

Abstract
Haplotype-resolved or phased genome assembly provides a complete picture of genomes and their complex genetic variations. However, current algorithms for phased assembly either do not generate chromosome-scale phasing or require pedigree information, which limits their application. We present a method named Diploid assembly (DipAsm) that uses long accurate reads and long-range conformation data for single individuals to generate a chromosome-scale phased assembly within a day. Applied to four public human genomes, PGP1, HG002, NA12878 and HG00733, DipAsm produced haplotype-resolved assemblies with contig NG50 up to 25 Mb and phased ~99.5% of heterozygous sites to 98–99% accuracy, outperforming other approaches in terms of both contiguity and phasing completeness. We demonstrate the importance of chromosome-scale phased assemblies for the discovery of structural variants (SVs), including thousands of new transposon insertions, and of highly polymorphic and medically important regions such as HLA and KIR. DipAsm will facilitate high-quality precision medicine and studies of individual haplotype variation and population diversity.
Humans contain two homologous copies of every chromosome and deriving the genome sequence of each copy is essential to correctly understand allele-specific DNA methylation and gene expression, and to analyze evolution, forensics, and genetic diseases1. However, traditional de novo assembly algorithms that reconstruct genome sequences often represent the sample as a haploid genome. For a diploid genome such as the human genome, this collapsed representation results in the loss of half of heterozygous variations in the genome, may introduce assembly errors in regions diverged between haplotypes and may lead to inflated assembly for species with high heterozygosity2. Several algorithms have been proposed to generate haplotype-resolved assemblies, also known as phased assemblies. Early efforts such as FALCON-Unzip3, Supernova4 and our previous work5 use relatively short-range sequence data for phasing and can only resolve haplotypes up to several megabases for human samples. These methods are unable to phase through centromeres or long repeats. FALCON-Phase6, which extends FALCON-Unzip, uses Hi-C to connect phased sequence blocks and can generate longer haplotypes, but it cannot achieve chromosome-long phasing. Trio binning7,8 is the only published method that can and assemble and phase entire chromosomes. It uses sequence reads from both parents to partition the offspring’s long reads and then assemble each partition separately. However, trio binning is unable to resolve regions heterozygous in all three samples in the trio and will leave such regions unphased. More importantly, parental samples are not always available, for example for samples caught in the wild or when parents are deceased. For mendelian diseases, de novo mutations in the offspring won’t be captured and phased with the parents if there are no other heterozygotes nearby. This limits the application of trio binning. Therefore, we currently lack methods that can accurately produce phased assembly for a single individual and keep pace with sequence technology innovations.
To overcome the limitations in the existing methods, we combined recent advances in long-read assembly and Hi-C based phasing to develop DipAsm, which accurately reconstructs the two haplotypes in a diploid individual using only PacBio’s long High-Fidelity (HiFi) reads9 and Hi-C data10, both at ~30-fold coverage, without any pedigree information (Fig. 1). Starting with an unphased Peregrine11 assembly scaffolded by 3D-DNA12 or HiRise13, our pipeline calls small variants with DeepVariant14, phases them with WhatsHap15 and HapCUT216, partitions the reads and assembles each partition independently with Peregrine again (Online Methods). Grouping contigs into chromosome-long scaffolds is necessary for WhatsHap and HiRise to phase entire chromosomes.
Fig 1.
