

Abstract
Interactions between chromatin-associated proteins and the histone landscape play major roles in dictating genome topology and gene expression. Cancer-specific fusion oncoproteins which display unique chromatin localization patterns often lack classical DNA-binding domains, presenting challenges in identifying mechanisms governing their site-specific chromatin targeting and function. Here we identify a minimal region of the human SS18-SSX fusion oncoprotein, the hallmark driver of synovial sarcoma (SS), that mediates a direct interaction between the mSWI/SNF complex and the nucleosome acidic patch. This binding results in altered mSWI/SNF composition and nucleosome engagement, driving cancer-specific mSWI/SNF complex targeting and gene expression. Further, the C-terminal region of SSX confers preferential affinity to repressed, H2AK119Ub-marked nucleosomes, underlying the selective targeting to polycomb-marked genomic regions and SS-specific dependency on PRC1 function. Together, our results describe a functional interplay between a key nucleosome binding hub and a histone modification that underlies the disease-specific recruitment of a major chromatin remodeling complex.
Introduction
A synchronous combination of histone reader domains, chromatin complex conformations, DNA-binding transcription factors (TFs), and other features are required to orchestrate the appropriate targeting of chromatin regulatory machinery in eukaryotic cells. Chromatin reader proteins play critical roles in mediating the engagement of regulatory proteins and protein complexes to specific features of nucleosomal architecture, often to facilitate site-specific catalytic activities. These include bromodomains which recognize acetylated lysines 1, PHD domains which recognize methylation and crotonylatation of histone tails 2,3, and increasingly appreciated, nucleosome acidic patch interacting domains of SNF2 helicase-based chromatin remodeling complexes 4–6. In parallel, TFs recognize their cognate DNA motifs genome-wide, and, when tethered to other proteins or protein complexes, such as chromatin remodeling complexes, can direct their global positioning on chromatin to achieve cell-, tissue- and cancer-specific gene expression programs. For example, TFs have been shown to tether transiently to the surfaces of mammalian SWI/SNF (BAF) ATP-dependent chromatin remodeling complexes to globally reposition them to sites enriched for specific TF DNA-binding motifs 7,8. Importantly, results of recent large-scale human genetic sequencing studies indicate that perturbations across each of the above classes of chromatin-bound factors represent frequent and recurrent events in human cancer 9–11, intellectual disability 12, and other disorders, with mutations ranging from point mutations and deletions to fusion proteins which alter target engagement and activity of chromatin regulatory complexes on the genome13–15.
It has remained elusive, however, how nuclear fusion oncoproteins that lack canonical TF DNA-binding or recognizable chromatin reader domains yield altered, region-specific targeting of chromatin regulatory proteins and protein complexes. For example, the SS18-SSX fusion oncoprotein involving the BAF complex subunit, SS18, and 78 amino acids of one of the SSX proteins normally expressed only in testes 16–19, is hallmark to 100% of cases of synovial sarcoma. Incorporation of SS18-SSX in to BAF complexes causes biochemical changes, such as the destabilization of the SMARCB1 (BAF47) subunit, and results in de novo BAF complex targeting to a highly cancer-specific set of sites, particularly, broad, polycomb-repressed regions at which polycomb complex occupancy is reduced and gene expression is activated 14. Although some studies have suggested SSX interactions with chromatin-associated factors20, the mechanism by which the site-specific binding and unique biochemical properties are achieved remains largely unknown.
Here we elucidate the mechanism by which the SS18-SSX oncogenic fusion protein engages with chromatin and directs BAF chromatin remodeling complexes to specialized target sites. Specifically, we find that SSX contains a basic region that directly binds the nucleosome acidic patch, altering BAF complex subunit configuration and activity. Further, SSX-nucleosome binding is augmented by the presence of ubiquitylated H2A (H2A K119Ub) on nucleosomes, preferential recognition of which requires a second, conserved region of SSX. These dual reader-like features of SSX underlie the highly disease-specific, hallmark chromatin remodeling complex targeting, gene expression, and functional dependencies in synovial sarcoma. Collectively, our studies reveal a novel mechanism of chromatin localization with important biological and disease implications.
