum (diluted 10-fold) and B cell supernatant (diluted 5-fold) IgM binding to tumor cells was assessed as previously described[9, 10]. Samples were washed and fixed in 1.5% buffered formaldehyde. Cells were analyzed using a BD LSR Fortessa X20 (Becton Dickinson).
ELISAs
Serum samples were diluted 1:100 in TBS containing 1% BSA (TBS-BSA) and analyzed for PC-specific IgM as previously described[29]. Diluted serum samples were added to Costar plates coated with 2 μg/mL PC(4)BSA (Biosearch Technologies) and blocked with TBS-BSA. Desialylated bovine submaxillary mucin (dBSM; MP Biologicals)-specific ELISAs were carried out as previously described[10]. Briefly, Nunc Maxisorb plates were coated with 10 μg/ml dBSM (in 0.1 M borate buffered saline overnight and then blocked with TBS-BSA), followed by incubation with serum diluted 1:100. Alkaline phosphatase-conjugated polyclonal goat anti-mouse IgM (Southern Biotech) and pNPP (Sigma) were used to detect bound IgM.
B cell purification and in vitro activation assays
B cells were isolated from peritoneal lavages using EasySep Pan B cell isolation (untouched) kit according to manufacturer’s instructions (StemCell), with supplementation of biotinylated anti-mouse CD23 and F4/80 during B cell isolation. Enriched B1 cells were further purified by incubation with biotinylated F4/80 and GR1 mAbs, followed by incubation with Dynal Biotin binder beads and magnetic removal. B1 cell purity was >92%. B cells were washed in DPBS and injected i.p. into recipients. Alternatively, B1 cells (5×105/ml) were cultured in 96 well plates in RPMI + 10% FCS containing 50 μM BME either alone with 10 μg/ml MPL and TDCM and 0.08 % squalene in triplicate or quadruplicate. On day 6, supernatants were collected and analyzed for IgM production and cells were stained and enumerated using Countbright beads (ThermoScientific) by flow cytometry.
Statistical analysis
Data are shown as means ± SEM with differences assessed using unpaired Student’s t test unless otherwise indicated. Differences in Kaplan-Meier survival curves were assessed using the Log Rank test.
Results
MPL/TDCM-induced increases in peritoneal monocytes and macrophages are normal or augmented in TLR4−/− and FcRγ−/− mice
The efficacy of MPL/TDCM treatment against peritoneal carcinomatosis depends on the production of tumor-reactive IgM by B-1a cells, classical complement activation[9], and Ly6C+/phagocytic cells[25]. To begin to elucidate the receptors and signaling pathways required for activation of these cells, we used FcRγ−/− mice which lack the capacity to signal through the TDCM receptor, Mincle, and TLR4−/− mice which lack the major receptor for MPL. Monocyte and macrophage numbers were significantly increased in the peritoneal cavities of WT and FcRγ−/− mice at 24 hours post treatment (Fig. 1A). Small peritoneal macrophages (SPMs) which arise from recruited monocytes were increased in WT and FcRγ−/− mice, although large peritoneal macrophages (LPMs), the resident macrophages of the peritoneal cavity, did not decrease in FcγR−/− mice as occurs in WT mice and SPM numbers FcRγ−/− mice were significantly increased over numbers in treated WT mice (Fig. 1B). The number of MHC II+ SPMs and monocytes were also increased in MPL/TDCM-treated FcRγ−/− mice and were elevated over increases found in WT mice (Fig. 1C). TLR4−/− mice exhibited similar increases in monocytes and macrophages (SPM) compared to WT mice (Fig. 1D) along with decreases in LPMs (Fig. 1E). TLR4−/− mice also had similar increases in activated MHC II+ SPM and monocytes (Fig. 1F). Thus, TLR4 and FcγR-mediated signaling are not individually required for recruitment of peritoneal monocytes/macrophages following MPL/TDCM treatment, suggesting that either signaling pathway is sufficient to compensate for the loss of the other. Nonetheless, it remains possible that functional (anti-tumor) responses of these cells could be impacted in the absence of either.
Figure 1. MPL/TDCM-induced accumulation of activated small peritoneal macrophages and monocytes is not reduced in FcγR−/− and TLR4−/− mice.
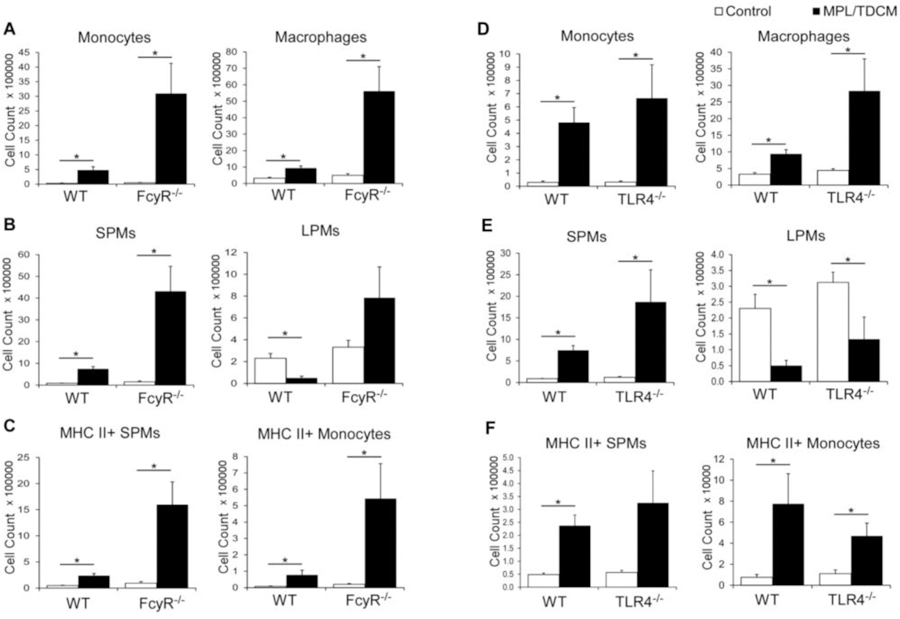
(A–F) Inflammatory monocytes (CD11b+ Ly6G− CD11c− B220− Ly6C+ SSClow) and macrophages (CD11b+ Ly6G− CD11c− B220− Ly6Clow SSClow) were enumerated in peritoneal cavities on day 1 post TA3-Ha cell challenge ± MPL/TDCM treatment. (A, D) Monocyte and macrophage numbers in WT, FcγR−/− and TLR4−/− mice. (B, E) Small peritoneal macrophages (SPM; CD11bmidF4/80mid) and large peritoneal macrophages (LPM; CD11bhiF4/80hi) were further quantified. (C, F) MHC II+ SPM and monocyte cell numbers on day 1 post-tumor challenge. Data representative of two experiments. Values represent means ± SEM. Asterisks (*) in A–F indicate significant differences between indicated groups (p<0.05; n=3–5 mice/group).