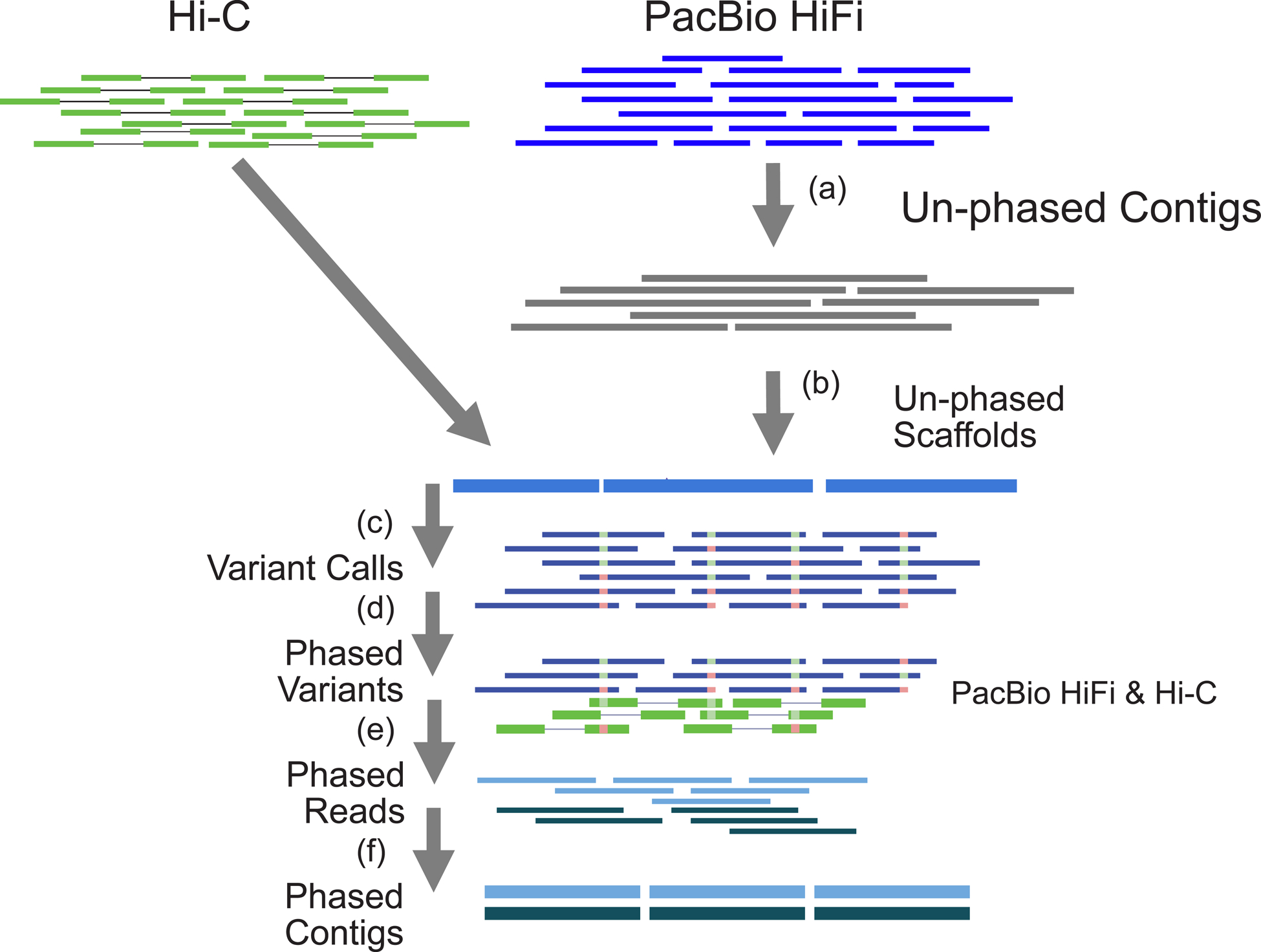
Outline of the phased assembly algorithm: DipAsm. (a) Assemble HiFi reads into unphased contigs using Peregrine. (b) Group and order contigs into scaffolds with Hi-C data using HiRise/3D-DNA. (c) Map HiFi reads to scaffolds and call heterozygous SNPs using DeepVariant. (d) Phase heterozygous SNP calls with both HiFi and Hi-C data using WhatsHap+HapCut2. (e) Partition reads based on their phase using WhatsHap. (f) Assemble partitioned reads into phased contigs using Peregrine.
We demonstrate our method on four human genomes: PGP1 from the Personal Genome Project, HG002 and NA12878 from the Genome In a Bottle dataset17,18 (GIAB) and HG00733 from the HGSVC project19. We produced HiFi data for the PGP1 genome and Hi-C data for HG002 and HG00733 and assembled the samples with DipAsm (Table 1). For HG002, we also generated a trio binning based assembly with Peregrine using parental Illumina reads (“Trio Peregrine” in Table 1), and obtained a published Trio Canu assembly9 for comparison (Table 1). All HG002 assemblies took the same HiFi data as input. For HG00733, we downloaded a FALCON-Phase assembly6 and a recent assembly assembled from HiFi and Strand-seq20. The Strand-seq assembly and our assembly use the same HiFi data. The FALCON-Phase assembly uses noisy Continuous Long Read (CLR) and a different Hi-C dataset.