Results
SS18-SSX-bound BAF complexes bind chromatin with uniquely high affinity
Recent studies have indicated that SS18-SSX-bound BAF complexes have specialized biochemical and chromatin localization properties 14,15. We sought to explore the underlying molecular recognition mechanisms driving these associations and activities. To this end, we expressed HA-tagged versions of either wild-type (WT) SS18 or SS18-SSX in HEK-293T cells and performed BAF complex purifications from soluble nuclear extract (NE) and nuclease-treated solubilized chromatin (CHR) (Fig. 1a). Strikingly, fusion oncoprotein SS18-SSX-bound BAF complexes preferentially eluted in the CHR material, in contrast to WT complexes, which eluted nearly completely in the soluble NE material, as expected from previous studies examining WT (and other loss-of-function mutant variants of) BAF complexes 9,21. Importantly, SS18-SSX-bound complexes captured near-stoichiometric amounts of core histone proteins H2A, H2B, H3, and H4 (Fig. 1a). We next subjected these complexes to mass-spectrometric (MS) analyses and found selective co-enrichment of histone peptides with HA-SS18-SSX, but not with HA-WT SS18 in the chromatin-bound fractions (Fig. 1b, Extended Data Fig. 1a–b). Notably, we captured peptides corresponding to the H2A K119Ub mark only in the purifications of SS18-SSX-bound complexes but not in SS18 WT complexes, in agreement with the visualization of this mark upon colloidal blue staining (Fig. 1a, Supplementary Table 1). SS18-SSX purifications most substantially enriched for ATPase subunits SMARCA4 and SMARCA2, BCL7A, ACTL6A, and beta-actin, consistent with the fact that SS18 is part of the ATPase module of mSWI/SNF complexes 21, while core module components, particularly SMARCB1 were less enriched compared to WT SS18 purifications (Fig. 1b, Extended Data Fig. 1c–d Supplementary Table 1). We did not detect binding to PRC1 components, as has been previously suggested 20 (Extended Data Fig. 1b).
Figure 1. SS18-SSX-containing BAF complexes exhibit increased affinity for chromatin.
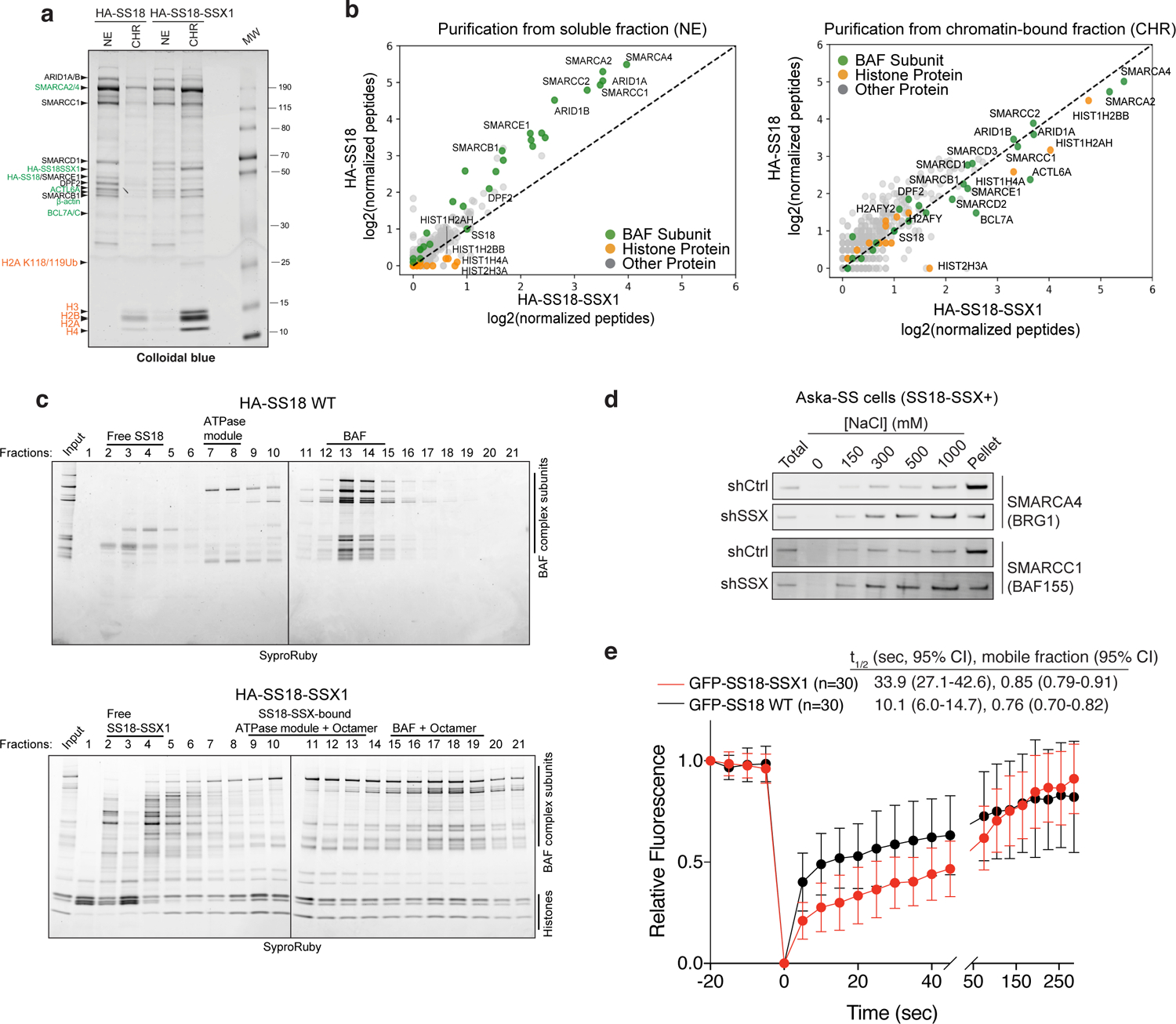
a, Colloidal blue staining of wild-type BAF complexes (from HA-SS18 WT-expressing 293T cells) and SS18-SSX-contaning BAF complexes (from HA-SS18-SSX1-expressing cells), purified from soluble nuclear extract (NE) and chromatin-bound (CHR) fractions. Equal amounts (by volume) of nuclei were fractionated as described in Methods. BAF complex subunits and histone proteins are identified at left.
b, MS spectral counts for BAF complex subunits (green) and histone proteins (orange) from HA-SS18 WT and HA-SS18-SSX purifications in (a). Peptide counts are log2 normalized to bait (SS18 peptides).