MPL/TDCM-induced increases in Ab-secreting B1 cells are intact in FcRγ−/−, but defective in TLR4−/− mice
MPL/TDCM treatment increases B1 cells in the peritoneal cavity by day 5 post treatment. CD5 expression on B1 cells decreases upon TLR agonist-induced activation and differentiation in this (data not shown) and other model systems [30, 31]. Although CD11b is also gradually downregulated with B1 cell differentiation to ASC, CD11b+CD19+ B1 cells remain detectable at day 5 post MPL/TDCM treatment. Peritoneal B1 cell frequencies and numbers along with dividing (Ki67+) peritoneal B1 cell frequencies and numbers, were significantly increased in treated WT and FcγR−/− mice, but not in TLR4−/− mice on d5 (Fig. 2A–B). B1 cell frequencies and numbers, as well as Ki67+ B1 cells, were also significantly increased in the spleens and visceral adipose tissue (VAT) of WT and FcγR−/− mice, but not TLR4−/− mice (Fig. 2D–E, 2G–H). Finally, B1 cell ASC numbers (enumerated as BLIMP1+CD138+CD11b+CD19+ B cells) were significantly increased in WT and FcγR−/− PerC, VAT, and spleen, but this was not observed in TLR4−/− mice (Fig. 2C, 2F, 2I).
Figure 2. B-1 cell activation, division, and ASC differentiation following MPL/TDCM treatment is impaired in the absence of TLR4 signaling.
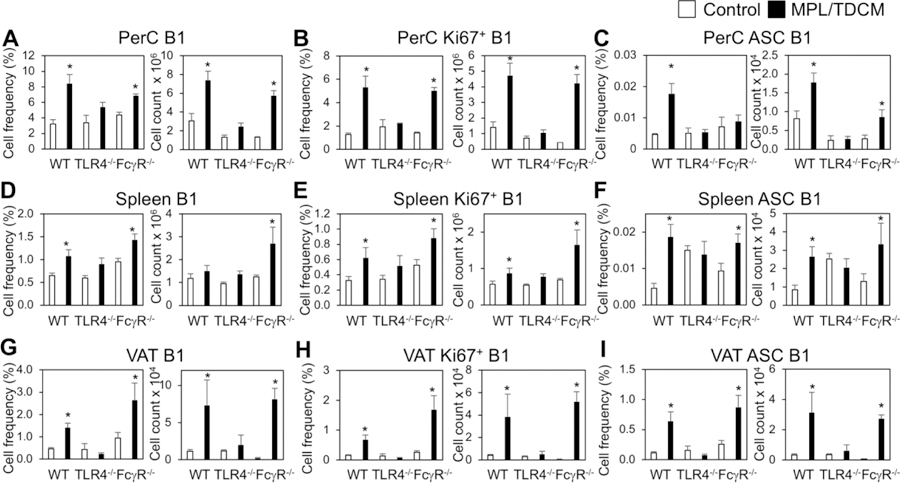
(A–I) WT, FcγR−/− and TLR4−/− mice were challenged with 2.5 × 104 TA3-Ha cells ± MPL/TDCM on day 0. On day 5, peritoneal lavage, spleen and visceral adipose tissue (VAT) were harvested and analyzed via flow cytometry. Graphs show B1 cell (CD19+CD11b+) frequencies and numbers in the peritoneal cavity (PerC, A) and spleen (Spl, D) and VAT (G); frequencies and numbers of dividing (Ki67+) B1 cells in peritoneal cavity (B), spleen (E) and VAT (H); and frequencies and numbers of BLIMP1+CD138+ B1 cells in peritoneal cavity (C), spleen (F), and VAT (I). Values represent means ± SEM. Asterisks (*) indicate significant differences between treated and untreated mice of the same genotype (n=3–5 mice/group).
We next examined the extent to which MPL/TDCM induced production of IgM reactive with TA3-Ha cells, a murine mammary adenocarcinoma which expresses high levels of epiglycanin bearing high levels of TACAs. FcγR−/− mice had higher pre-existing levels of TA3-Ha-reactive serum IgM relative to WT mice (Fig. 3A). FcγR−/− mice also had significantly higher levels of TA3-Ha-reactive IgM in response to MPL/TDCM treatment relative to WT mice. However, the overall increase in IgM binding relative to baseline levels was similar between FcγR−/− and WT mice (Fig. 3C). Interestingly, TLR4−/− mice also had significantly higher pre-existing levels of serum IgM reactive with TA3-Ha cells relative to WT mice (Fig. 3B). However, MPL/TDCM treatment did not elicit further increases in TA3-Ha-reactive IgM in TLR4−/− mice (Fig. 3B–C). Collectively, our findings demonstrate a critical role for TLR4, but not FcγR, in eliciting in vivo B1 cell activation, expansion, ASC differentiation and production of TA3-Ha-reactive IgM in response to MPL/TDCM treatment.
Figure 3. MPL/TDCM-induced anti-tumor IgM responses are independent of FcγR, but dependent on TLR4.