Table 1.
Assembly statistics. HiFi read N50: 50% of HiFi reads are longer than this number. Contig NG50: minimum contig length needed to cover 50% of the known genome (GRCh38). Contig NGA50: similar to NG50, but based on contig alignment lengths to GRCh38 instead of contig sizes. Phasing switch error rate: percent adjacent SNP pairs are wrongly phased. Phasing hamming error rate: percent SNPs wrongly phased in comparison to true phases.
| Sample | HG002 (NA24385) | NA12878 | PGP1 | HG00733 | ||||
|---|---|---|---|---|---|---|---|---|
| Long-read coverage | 29.7 (HiFi) | 30.1 (HiFi) | 23.9 (HiFi) | 33.4 (HiFi) | 93.0 (CLR) | |||
| Long read N50 (bp) | 13,480 | 10,004 | 12,974 | 11,769 | 33,090 | |||
| Hi-C read coverage | 38.5 | 44.8 | 261.7 | 35.5 | 67.1 | |||
| Assembly algorithm | Trio Canu | Trio Peregrine | DipAsm | DipAsm | DipAsm | DipAsm | Strand-seq | Falcon-Phase |
| Scaffolding | 3D-DNA | HiRise | HiRise | 3D-DNA | ||||
| Paternal / maternal contig size (Gbp) | 2.96 / 3.04 | 2.81 / 2.88 | 2.98 / 2.97 | 2.97 / 2.97 | 2.98 / 2.98 | 2.93 / 2.93 | 2.90 / 2.90 | 2.89 / 2.89 |
| Paternal / maternal contig NG50 (Mbp) | 15.5 / 18.3 | 16.6 / 15.2 | 25.2 / 24.3 | 19.6 / 18.7 | 15.1 / 18.4 | 25.2 / 26.2 | 28.5 / 23.6 | 22.3 / 22.3 |
| Paternal / maternal contig NGA50 (Mbp) | 10.2 / 12.8 | 11.0 / 10.6 | 14.3 / 13.5 | 12.7 / 12.1 | 10.3 / 11.0 | 16.0 / 16.6 | 15.8 / 15.8 | 14.3 / 13.7 |
| Phasing switch / hamming error rate (%) | 0.38 / 0.23 | 0.38 / 0.31 | 0.50 / 0.49 | 0.15 / 2.13 | 0.21 / 1.63 | 0.16 / 0.60 | 0.30 / 0.70 | 0.43 / 35.8 |
| SNP / INDEL false positive rate (×10⁻⁶) | 1.9 / 31.6 | 2.6 / 32.0 | 2.4 / 27.7 | 2.0 / 4.2 | ||||
| SNP / INDEL false negative rate (%) | 4.31 / 5.85 | 3.28 / 5.00 | 0.36 / 2.09 | 0.56 / 1.22 | 3.32 / - | 4.00 / - | 7.89 / - | |
| SV sensitivity / precision (%) | 90.7 / 92.8 | 90.6 / 92.6 | 93.4 / 92.6 | |||||
From sample HG002, we generated a phased de novo assembly of 5.95 gigabases (Gb) in total, including both parental haplotypes. Half of the assembly is contained in contigs of length ~25Mb (i.e. N50), achieving better contiguity than trio binning based assemblies. The scaffold N50 for each parent is >130 Mb. In comparison to GIAB’s SNPs phased by trio, our phasing disagrees only at 0.49% of heterozygous SNPs. This low hamming error rate over the whole genome suggests we have phased almost every chromosome into maternal and paternal haplotypes, and that the switch errors that occur only cause small local errors in phasing of a small fraction of variants.