c, SDS-PAGE of 10–30% glycerol gradient sedimentation performed on purified HA-SS18 WT and HA-SS18-SSX1 fractions from HEK-293T cells. BAF complex subunits and histone proteins are indicated. SyproRuby staining was used for visualization.
d, Immunoblot of SMARCA4 and SMARCC1 performed on Aska SS cells in shCtrl (control, non-targeting hairpin shRNA) and shSSX (shRNA targeted to SSX) conditions following differential salt extraction (0–1000 mM NaCl).
e, FRAP studies performed on HEK293T cells expressing either GFP-SS18 WT or GFP-SS18-SSX1. Recovery kinetics were recorded and the recovery half-times were 10.1 and 33.9 seconds for GFP-SS18 WT and GFP-SS18-SSX1, respectively. Values represent mean ± s.d. of n=30 cells per condition collected from two biological replicates. t1/2, 95% CI and mobile fraction values were determined by fitting a curve to the data using nonlinear regression. Data for panels a and b are presented in Supplementary Table 1. Uncropped gel images for all panels a, c and d are presented in Supplementary Figure 1. Raw binding data for panels e,h are presented in Source Data.
Purification of SS18-SSX-bound complexes followed by density sedimentation using 10–30% glycerol gradients revealed larger-sized fusion-containing BAF complexes migrating in fractions 15–19, compared to WT SS18-bound complexes in fractions 13–14, as expected 21, suggesting high-affinity, stable binding of SS18-SSX-bound BAF complexes to the full histone octamer (Fig. 1c, Extended Data Fig. 1e). In addition, we observed histones bound to the ATPase module components in isolation as well as to free SS18-SSX in fractions 9–13 and 2–4, respectively. These results suggest that fusion-containing BAF complexes bind nucleosomes more strongly than WT SS18-containing BAF complexes which did not copurify with nucleosomes in ultracentrifugation experiments. Finally, to determine the relative chromatin affinities of WT BAF complexes versus SS18-SSX-containing BAF complexes, we performed differential salt extraction in both SS cell lines and HEK-293T cells expressing SS18-SSX (Fig. 1d, Extended Data Fig. 1f–g). We observed normal extraction profiles for WT complexes (elution at 300–500 mM NaCl), consistent with previous findings 22,23however, fusion-containing complexes remained insoluble in up to 1M NaCl. In support of these findings, fluorescence recovery after photobleaching (FRAP) experiments in HEK-293T cells infected with either GFP-SS18 or GFP-SS18-SSX revealed substantially increased chromatin residency times for SS18-SSX-bound BAF complexes (Fig. 1e, Extended Data Fig. 1h). Taken together, these findings indicate an unexpected, uniquely high-affinity conjugation of SS18-SSX-bound BAF complexes to nucleosomes, a property specific to this disease-associated BAF complex perturbation, suggesting this as a feature that may underlie the site-specific targeting of SS18-SSX complexes on chromatin.