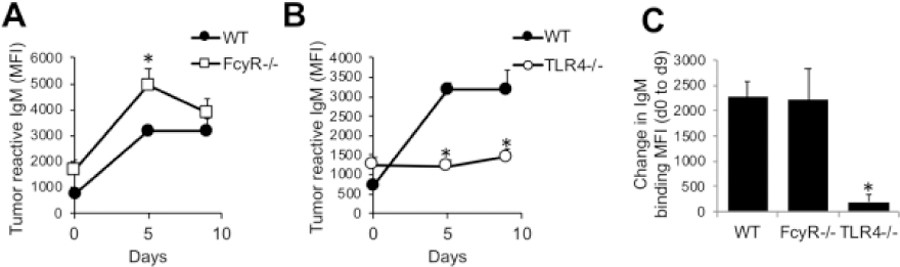
(A–C) WT and FcγR−/− (A, C) and TLR4−/− (B, C) mice were treated with MPL/TDCM i.p., with IgM binding (MFI) to TA3-Ha cells examined for serum harvested on d0, 5, and 9 using flow cytometry. Values in C indicate relative changes in IgM MFI values at d9 versus those obtained for d0. Values represent means ± SEM. Asterisks (*) indicate significant differences from WT mice (p<0.05, n=5–7 mice/group).
MPL/TDCM-elicited protection depends on TRIF signaling
We next examined the role of TLR4 and its signaling components in MPL/TDCM-mediated protection against TA3-Ha challenge. Unexpectedly, TLR4−/− mice were relatively resistant to TA3-Ha tumor challenge, with only ~50% of mice developing peritoneal carcinomatosis (Fig. 4A). It is not clear whether altered inflammation in the peritoneal cavity of these mice influences this resistance. MPL/TDCM slightly improved survival in these mice, although this was not significant (p=0.1). In contrast, naïve FcγR−/− mice were fully susceptible to TA3-Ha challenge but were offered significant, although partial, protection by MPL/TDCM treatment (~50% survival; Fig. 4E).
Figure 4. MPL/TDCM does not elicit significant protection against TA3-Ha challenge in TLR4−/− and TRIF−/− mice.
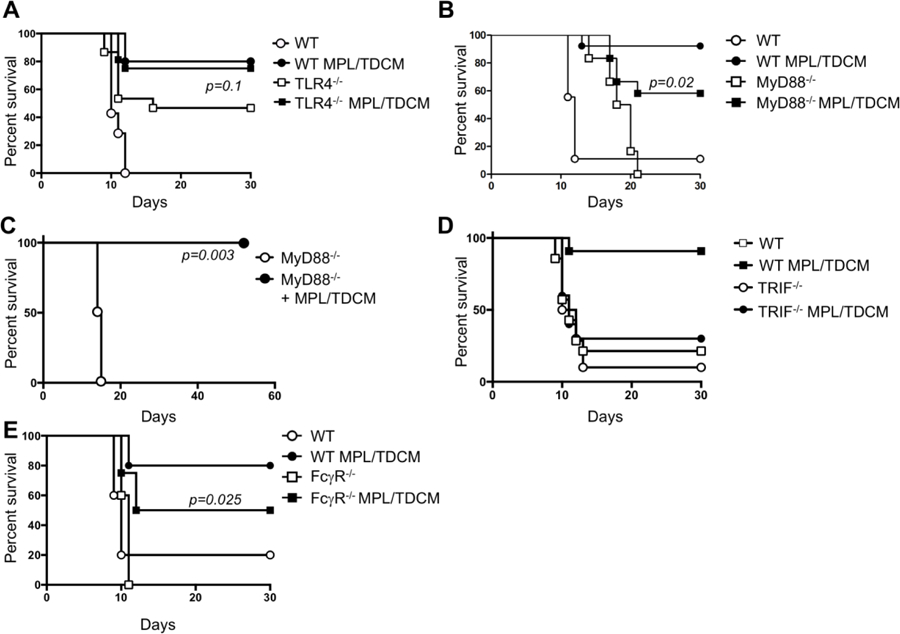
WT (A–E), TLR4−/− (B, n≥15/group), MyD88−/− (C, n≥6/group and D, n≥7/group), TRIF−/− (D, n≥10/group), and FcγR−/− mice (E, n≥8/group) mice were challenged with 2.5 × 104 TA3-Ha cells ± MPL/TDCM on d0 and monitored for morbidity requiring euthanasia. In D, MyD88−/− were reared on antibiotic water. Experiments were repeated at least once, with similar results obtained. In A–E, p values indicate Log-rank results comparing survival curves of untreated and treated knockout mice. In A, B, D, and E, survival in treated WT mice was significantly greater (p<0.05) than in non-treated WT mice.
TLR4 can signal through MyD88- and TRIF-dependent pathways. We therefore assessed the effect of MyD88 and TRIF deficiency on MPL/TDCM-elicited protection. Untreated MyD88−/− mice had prolonged survival relative to WT mice, but eventually succumbed to ascites development (Fig. 4B). MPL/TDCM treatment provided significant protection in MyD88−/− mice, improving survival from 0% to 60%. Untreated MyD88−/− mice born and raised on antibiotic water (bactrim) exhibited survival curves similar to WT mice (Fig. 4C), suggesting microbial differences in these mice may have contributed to their enhanced survival. MPL/TDCM treatment completely protected these antibiotic-reared MyD88−/− mice during TA3-Ha challenge. In contrast, MPL/TDCM did not significantly protect TRIF−/− mice against challenge, with ~30% survival in treated TRIF−/− mice vs. 90% survival in WT mice (Fig. 4D). This finding is consistent with the known role for MPL in skewing TLR4 induction of the MyD88-independent, TRIF-dependent signaling pathway[32].
TRIF signaling is essential for B1 cell activation, proliferation, and differentiation into tumor-reactive IgM-producing cells
Given the above result, we assessed the capacity of MPL/TDCM to elicit tumor-reactive IgM production in TRIF−/− mice. Similar to results with TLR4−/− mice, this treatment did not significantly increase TA3-Ha reactive IgM levels in TRIF−/− mice (Fig. 5A). A similar trend was found for serum IgM reactivity with desialylated bovine submaxillary mucin (dBSM) which shares tumor-associated antigens, such as Tn and TF, with the epiglycanin mucin expressed at high levels by TA3-Ha cells[9, 10, 33, 34]. MPL/TDCM elicited significant increases in dBSM-reactive IgM in WT mice, but responses were not elicited in TLR4−/− mice and were also significantly lower in TRIF−/− mice (Fig. 5B). In contrast to results with tumor-related Ags, MPL/TDCM treatment induced WT levels of PC-specific IgM, a common specificity of peritoneal B1a cells[29, 35], in TRIF−/−, but not TLR4−/− mice (Fig. 5C). Collectively, these results indicate TRIF expression is differentially required for MPL/TDCM-induced tumor-reactive vs. PC-reactive IgM production, although TLR4 is essential for both.