To evaluate the consensus accuracy of our assembly, we ran the dipcall pipeline21 to align the phased contigs of HG002 against the human reference genome, called SNPs and short insertions and deletions (INDELs) from the alignment and then compared the assembly-based variant calls to the GIAB calls. Out of the 2.36Gb confident regions in GIAB, our de novo assembly yields 5,753 false SNP alleles (0.19% of called SNPs) and 65,302 false INDEL alleles (11.86% of called INDELs). 77% of INDEL errors are 1bp deletions, consistent with the previous observation that 1bp deletion is the major error mode for this dataset9. On the assumption that false positive calls are all consensus errors, not structural assembly errors or contig alignment errors, this gives a per-base error rate of 1.5×10−5 [=(5753+65392)/(2×2.36×109)] or Q48 in the Phred scale. Notably, our de novo assembly achieves a consensus accuracy comparable to the Arrow-polished TrioCanu assembly. This suggests signal-based Arrow polishing may not be necessary for HiFi data.
The comparison to the GIAB truth data also reveals the phasing power. During assembly, failing to partition reads in heterozygous regions leads to the loss of heterozygotes and thus the elevated false negative rate in Table 1. On this metric, our Hi-C based assemblies only miss 0.4% of heterozygous SNPs, ~8 times better than trio binning based assemblies. Trio binning is less powerful potentially because it is unable to phase a heterozygote when all individuals in a trio are heterozygous at the same site. In addition, trio binning breaks short reads into k-mers, which also reduces power in comparison to mapping full-length paired-end Hi-C reads in our pipeline.
The dipcall pipeline outputs phased long INDELs along with small variants. Evaluated against the GIAB SV truth set22 (version 0.6) with Truvari v1.3.2, our de novo assembly based callset shows sensitivity 93.4% and precision 92.6% (Table 1). The sensitivity of trio binning based callsets is ~3% lower, consistent with their lower sensitivity on small variants. Nearly all of the putative false positive calls are low-complexity sequences. We manually inspected some of these false positive calls from the de novo assembly. In many cases, our long INDEL calls are apparent in both HiFi read alignment and contig alignment but they are often split into multiple INDEL calls that sum to the same length as the GIAB call. Current SV benchmarking tools are unable to match SVs between vcf files when SVs are represented as multiple events in the VCF22. Therefore, our precision is likely substantially higher than 92.6% within the GIAB SV benchmark regions.