A minimal 34-aa region of SSX is necessary and sufficient for direct binding to repressive nucleosomes and SS18-SSX-mediated oncogenic functions
We next sought to determine whether the 78 residues of SSX in isolation (not fused to the SS18 subunit and hence not part of BAF complexes) could directly bind nucleosomes and could be responsible for conferring the unique affinity and nucleosome binding properties of the SS18-SSX fusion protein. Indeed, pull-down experiments revealed that the C-terminal 78 residues of SSX (aa 111–188) were sufficient for its nucleosomal interactions (Fig. 2a, Extended Data Fig. 2a–b). In addition, we found that binding to mammalian nucleosomes (purified via MNase digestion of HEK-293T cell chromatin and hence representing the diverse array of histone variants and modifications) was stronger than binding to recombinant, unmodified nucleosomes (Extended Data Fig. 2c–d), indicating that a mammalian histone modification might provide added affinity and site specificity. In agreement with this, targeted quantitative mass-spectrometry (MS) analysis of SSX-bound mammalian nucleosomes (pooled, purified by MNase digestion from HEK-293T cells, containing the full diversity of histone marks) revealed strong enrichment of nucleosomes decorated with known repressive histone marks and depletion of nucleosomes marked with known activation marks (Fig. 2b, Extended Data Fig. 2e–h, Supplementary Table 2). For example, we detected SSX-mediated enrichment of nucleosomes decorated with repressive marks such as H3K27me3 and H3K9me3, and SSX-mediated depletion of nucleosomes decorated with activating marks such as H4 lysine acetylation and H3K4me2/3 (while nucleosomes containing unmodified H4 and H3 were enriched). Further, immunofluorescence (IF) analyses revealed strong colocalization of SS18-SSX as well as SSX in isolation (SSX aa 1–188, as expressed in testes) to Barr bodies marked with repressive PRC1 and PRC2 complexes and their marks (Fig. 2c, Extended Data Fig. 3a–b).
Figure 2. Conserved basic and acidic regions within a minimal SSX domain are necessary and sufficient for nucleosome binding and BAF complex recruitment and activity.