Figure 5. MPL/TDCM-induced in vivo anti-tumor IgM responses are dependent on TLR4 and TRIF signaling.
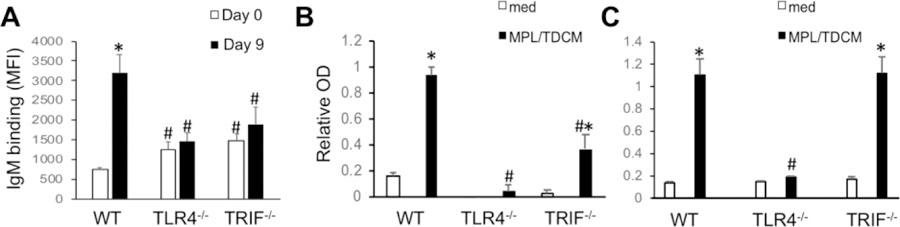
(A–C) WT, TLR4−/− and TRIF−/− mice were treated with MPL/TDCM i.p., with IgM binding to TA3-Ha cells (A), dBSM (B), and phosphorylcholine (PC) examined for serum harvested on d0 and 9 using flow cytometry (A) and ELISAs (B–C). Values represent means ± SEM. Asterisks (*) indicate significant differences between values for medium and stimulated cultures of the same genotype, and hashtags (#) indicate differences between WT values for the same condition(p<0.05, n≥5 mice/group). Results representative of 2 independent experiments.
To determine whether TLR4- or TRIF -intrinsic signaling was required for B1 cell effector functions, we isolated B1 cells and stimulated them with MPL/TDCM in vitro. Six days post culture we analyzed their activation, proliferation, differentiation to ASC, and secretion of tumor-reactive IgM. As shown in Fig. 6A–B, WT, but not TLR4−/− or TRIF−/− B1 cells upregulated CD86 and proliferated (cell yield) in response to MPL/TDCM. Interestingly, WT and TRIF−/−, but not TLR4−/−B1 cells downregulated surface IgM and MHC class II following MPL/TDCM stimulation (Fig. 6C), which was perhaps associated with ASC differentiation. Indeed, WT B1 cells treated with MPL/TDCM exhibited significant increases in the number of CD138+ cells, indicative of ASC differentiation, with nearly a 10-fold expansion relative to B1 cells cultured in media (Fig. 6D). TLR4−/− B1 cells exhibited no increase whereas a doubling was observed in TRIF−/− cells. This was associated with a significant increase in CD138hi, but not CD138 intermediate cells in TRIF−/− B1 cell cultures. This could perhaps be explained by the lack of proliferation in TRIF−/− B1 cells but terminal ASC differentiation by a subset of B cells in response to MPL/TDCM.
Figure 6. MPL/TDCM-induced B1 cell activation, proliferation, ASC differentiation, and production of tumor-reactive IgM are dependent on TLR4 and TRIF signaling.
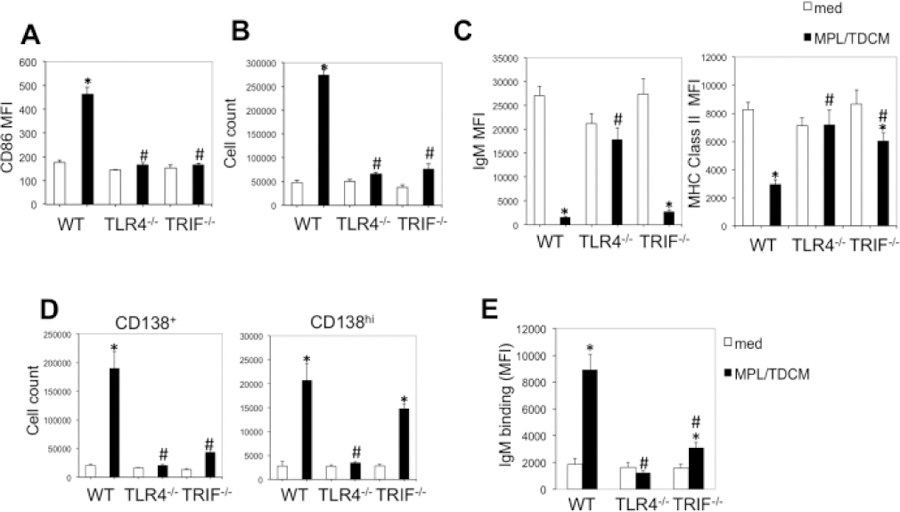
Purified peritoneal B1 cells from WT, TLR4−/− and TRIF−/− mice were cultured in medium alone or with MPL/TDCM + squalene. On day 6, CD86 upregulation (A), cell yield (B), surface IgM and MHC class II expression (C), and CD138+ and CD138hi B cell yields (D) were assessed by flow cytometry. (E) TA3-Ha-reactive IgM was detected in supernatants of cultured B1 cells using flow cytometry. Asterisks (*) indicate significant differences between medium cultures of the same genotype and hashtags (#) indicate differences between WT values (p<0.05). Results representative of 3 independent experiments.
Notably, there was significant production of tumor-reactive IgM by WT B1 cells cultured with MPL/TDCM (5-fold induction over cells cultured in media; Fig. 6E). TLR4−/− B1 cells were not induced to secrete tumor-reactive IgM. Finally, TRIF−/− B1 cells, some of which were induced to differentiate into CD138hi cells but not proliferate, secreted a moderate amount of tumor-reactive IgM (1.7-fold over media cultures). Collectively, this reveals a major role for TLR4- and TRIF-mediated signaling in driving B1 cell activation, proliferation, and differentiation into tumor-reactive IgM secreting B cells.