We additionally ran RepeatMasker23 on SV insertion sequences (9.1 Mb in total length) and discovered that 831, 540, and 2,303 of these are within LINEs, LTRs, and SINEs, respectively. There are 123 microsatellites, 3,582 simple repeats and 270 low-complexity sequences. We also found 21 inversions relative to the reference genome in these HG002 haplotigs (max length 25 kb, average length 5kb). A subset of SVs called from our haplotype assemblies are analyzed in Fig. 2b.
Fig 2:
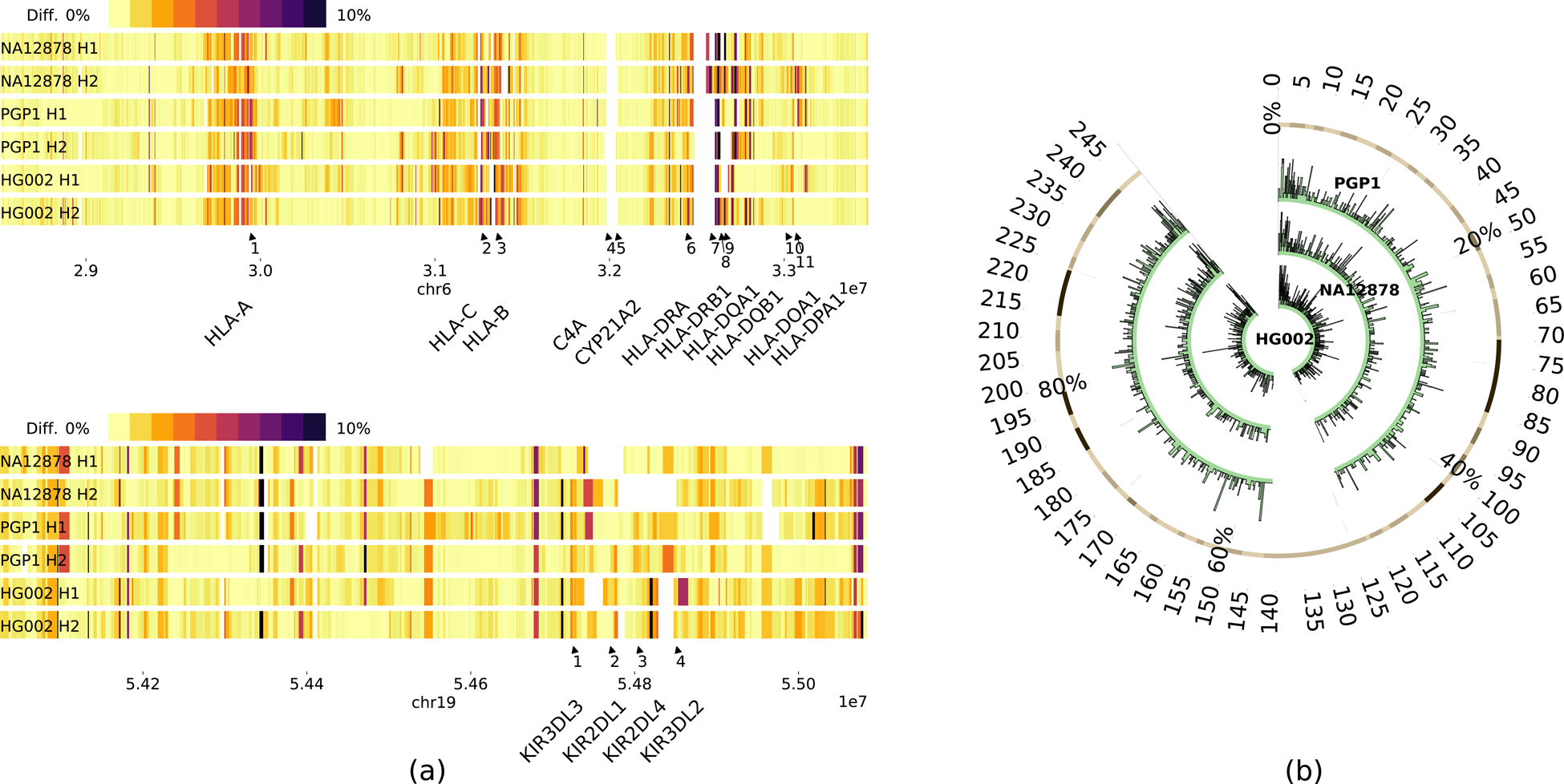
Applications of phased assemblies. (a) Local sequence divergence in comparison to the HLA (top) and the KIR (bottom) regions in GRCh38. (b) SV density per 100kb on chr1 over HG002 (inner), NA12878 (middle) and PGP1 (outer).
Our HG00733 assembly has similar contiguity to the Strand-seq assembly. Evaluated against the phased SNP calls generated by the HGSVC project19, our assembly has slightly lower phasing error rate and phases more heterozygous SNPs. It is worth noting that the HGSVC calls are not curated. Some of the false negatives in the table may be false positives by HGSVC. We also cannot estimate false positive rates as HGSVC does not provide confident regions. Both our and Strand-seq assemblies can phase entire chromosomes. The FALCON-Phase assembly cannot, indicated by the 35.8% hamming error rate. This assembly swaps large blocks of haplotypes between the two phases.
We assembled two other human genomes, NA12878 and PGP1, with DipAsm. We can achieve chromosome-long phasing albeit the shorter read length of NA12878 and the lower read coverage of PGP1. Compared again to GIAB, the NA12878 assembly has even better consensus accuracy, measured at Q55 in GIAB’s confident regions. Notably, the raw HiFi base quality of NA12878 and HG002 are about the same. To understand why NA12878 has better consensus, we counted distinct 31-mers in both assemblies and HiFi reads. We found for NA12878, 3.63% of 31-mers occurring ≥3 times in reads are absent from the assembly, but for HG002, the percentage rises to 6.35%. Given that the completeness of NA12878 and HG002 are about the same, the higher percentage suggests there are more recurrent sequencing errors in HG002, which could explain the lower consensus accuracy of HG002.