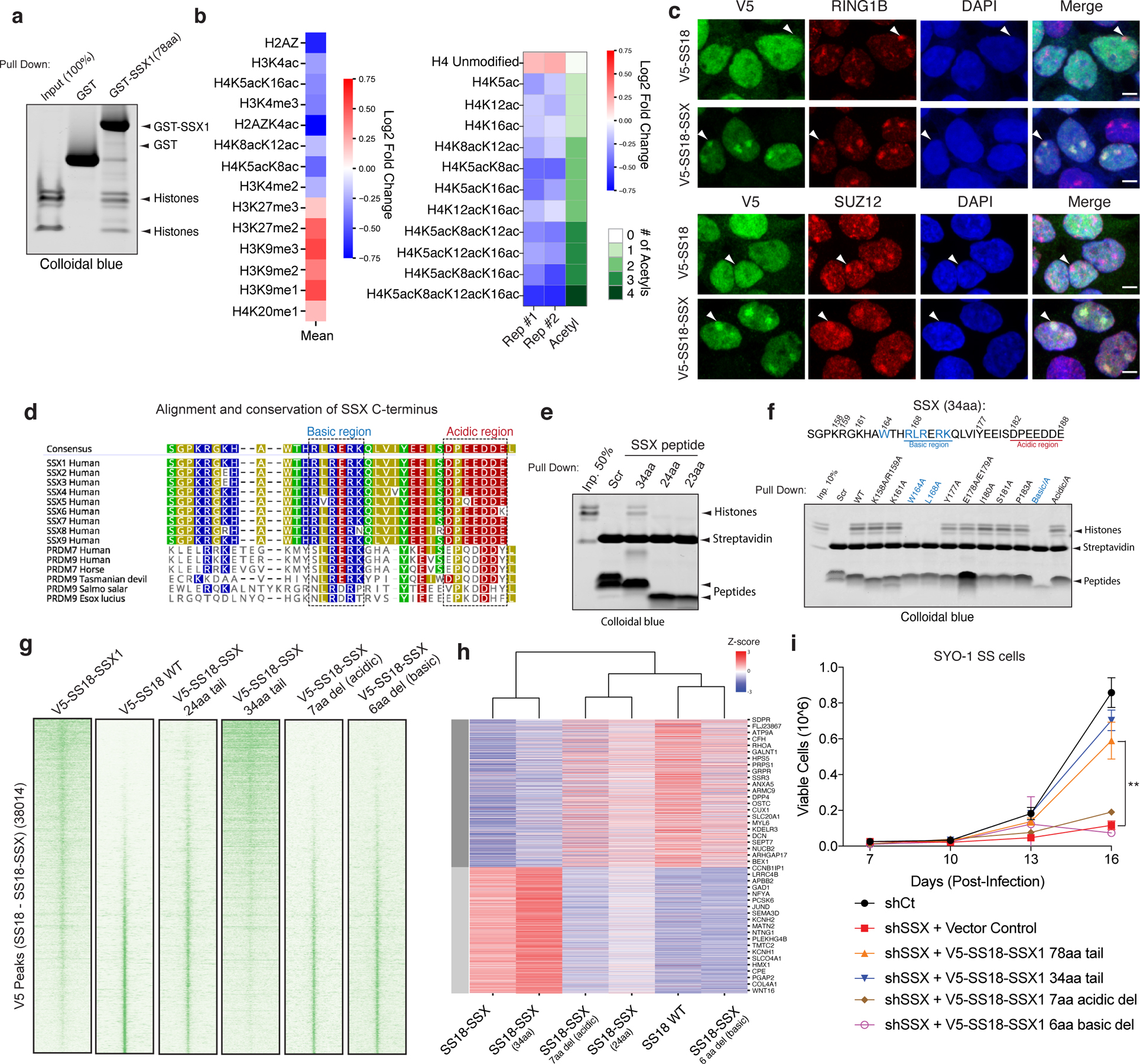
a, GST (control) and GST-SSX1 (78aa) purified recombinant proteins incubated with mammalian mononucleosomes (purified by MNase digestion) were captured using glutathione resin and visualized using colloidal blue.
b, Quantitative MS analysis of MBP pull-down experiments using the MBP-SSX 78aa protein and endogenous mammalian nucleosomes purified using MNase digestion from 293T cells. Log2 (FC) calculated relative to input sample. Red, enriched; blue, depleted. Raw data are presented in Supplementary Table 2.
c, IF analysis of V5-tagged SS18 and SS18-SSX relative to RING1B and SUZ12 in 293T cells. Arrows indicate Barr bodies. Scale bar=5μm.
d, SSX1 protein sequence alignment across species and compared to related PRDM7, PRDM9 proteins. Highly conserved basic (blue) and acidic (red) regions are indicated.
e, Pull-down experiments with biotinylated SSX peptides (scrambled (aa155–188), SSX 34aa (aa155–188), SSX 24aa (aa164–188) and SSX 23aa (aa165–188) incubated with mammalian mononucleosomes and visualized with colloidal blue.
f, Pull-down experiments of N-terminally biotinylated SSX peptides including scrambled control, WT and mutant variants (single alanine substitutions as well as regional substitutions (i.e. Basic-A, basic region RLRERK-->AAAAAA; Acidic-A, acidic region DPEEDDE-->AAAAAAA) incubated with mammalian mononucleosomes, visualized with colloidal blue.
g, ChIP-seq density heatmaps reflecting chromatin occupancy of V5-SS18-SSX1, V5-SS18, V5-SS18-SSX (24aa) and V5-SS18-SSX (34aa) over all V5 Peaks (38,014 total peaks).
h, Heatmap reflecting top 5% upregulated and downregulated genes (Z-score) by RNA-seq for each condition.
i, Proliferation of SYO-1 SS cells infected with either control hairpin (shCt) or shSSX with overexpression of empty vector control, SS18-SSX 78aa or SS18-SSX 34aa variants. Error bars represent mean ± s.d. of n=3 independent experimental replicates; ** p<0.01 determined from a two-tailed t-test. Raw binding data is presented in Source Data. Uncropped gel images for panels a, e and f are presented in Supplementary Figure 1.