TLR4 and TRIF signaling is essential for B1-mediated protection against peritoneal carcinomatosis
Our previous studies demonstrated that MPL/TDCM treatment of mice lacking B cells or CD19 (CD19−/−) did not provide protection against TA3-Ha-induced peritoneal carcinomatosis [9]. However, adoptive transfers of B1a cells into these mice restored MPL/TDCM-elicited protection. We therefore adoptively transferred purified B1 cells from WT, TLR4−/−, and TRIF−/− mice into CD19−/− mice which lack the protective effects of B1 cells. WT B1 cells reconstituted protection in CD19−/−mice as expected (Fig. 7A–B; p=0.003). However, B1 cells lacking TLR4 or TRIF did not reconstitute protection in MPL/TDCM-treated CD19−/− mice (Fig. 7C–D). Thus, B1 cell-intrinsic TLR4 and TRIF expression is critical for protection elicited by MPL/TDCM treatment.
Figure 7. B1 cell-intrinsic TLR4 and TRIF expression is required for B1 cell-elicited protection against peritoneal carcinomatosis.
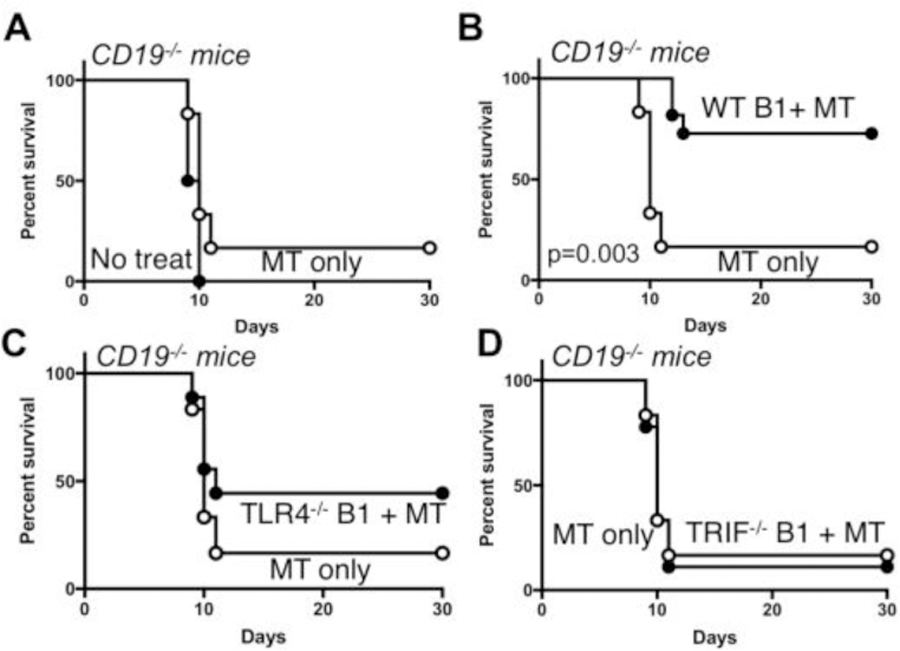
(A–D) CD19−/− mice were challenged with 2×104 TA3-Ha cells i.p.± MPL/TDCM treatment on d0 and monitored for survival (p<0.05 shown for Log rank test; n=6–11 mice per group). In B–D, CD19−/− mice were reconstituted with 2×106 purified peritoneal B1 cells i.p. one day before challenge. Results representative of 2 independent experiments. In B, the p value indicates Log-rank results.
Discussion
B1 cells play a key role in MPL/TDCM-elicited protection against mouse models of peritoneal carcinomatosis through their production of tumor-reactive IgM[9], which when combined with complement and the anti-tumor functions of peritoneal phagocytes, supports highly effective tumor cell killing[25]. Using mice lacking TLR4 and FcRγ expression, we determined that the recruitment of monocytes and SPM is intact in the absence of either signaling pathway, suggesting redundancy in the MPL- and TDCM-induced signals required to increase these myeloid cells in the peritoneal cavity. In contrast, MPL/TDCM-induced increases in B1 cell division, ASC differentiation, and tumor-reactive IgM production were largely dependent upon TLR4, as opposed to FcRγ, signaling. This indicates MPL, as opposed to TDCM, drives B1 cell production of tumor-reactive IgM. Importantly, our study identified a critical role for TRIF in TLR4-elicited tumor-reactive IgM production by B1 cells and protection against peritoneal carcinomatosis. Thereby, these findings reveal key pathways through which peritoneal B1 cells and monocytes/macrophages are activated by MPL/TDCM treatment to participate in tumor cell killing.
TLR4 was essential for MPL/TDCM-induced tumor reactive IgM responses in vivo and B1 cell activation, proliferation, and Ab production in vitro. TLR4 signaling was not necessary for development of B1a or B1b cells, consistent with what has been previously reported [30, 36]. However, it was necessary for inducing key changes required for B1 cell anti-tumor responses in vivo. This includes the division and increased numbers of B1 cells in the peritoneal cavity, spleen and VAT, as well as Ab secretion, as evidenced by increases in CD138+BLIMP1+ B1 cells in the peritoneal cavity, spleen, and VAT. The requirement for TLR4 in eliciting these responses, along with tumor-reactive and PC-specific IgM production, is perhaps not unexpected given the known sensitivity of murine B1 cells to TLR4 agonists, such as LPS [37]. LPS elicits significant division and Ab secretion by these cells both in vivo and in vitro[37, 38]. Although the effects of MPL on these cells have been studied to a lesser agree, our results show the commercial adjuvant preparation containing MPL, TDCM, and squalene promotes significant B1 activation, expansion, and differentiation to ASC via TLR4-dependent activation.