The Human Leukocyte Antigen region (HLA) and the Killer-cell Immunoglobulin-like Receptor region (KIR) are among the most polymorphic regions in the human genome. Our phased assemblies can reconstruct most of these regions with two contigs for each haplotype. Based on the pattern of local sequence divergence (Fig. 2a), we can see the two haplotypes in each individual are distinct from one another. Such regions can only be faithfully assembled when we phase through the entire regions.
We present a method to generate a phased assembly for a single human individual or potentially a diploid sample of other species. It accurately produces chromosome-long phasing using only two types of input data: HiFi and Hi-C. In comparison to other published single-sample phased assembly algorithms, ours is the only method capable of chromosome-long phasing. While Strand-seq, in combination with HiFi, has recently been used to phase entire chromosomes as well20, Hi-C is easier to produce and more widely used. In comparison to trio binning, our method is not restricted to samples having pedigree data and can phase de novo mutations. It gives more contiguous assembly and phases a larger fraction of the genome for human samples. Meanwhile, our assembly strategy is not without limitations. First, relying on accurate SNP calls from long reads and using Peregrine for assembly, our pipeline does not work with noisy long reads at present. It is possible to switch to a noisy read assembler and to add Illumina data for SNP calling, but the assembly accuracy may be reduced due to the elevated sequencing error rate. Second, starting with an unphased assembly, we may miss highly heterozygous regions involving long SVs as demonstrated in our previous works on small genomes5,8. A potential solution is to retain heterozygous events in the initial assembly graph and to scaffold and dissect these events later to generate a phased assembly. Nevertheless, our improved de novo method sets a milestone. Its ability to generate phased assemblies without using a reference sequence will enable the unbiased characterization of human genome diversity and construction of a comprehensive human pangenome, which are currently goals of the Human Genome Reference Project. The ability to accurately resolve highly polymorphic regions of biological importance such as MHC and KIR, will further the goals of precision medicine.
Methods
PacBio CCS sequencing for PGP-1.
Library Preparation: Genomic DNA was converted into a SMRTbell™ library as previously described9 but with a few modifications to generate slightly larger inserts. Specifically, genomic DNA was sheared using the MegaruptorR from Diagenode with the 30kb shearing protocol using a long hydropore cartridge. Prior to library preparation, the size distribution of the sheared DNA was characterized on the Agilent Femto Pulse System. A sequencing library was constructed from this sheared genomic DNA using the SMRTbell™ Template Prep Kit v 1.0 (Pacific Biosciences Ref. No. 100-259-100). In order to tighten the size distribution of the SMRTbell™ library, library was size fractionated using SageELF System from Sage Science. Approximately 4μg of SMRTbell™ Library, prepared with loading solution/Marker40. After which, the sample was loaded onto a 0.75% agarose 10kb-40kb gel cassette and size fractionated using a run target size of 7000bp set for elution well 12. A total of 8μg was fractionated on two cassettes. Fractions having the desired size distribution ranges were identified on the Agilent Femto Pulse System. Fractions centered at 11kb were pooled to generate an 11kn library and fractions centered at 16 kb were pooled to create a 16kb library. Both libraries were used for sequencing.
Sequencing: Sequencing reactions were performed on the PacBio Sequel System with the Sequel Sequencing Kit 3.0 chemistry. The samples were pre-extended without exposure to illumination for 12 hours to enable the polymerase enzymes to transition into the highly processive strand-displacing state and sequencing data was collected for 24 hours to ensure maximal yield of high-quality HiFi reads. In addition, sequencing reactions were also performed on the PacBio Sequel II System using the Sequel II Sequencing Kit 1.0 chemistry. On the Sequel II system the data collection was extended to 30 hours to ensure suitable amounts of data.
Hi-C sequencing for HG002 and HG00733.