SSX-like protein sequences are only found in mammalian SSX family proteins (human SSX1–9) and members of the vertebrate-specific PRDM7 and PRDM9 methyltransferases. We identified a 34aa region of SSX (SSX aa155–188) that is highly conserved across vertebrate species of SSX (putative PFAM SSXRD domain) and is similar to that of PRDM7 and PRDM9 proteins (Fig. 2d). Pull-down experiments using biotinylated peptides corresponding to this region indicated it was sufficient for SSX nucleosome binding, while shorter 23- (SSX aa166–188) and 24- (SSX aa165–188) residue peptides (lacking the W164 residue) failed to do so (Fig. 2e). This SSX-nucleosome interaction was specific as biotin pulldowns were outcompeted by addition of unlabeled SSX 34-residue peptide and could not be competed by a scrambled control peptide corresponding to the same SSX 34aa region (Extended Data Fig. 3c–d).
To define whether SSX 34-residue peptide could be used as a probe for repressive Barr bodies or polycomb bodies in cells, we implemented a peptide hybridization approach performed on methanol-fixed (non-crosslinked) IMR90 fibroblasts incubated with biotinylated SSX peptides and subsequently co-stained with the Barr body marker H2A K119Ub. We observed clear labeling of Barr bodies suggesting an innate ability of the SSX 34 residue region to selectively localize to repressed chromatin regions (Extended Data Fig. 3e). 34aa regions corresponding to most human SSX proteins exhibited interactions with nucleosomes, while shorter SSX-like sequences found in in PRDM7 or PRDM9 proteins lacking the W164 and first R residues of the basic region (R167) failed to do so, suggesting a newly evolved, mammalian-specific function of this full protein region (Extended Data Fig. 3f–g). Finally, to identify residues important for nucleosome binding, we designed a library of 34-residue SSX peptides containing alanine substitutions in either single conserved residues or alanine substitutions across the full basic and acidic regions. Importantly, these experiments revealed that the core residues of the 6-aa basic region of the SSX (RLRERK) were required, as single residue and full region alanine substitutions in this region completely abrogated SSX-nucleosome binding (Fig. 2f).
To determine whether these minimal regions were sufficient for the genome-wide targeting of fully-formed, endogenous SS18-SSX-containing BAF complexes in cells, we expressed either WT SS18, SS18-SSX, or SS18 fused to a range of mutant SSX variants for lentiviral infection in to CRL7250 human fibroblasts. ChIP-seq experiments revealed that the 34aa SSX tail fused to SS18 was sufficient to achieve SS18-SSX targeting, while the 24aa fusion was unable to do so (Fig. 2g, Extended Data Fig. 4a). Notably, deletion of either the basic or the acidic conserved regions of SSX resulted in complete loss of oncogenic fusion complex targeting, suggesting that both of these regions are required for SS18-SSX-specific properties. These findings were consistent with biochemical results suggesting that the full 34aa tail is needed to confer tight affinity of SS18-SSX to chromatin in cells (Extended Data Fig. 4b). Importantly, these changes in chromatin targeting resulted in corresponding changes in gene expression by RNA-seq, as evidenced by clustering of the transcriptional profiles of the 34-residue tail fusion with the full SS18-SSX fusion (78-aa fusion tail), while deletion of either basic or acidic conserved regions or 24aa SSX tail variants clustered with SS18 WT gene expression profiles (Fig. 2h). These findings were further corroborated using IF for SS18-SSX Barr body localization (Extended Data Fig. 4c) as well as beta-galactosidase senescence assays in IMR90 fibroblasts performed across SS18-SSX and SSX (alone) variants (Extended Data Fig. 4d). Finally, both SS18-SSX −78aa and −34aa minimal fusions rescued proliferation in synovial sarcoma cell lines that are well-established to be dependent on the function of SS18-SSX and bearing shRNA-mediated KD of the endogenous SS18-SSX fusion. Taken together, these data suggest that the 34aa minimal region of SSX that contains the conserved basic and acidic regions, is responsible for the maintenance of oncogenic gene expression and proliferation in SS cell lines driven by the SS18-SSX fusion oncoprotein (Fig. 2i, Extended Data Fig. 4e).