Despite the requirement for TLR4 in driving B1 cell anti-tumor natural IgM responses in response to MPL/TDCM, naïve TLR4−/− mice had significantly higher levels of tumor-reactive IgM compared to WT mice and due to this and/or other reasons, such as an altered inflammatory state due to microbiota changes[39], had reduced susceptibility to TA3-Ha cell challenge. Nonetheless, MPL/TDCM treatment did not significantly improve survival in these mice, in line with the observation that TLR4 was required for tumor-reactive IgM production in response to treatment. Unexpectedly, naive FcγR−/− mice also showed increased tumor-reactive IgM levels, but in contrast to TLR4−/− mice, responded to MPL/TDCM treatment by producing significant increases in tumor-reactive IgM. It is unclear as to why naïve FcγR−/− mice also have increased tumor-reactive IgM, but it is interesting to note that splenic B1 ASC numbers were increased in the spleens of both strains of naïve mice, suggesting potential dysregulation of the B1 cell compartment. Further studies to investigate this possibility are warranted. Although naïve FcγR−/− mice had higher levels of tumor reactive IgM, this was not sufficient to confer resistance to TA3-Ha challenge in the absence of treatment. Nonetheless, treatment did produce significant protection in FcγR−/− mice, albeit to a lesser extent to WT mice, indicating FcγR-dependent signaling is required for optimal protection elicited by MPL/TDCM treatment. However, it is important to take into consideration that LPM and SPM were much higher in FcγR−/− versus WT mice following treatment. This is perhaps not unexpected as Mincle deletion or knockdown results in significant hyperresponsiveness to LPS in vitro and heightened LPS-mediated inflammation in vivo [40]. Although hyperreponsive macrophages could be hypothesized to provide significantly increased tumor cell-killing in the presence of tumor-reactive IgM, it is also possible that Mincle/MCL signals dampen selective TLR4-induced signals that are counterproductive to the anti-tumor response. Although this work focused primarily on MPL-elicited signals in B1 cells, an understanding of the critical signals elicited by TDCM and MPL on other cells remain to be elucidated. Indeed, whether MPL or TCDM are capable of directly activating recruited monocytes/SPM either prior to, or after trafficking into the cavity hours to days post treatment, remains to be established.
MPL mediates immune responses through activation of the TLR4/MD-2 coreceptor, which can signal via MyD88-dependent and -independent pathways[26]. Interestingly, MyD88−/− mice, although susceptible to TA3-Ha challenge, showed significantly delayed development of ascites and morbidity. MyD88−/− lack most functional TLR signaling as well as IL1 receptor family signaling and thereby have significant pre-existing alterations that may confound conclusions drawn regarding signaling through particular TLR[36, 41]. The fact that antibiotic treatment from birth normalized ascites development in MyD88−/− mice suggests that the delayed ascites development in these mice may have been due to altered bacterial species, load, and/or the inflammation associated with these changes as we hypothesize to occur in TLR4−/− mice.
In contrast to results with MyD88−/− mice, TRIF−/− mice did not show resistance or delayed development of ascites in response to TA3-Ha challenge. Moreover, they were not protected by MPL/TDCM treatment and their B1 cells failed to adequately respond to MPL/TDCM by dividing, differentiating, and producing tumor reactive IgM. E. coli-derived LPS selectively induces the MyD88-dependent signaling pathway while Salmonella minnesota LPS is reported to activate the MyD88-independent pathway[42]. Consistent with this, the immunostimulatory activity of S. minnesota MPL, the detoxified derivative of lipid A, has been shown to be largely driven by the TRIF-pathway[32]. Indeed, inclusion of MPL in peptide-loaded liposomes was previously reported to drive TLR4- and TRIF-dependent, but MyD88-independent stimulation of T cell–independent isotype switching in mice [43]. Another study showed that MPL induces B1 cells to downregulate integrins in the absence of MyD88 expression, which is required for migration out of the peritoneal cavity[44]. Thus, several studies support S. minnesota MPL preferentially stimulates TRIF-dependent signaling. Interestingly, however, in our previous study in which MPL/TDCM was used as an adjuvant to boost T cell-independent Ab responses to pneumococcal polysaccharides, we found that B cell-intrinsic MyD88-, but not TRIF-dependent signaling, was required for optimal adjuvant effects [45]. Taken together, these results suggest it is possible that different B cell subsets (ie., B1a versus B1b/marginal zone B cells) may have distinct TLR4 signaling responses that may be further modulated by signaling via other surface receptors, including the B cell receptor. Indeed, while TLR4−/− mice lacked production of tumor- and PC-reactive IgM in response to MPL/TDCM, TRIF−/−mice produced WT levels of PC-reactive IgM but had significant defects in the production of tumor-reactive IgM. Of note, although TRIF−/− B1 cells did not expand in response to MPL/TDCM in vitro, they downregulated IgM and differentiated into CD138hi ASC, in contrast to TLR4−/− B cells, suggesting that TRIF-independent signals induced by MPL/TDCM support terminal differentiation of a subset of B1 cells.
Currently, it is not clear how tumor-reactive B1 cell development is regulated. Development of B1 cells with reactivities against some self-Ags, such as PC, appears to be independent of flora and exogenous Ags[46], whereas development of others may be dependent on the microbiota [47]. Nonetheless, Ag exposure, microbiota, and cytokines can support expansion and Ab production by these B1 cells[29, 47]. Recent work by Barton and colleagues suggests TLR2 and TLR4 regulate microbiota-reactive B1a Ab responses, whereas Unc93b1-dependent (endosomal) TLRs regulate self reactivities against phosphatidylcholine [36]. Whether B1 cells with TACA-specificities are similarly regulated remains to be determined. Defective tumor-reactive IgM production in TLR4−/− and TRIF−/−mice appears to be due to defective signaling in direct response to MPL/TDCM, although we have not yet addressed whether select tumor-Ag-related specificities are altered or lacking in these mice.
The requirement for TLR4 expression by B1 cells in order to stimulate their anti-tumor functions likely renders this PAMP unsuitable for use as a B1-cell activating therapy in peritoneal carcinomatosis patients, given that human B cells do not express appreciable levels of TLR4[48] and are not responsive to LPS. The only other TLR known to elicit TRIF signaling, TLR3, is also lacking from most human B cells [48]. That said, it is important to note that E. coli-derived LPS, which may alternatively elicit MyD88-dependent Ab secretion, promotes protection against intraperitoneal TA3-Ha challenge[9]. Thus, these foundational studies suggest there may be promise in using other TLR agonists, or other PAMPs for that matter, to activate human B cells to secrete protective tumor-reactive Ab. Our work in NHP clearly shows primates harbor peritoneal B cells with the phenotype and functional characteristics of mouse B1 cells [5, 6] and thereby supports the exciting possibility that human peritoneal B cells could also be leveraged to provide protection against peritoneal cancers.