A Hi-C library was generated on HG002 and HG00733 by Arima Genomics using a modified version of the Arima-HiC kit. Briefly, the current Arima-HiC kit (P/N: A510008) utilizes 2 restriction enzymes for simultaneous chromatin digestion. In the modified protocol, 4 restriction enzymes were deployed to enable more uniform per base coverage of the genome while maintaining the highest long-range contiguity signal, thereby benefiting analyses such as variant discovery, base polishing, scaffolding, and phasing. After the modified chromatin digestion, digested ends were labelled, proximally ligated, and then proximally-ligated DNA was purified. After the modified Arima-HiC protocol, Illumina-compatible sequencing libraries were prepared by first shearing purified Arima-HiC ligation products and then size-selecting DNA fragments using SPRI beads. The size-selected fragments containing ligation junctions were enriched using Enrichment Beads provided in the Arima-HiC kit, and converted into Illumina-compatible sequencing libraries using the Swift Accel-NGS 2S Plus kit (P/N: 21024) reagents. After adapter ligation, DNA was PCR amplified and purified using SPRI beads. The purified DNA underwent standard QC (qPCR and Bioanalyzer) and sequenced on the HiSeq X following manufacturer’s protocols.
Phased sequence assembly.
We ran Peregrine v0.1.5.2 with the following command line: “peregrine asm reads.lst 24 24 24 24 24 24 24 24 24 --with-consensus --shimmer-r 3 --best_n_ovlp 8 --output asm”, where file “reads.lst” gives the list of input read files and directory “asm” holds the output assembly. We mapped Hi-C reads to contigs with BWA-MEM v0.7.17 and scaffolded the Peregrine contigs with juicer v1.5 and 3D-DNA v180922. We preprocessed data with “juicer.sh -d juicer -p chrom.sizes -y cut-sites.txt -z contigs.fa -D”, where file “cut-sites.txt” was generated using the generate_site_positions_Arima.py script which outputs merged_nodups.txt. The scaffolds were produced with “run-asm-pipeline.sh -m haploid contigs.fa merged_nodups.txt”. We then called small variants using DeepVariant v0.8.0 with the pretrained “PACBIO” model. We mapped Hi-C reads to the scaffolds and ran HapCUT2 v1.1 over heterozygous SNP sites to obtain sparse phasing at the chromosome scale. The resulting haplotypes were then combined with PacBio HiFi data using WhatsHap v0.18 with the default parameters to generate fine-scale chromosome-long phasing. We partitioned HiFi reads based on the phases of SNPs residing on these reads, and ran Peregrine again for reads on the same haplotype from the same scaffold. This gives the final phased assembly.
Evaluating variant calling accuracy.
For GIAB samples HG002 and NA12878, we compared small variant calls to GIAB v3.3.2 with RTG’s vcfeval v3.8.4. We extracted allelic errors with the “hapdip.js rtgeval” script from the syndip pipeline21. For sample HG002, we used Truvari v1.3.2 to evaluate long INDEL accuracy against GIAB-SV v0.6. We specified option “--passonly --multimatch” to skip filtered calls in the GIAB VCF and to allow base calls to match multiple comparison calls and vice versa. Increasing evaluation distance from the default 500 to 1000 with “-r 1000” only mildly improves the precision from 92.6% to 93.3%.
Reporting Summary
Further information on research design is available in the Life Sciences Reporting Summary in this article.
Data availability.
HG002 HiFi reads and the 250bp parental short reads were acquired from the GIAB ftp site ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/. HG002 Hi-C reads (AC:SRR11016318), HG00733 Hi-C (AC:SRR11347815) and PGP1 HiFi reads (AC:SRR11016319) sequenced by us were deposited to SRA. NA12878 HiFi (AC:SRX5780566) and Hi-C reads (AC:SRR6675327), and PGP1 Hi-C reads26 (AC:SRP173234) were downloaded from SRA. The HG00733 Falcon-Phase assembly was obtained from NCBI (AC:GCA_003634875.1). Other assemblies and assembly-based variant calls used in this work are publicly available at ftp://ftp.dfci.harvard.edu/pub/hli/whdenovo/. HG00733 phased SNP calls were downloaded from ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20160704_whatshap_strandseq_10X_phased_SNPs/PUR/.
Code availability.
The complete pipeline is available at https://github.com/shilpagarg/DipAsm
Acknowledgements.
We are grateful to S. Koren and A. Phillippy for providing the Arrow-polished TrioCanu assembly of HG002. We thank A. English for suggesting appropriate Truvari parameters, and thank C-Z. Zhang for discussions at an early stage of this work. The authors thank Olga Dudchenko for insightful discussions. This study was supported by US National Institutes of Health (grant R01HG010040 and U01HG010971 to H.L., K99HG010906 to S.G., RM1HG008525 to G.M.C. and J.A. and UM1HG008898 to F.J.S.).