An RLR motif within the SSX basic region competes with SMARCB1 for nucleosome acidic patch binding
Using systematic mutagenesis on the SSX 34-residue region, we found that single residue perturbations to the basic region, which includes a Kaposi’s sarcoma-associated herpesvirus (KSHV) LANA-like RLR motif, resulted in complete loss of nucleosome binding (Fig. 3a). These data suggested that this highly basic region binds directly to the H2A-H2B acidic patch of the nucleosome. To identify the specific sites involved in acidic patch engagement, we introduced reactive diazirine probes at various residues within the nucleosome acidic patch and performed photocrosslinking studies 6 with SSX 34-residue peptides (Fig. 3b, Extended Data Fig. 5a–b). Histone-SSX crosslinks were identified at several positions across the extended acidic patch region, most prominently at positions H2A E56 and H2B E113, which importantly, were substantially reduced when key RLR basic residues in SSX were mutated (Fig. 3b–c). To probe this further, we assembled nucleosomes containing H2A mutant variants D90N, E92K, and E113K which disrupt the integrity of the acidic patch for GST-SSX pull down experiments. These experiments showed near complete loss of SSX binding to acidic patch-mutant nucleosomes, indicating the importance of this highly conserved and important docking site for the SSX-chromatin interaction (Fig. 3d (homotypic), Extended Data Fig. 5c (heterotypic)). These results were further corroborated by the fact that we observed direct nucleosome binding competition between LANA peptide and SSX (Fig. 3e, Extended Data Fig. 5d–e ), as the LANA peptide is well-established to bind the nucleosome acidic patch 24. In cells, single-residue mutations within the nucleosome acidic patch binding region of SSX (SSX R169A as well as W164A) resulted in attenuation of SS18-SSX-specific BAF complex chromatin occupancy, recruitment to Barr bodies, gene expression activation, and proliferative maintenance in SS cell lines (Fig. 3f, Extended Data Fig. 6a–g). Taken together, these data establish the role for the basic region, specifically the RLR motif, in mediating SS18-SSX-nucleosome binding, in conferring SS18-SSX-containing BAF complex chromatin binding properties, as well as function in gene expression and proliferative maintenance.
Figure 3. The SSX basic region outcompetes the SMARCB1 C-terminal alpha-helical domain for nucleosome acidic patch binding.