Acknowledgments
Declarations:
Funding: This work was supported by DOD grant W81XWH-15-1-0585 and NIH NIAID/NIH R01AI18876 awarded to KMH. Shared resources support was provided by NCI-CCSG grant P30CA012197. AMD was supported was in part by National Institutes of Health Training Grant AI007401.
Abbreviations:
- Ab
Antibody
- Abs
Antibodies
- ASC
Antibody Secreting Cell
- CLR
C Type Lectin Receptor
- dBSM
Desialylated Bovine Submaxillary Mucin
- FcRγ
Fc Receptor Common Gamma Chain
- Ig
Immunoglobulin
- LPS
Lipopolysaccharide
- MCL
Macrophage C-type Lectin
- MPL
Monophosphoryl Lipid A
- MyD88
Myeloid Differentiation Factor 88
- SPM
Small Peritoneal Macrophage
- TACA
Tumor Associated Carbohydrate Antigen
- TDCM
Trehalose Dicorynomycolate
- TLR
Toll-like Receptor
- TRIF
TIR domain-containing adaptor-inducing IFN-β
- VAT
Visceral Adipose Tissue
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest: The authors have no conflicts to disclose.
Ethical approval: Animal studies were approved by Wake Forest Animal Care and Use Committee.
References
- 1.Lambert LA, Wiseman J (2018) Palliative Management of Peritoneal Metastases. Ann Surg Oncol. 25: 2165–2171. [DOI] [PubMed] [Google Scholar]
- 2.Cabral FC, Krajewski KM, Kim KW, Ramaiya NH, Jagannathan JP (2013) Peritoneal lymphomatosis: CT and PET/CT findings and how to differentiate between carcinomatosis and sarcomatosis. Cancer Imaging. 13: 162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morano WF, Aggarwal A, Love P, Richard SD, Esquivel J, Bowne WB (2016) Intraperitoneal immunotherapy: historical perspectives and modern therapy. Cancer Gene Ther. 23: 373–381. [DOI] [PubMed] [Google Scholar]
- 4.Capobianco A, Cottone L, Monno A, Manfredi AA, Rovere-Querini P (2017) The peritoneum: healing, immunity, and diseases. J Pathol. 243: 137–147. [DOI] [PubMed] [Google Scholar]
- 5.Haas KM (2015) B-1 lymphocytes in mice and nonhuman primates. Ann N Y Acad Sci 1362: 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yammani RD, Haas KM (2013) Primate B-1 cells generate antigen-specific B cell responses to T cell-independent type 2 antigens. J Immunol. 190: 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffin DO, Holodick NE, Rothstein TL (2011) Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70-. J Exp Med. 208: 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baumgarth N (2011) The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. 11: 34–46. [DOI] [PubMed] [Google Scholar]
- 9.Haro MA, Dyevoich AM, Phipps JP, Haas KM (2019) Activation of B-1 cells promotes potent tumor cell killing in the peritoneal cavity. Cancer Res. 79: 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haro MA, Littrell CA, Yin Z, Huang X, Haas KM (2016) PD-1 Suppresses Development of Humoral Responses That Protect against Tn-Bearing Tumors. Cancer Immunol Res. 4: 1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez-Zhurbenko N, Rabade-Chediak M, Martinez D, Grinan T, Hernandez AM (2015) Anti-NeuGcGM3 reactivity: a possible role of natural antibodies and B-1 cells in tumor immunosurveillance. Ann N Y Acad Sci. 1362: 224–38. [DOI] [PubMed] [Google Scholar]
- 12.Vollmers HP, Brandlein S (2009) Natural antibodies and cancer. N Biotechnol. 25: 294–298. [DOI] [PubMed] [Google Scholar]
- 13.Kieber-Emmons T, Monzavi-Karbassi B, Hutchins LF, Pennisi A, Makhoul I (2017) Harnessing benefit from targeting tumor associated carbohydrate antigens. Hum Vaccin Immunother. 13: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz-Zaragoza M, Hernandez-Avila R, Viedma-Rodriguez R, Arenas-Aranda D, Ostoa-Saloma P (2015) Natural and adaptive IgM antibodies in the recognition of tumor-associated antigens of breast cancer (Review). Oncol Rep. 34: 1106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15., Varambally S, Bar-Dayan Y, Bayry J, Lacroix-Desmazes S, Horn M, Sorel M, Bar-Dayan Y, Ruberti G et al. (2004) Natural human polyreactive IgM induce apoptosis of lymphoid cell lines and human peripheral blood mononuclear cells. Int Immunol. 16: 517–24. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi T, Johnson TD, Nishinaka Y, Morton DL, Irie RF (1999) IgM anti-ganglioside antibodies induced by melanoma cell vaccine correlate with survival of melanoma patients. J Invest Dermatol. 112: 205–209. [DOI] [PubMed] [Google Scholar]
- 17.Hsueh EC, Gupta RK, Qi K, Morton DL (1998) Correlation of specific immune responses with survival in melanoma patients with distant metastases receiving polyvalent melanoma cell vaccine. J Clin Oncol. 16: 2913–2920. [DOI] [PubMed] [Google Scholar]
- 18.Livingston PO, Wong GY, Adluri S, Tao Y, Padavan M, Parente R, Hanlon C, Calves MJ et al. (1994) Improved survival in stage III melanoma patients with GM2 antibodies: a randomized trial of adjuvant vaccination with GM2 ganglioside. J Clin Oncol. 12: 1036–1044. [DOI] [PubMed] [Google Scholar]
- 19.Fremd C, Stefanovic S, Beckhove P, Pritsch M, Lim H, Wallwiener M, Heil J, Golatta M et al. (2016) Mucin 1-specific B cell immune responses and their impact on overall survival in breast cancer patients. Oncoimmunology. 5: e1057387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matlack R, Yeh K, Rosini L, Gonzalez D, Taylor J, Silberman D, Pennello A, Riggs J (2006) Peritoneal macrophages suppress T-cell activation by amino acid catabolism. Immunology. 117: 386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stein SH, Phipps RP (1990) Macrophage-secreted prostaglandin E2 potentiates immune complex-induced B cell unresponsiveness. Eur J Immunol. 20: 403–7. [DOI] [PubMed] [Google Scholar]
- 22.Hamilton MJ, Antignano F, von Rossum A, Boucher JL, Bennewith KL, Krystal G (2010) TLR agonists that induce IFN-beta abrogate resident macrophage suppression of T cells. J Immunol. 185: 4545–53. [DOI] [PubMed] [Google Scholar]