Footnotes
Competing interests. F.J.S. obtained a Pacbio SMRT grant in 2019 and had multiple travels sponsored by Pacific Biosciences and Oxford Nanopore Technologies. E.H. and P.P. are employees of Pacific Biosciences. C-S.C. and A.F. are employees of DNAnexus. A.S., X.Z. and S.M. are employees of Arima Genomics. J.G. and J.M. are employees of Dovetail Genomics. A.C. is an employee of Google. H.L. is a consultant of Integrated DNA Technologies, Inc and on the Scientific Advisory Boards of Sentieon, Inc, BGI and OrigiMed. G.M.C. is a co-founder of Editas Medicine and has other financial interests listed at arep.med.harvard.edu/gmc/tech.html.
References
- 1.Tewhey R, Bansal V, Torkamani A, Topol EJ & Schork NJ The importance of phase information for human genomics. Nat. Rev. Genet 12, 215–223 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vinson JP et al. Assembly of polymorphic genomes: algorithms and application to Ciona savignyi. Genome Res. 15, 1127–1135 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chin C-S et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weisenfeld NI, Kumar V, Shah P, Church DM & Jaffe DB Direct determination of diploid genome sequences. Genome Res. 27, 757–767 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garg S et al. A graph-based approach to diploid genome assembly. Bioinformatics 34, i105–i114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kronenberg ZN et al. Extended haplotype phasing of de novo genome assemblies with FALCON-Phase. bioRxiv 327064 (2018) doi: 10.1101/327064. [DOI] [Google Scholar]
- 7.Koren S et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol (2018) doi: 10.1038/nbt.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garg S et al. A haplotype-aware de novo assembly of related individuals using pedigree sequence graph. Bioinformatics (2019) doi: 10.1093/bioinformatics/btz942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burton JN et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol 31, 1119–1125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin C-S & Khalak A Human Genome Assembly in 100 Minutes. bioRxiv 705616 (2019) doi: 10.1101/705616. [DOI] [Google Scholar]
- 12.Dudchenko O et al. De novo assembly of the genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Putnam NH et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poplin R et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol 36, 983–987 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Martin M et al. WhatsHap: fast and accurate read-based phasing. bioRxiv 085050 (2016) doi: 10.1101/085050. [DOI] [Google Scholar]
- 16.Edge P, Bafna V & Bansal V HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res. 27, 801–812 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zook JM et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol 32, 246–251 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Zook JM et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol 37, 561–566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10, 1784 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porubsky D et al. A fully phased accurate assembly of an individual human genome. bioRxiv 855049 (2019) doi: 10.1101/855049. [DOI] [Google Scholar]
- 21.Li H et al. A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat. Methods 15, 595–597 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zook JM et al. A robust benchmark for germline structural variant detection. bioRxiv 664623 (2019) doi: 10.1101/664623. [DOI] [Google Scholar]
- 23.Smit AFA and Hubley R and Green P. RepeatMasker Open-4.0 http://www.repeatmasker.org (2015).
- 24.Nir G et al. Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet. 14, e1007872 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
HG002 HiFi reads and the 250bp parental short reads were acquired from the GIAB ftp site ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/. HG002 Hi-C reads (AC:SRR11016318), HG00733 Hi-C (AC:SRR11347815) and PGP1 HiFi reads (AC:SRR11016319) sequenced by us were deposited to SRA. NA12878 HiFi (AC:SRX5780566) and Hi-C reads (AC:SRR6675327), and PGP1 Hi-C reads26 (AC:SRP173234) were downloaded from SRA. The HG00733 Falcon-Phase assembly was obtained from NCBI (AC:GCA_003634875.1). Other assemblies and assembly-based variant calls used in this work are publicly available at ftp://ftp.dfci.harvard.edu/pub/hli/whdenovo/. HG00733 phased SNP calls were downloaded from ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20160704_whatshap_strandseq_10X_phased_SNPs/PUR/.