- 23.Goldman N, Valiuskyte K, Londregan J, Swider A, Somerville J, Riggs JE (2017) Macrophage regulation of B cell proliferation. Cell Immunol. 314: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maseda D, Candando KM, Smith SH, Kalampokis I, Weaver CT, Plevy SE, Poe JC, Tedder TF (2013) Peritoneal cavity regulatory B cells (B10 cells) modulate IFN-gamma+CD4+ T cell numbers during colitis development in mice. J Immunol. 191: 2780–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dyevoich AM, Haas KM (2020) Type I IFN, Ly6C+ cells, and phagocytes support suppression of peritoneal carcinomatosis elicited by a TLR and CLR agonist combination. Mol Cancer Ther. In press. [DOI] [PMC free article] [PubMed]
- 26.Barton GM, Kagan JC (2009) A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 9: 535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu X, Nagata M, Yamasaki S (2018) Mincle: 20 years of a versatile sensor of insults. Int Immunol. 30: 233–239. [DOI] [PubMed] [Google Scholar]
- 28.Cain DW, O’Koren EG, Kan MJ, Womble M, Sempowski GD, Hopper K, Gunn MD, Kelsoe G (2013) Identification of a tissue-specific, C/EBPbeta-dependent pathway of differentiation for murine peritoneal macrophages. J Immunol. 191: 4665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKay JT, Haro MA, Daly CA, Yammani RD, Pang B, Swords WE, Haas KM (2017) PD-L2 Regulates B-1 Cell Antibody Production against Phosphorylcholine through an IL-5-Dependent Mechanism. J Immunol. 199: 2020–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savage HP, Klasener K, Smith FL, Luo Z, Reth M, Baumgarth N (2019) TLR induces reorganization of the IgM-BCR complex regulating murine B-1 cell responses to infections. Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haas KM (2011) Programmed cell death 1 suppresses B-1b cell expansion and long-lived IgG production in response to T cell-independent type 2 antigens. J Immunol. 187: 5183–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC (2007) The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 316: 1628–32. [DOI] [PubMed] [Google Scholar]
- 33.Fung PY, Longenecker BM (1991) Specific immunosuppressive activity of epiglycanin, a mucin-like glycoprotein secreted by a murine mammary adenocarcinoma (TA3-HA). Cancer Res. 51: 1170–6. [PubMed] [Google Scholar]
- 34.Van den Eijnden DH, Evans NA, Codington JF, Reinhold V, Silber C, Jeanloz RW (1979) Chemical structure of epiglycanin, the major glycoprotein of the TA3-Ha ascites cell. The carbohydrate chains. J Biol Chem. 254: 12153–9. [PubMed] [Google Scholar]
- 35.Haas KM, Poe JC, Steeber DA, Tedder TF (2005) B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 23: 7–18. [DOI] [PubMed] [Google Scholar]
- 36.Kreuk LS, Koch MA, Slayden LC, Lind NA, Chu S, Savage HP, Kantor AB, Baumgarth N et al. (2019) B cell receptor and Toll-like receptor signaling coordinate to control distinct B-1 responses to both self and the microbiota. Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Genestier L, Taillardet M, Mondiere P, Gheit H, Bella C, Defrance T (2007) TLR agonists selectively promote terminal plasma cell differentiation of B cell subsets specialized in thymus-independent responses. J Immunol. 178: 7779–86. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y, Tung JW, Ghosn EE, Herzenberg LA (2007) Division and differentiation of natural antibody-producing cells in mouse spleen. Proc Natl Acad Sci U S A. 104: 4542–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao L, Chen B, Feng D, Yang T, Li T, Chen J (2019) TLR4 May Be Involved in the Regulation of Colonic Mucosal Microbiota by Vitamin A. Front Microbiol. 10: 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greco SH, Mahmood SK, Vahle AK, Ochi A, Batel J, Deutsch M, Barilla R, Seifert L et al. (2016) Mincle suppresses Toll-like receptor 4 activation. J Leukoc Biol. 100: 185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sivick KE, Arpaia N, Reiner GL, Lee BL, Russell BR, Barton GM (2014) Toll-like receptor-deficient mice reveal how innate immune signaling influences Salmonella virulence strategies. Cell Host Microbe. 15: 203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zughaier SM, Zimmer SM, Datta A, Carlson RW, Stephens DS (2005) Differential induction of the toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect Immun. 73: 2940–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pihlgren M, Silva AB, Madani R, Giriens V, Waeckerle-Men Y, Fettelschoss A, Hickman DT, Lopez-Deber MP et al. (2013) TLR4- and TRIF-dependent stimulation of B lymphocytes by peptide liposomes enables T cell-independent isotype switch in mice. Blood. 121: 85–94. [DOI] [PubMed] [Google Scholar]
- 44.Ha SA, Tsuji M, Suzuki K, Meek B, Yasuda N, Kaisho T, Fagarasan S (2006) Regulation of B1 cell migration by signals through Toll-like receptors. J Exp Med. 203: 2541–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phipps JP, Haas KM (2019) An Adjuvant that Increases Protective Antibody Responses to Polysaccharide Antigens and Enables Recall Responses. J Infect Dis 219: 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sigal NH, Gearhart PJ, Klinman NR (1975) The frequency of phosphorylcholine-specific B cells in conventional and germfree BALB/C mice. J Immunol. 114: 1354–8. [PubMed] [Google Scholar]
- 47.New JS, King RG, Kearney JF (2016) Manipulation of the glycan-specific natural antibody repertoire for immunotherapy. Immunol Rev. 270: 32–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bekeredjian-Ding I, Jego G (2009) Toll-like receptors--sentries in the B-cell response. Immunology. 128: 311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]