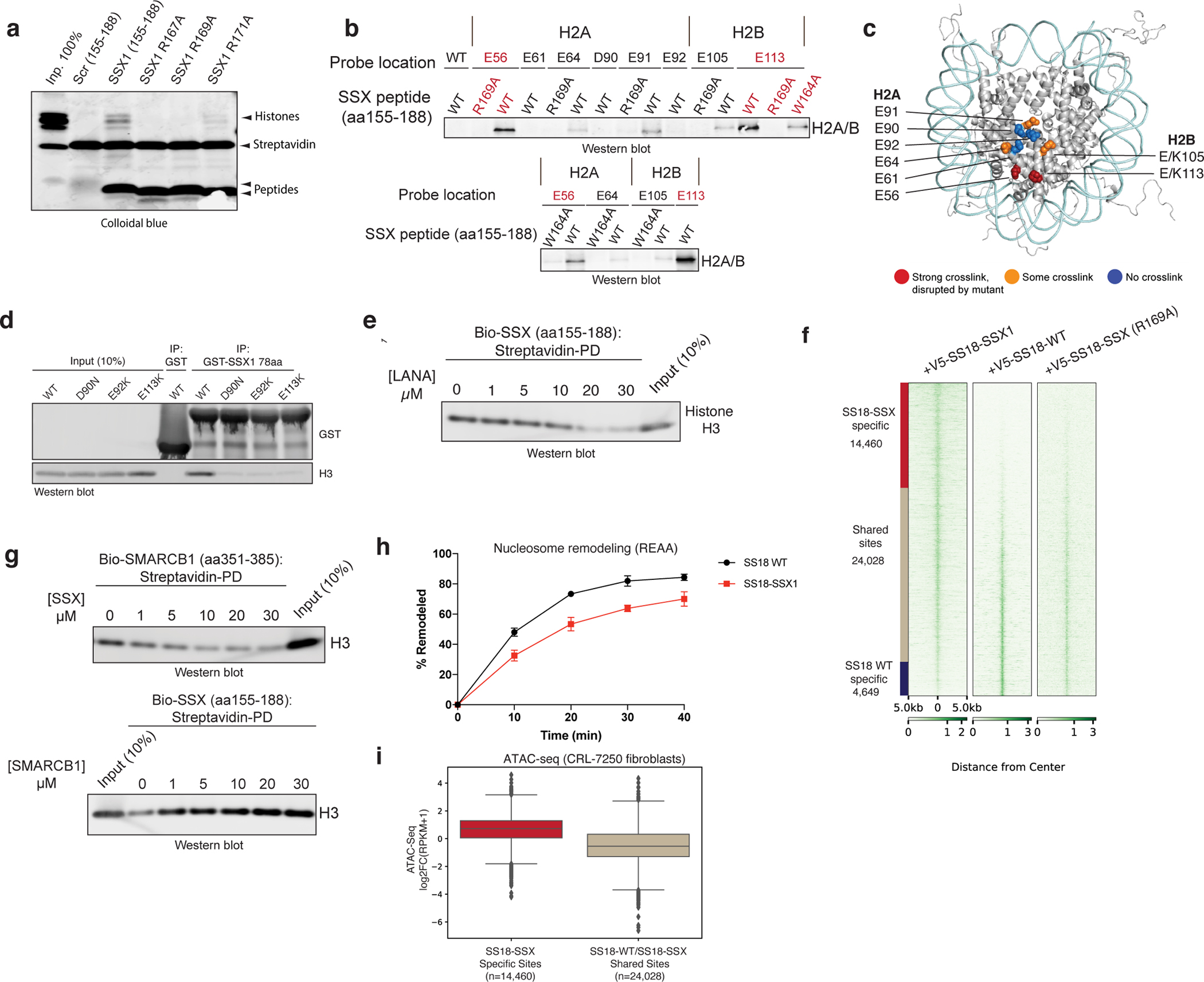
a, Incubation of biotinylated SSX peptides (aa 155–188) in either WT or RLR motif-mutant forms (R167A, R169A, R171A) with nucleosomes. Proteins are visualized by Colloidal blue staining.
b, Western blots of photocrosslinking assays performed with reactive diazirine probes localized to the indicated nucleosome acidic patch residues. Red labels indicate strongest binding to H2A E56 and H2B E113..
c, SSX binding sites mapped on nucleosome structure [PDB: 1KX5]. Acidic patch crosslinked sites are labeled.
d, Pull-down assays of GST-SSX 78aa tail with either WT or the indicated acidic patch mutant nucleosomes (D90N, E92K, and E113K). Binding is visualized by histone H3 immunoblotting.
e, LANA peptide competition experiment with SSX 34aa biotinylated peptide bound to nucleosomes.
f, V5 ChIP-seq heat map reflecting genome-wide localization of V5-tagged SS18-SSX, SS18 WT and SS18-SSX RLR-->RLA (R169A) mutant in CRL7250 fibroblasts.
g, Reciprocal competition experiments performed with either SMARCB1 C-terminal alpha helical domain bound to nucleosomes or SSX 34aa bound to nucleosomes and competed with indicated peptide. Binding is visualized by histone H3 immunoblotting.
h, REAA nucleosome remodeling assay performed with BAF complexes containing either WT SS18 or SS18-SSX. A 0–40 min time course was performed at 37°C; BAF complex capture performed using ARID1A IP. Error bars represent mean ± s.d. of n=3 experimental replicates.
i, Box and whisker plot depicting ATAC-seq DNA accessibility (log2FC(RPKM+1) performed in CRL7250 fibroblasts over SS18-SSX-specific sites and SS18 WT-SS18-SSX shared sites, defined in Fig. 3f. Boxes represent the interquartile range (IQR); horizontal bar represents median; minima and maxima shown extend 1.5 × IQR from the box. Raw data are presented in Source Data. Uncropped gel images for panels a, d, e, g are presented in Supplementary Figure 1.
We previously demonstrated that upon SS18-SSX expression and incorporation in to BAF complexes, the SMARCB1 (BAF47) subunit of BAF complexes, part of the core module21, is destabilized and proteasomally degraded 15 (see also Extended Data Fig. 7a,b, Supplementary Table 3). Intriguingly, using pull down competition assays, we found that SSX competed with the recently-identified SMARCB1 C-terminal alpha helix (aa351–385) region 25,26 for nucleosome acidic patch binding (Fig. 3g) 25,26. However, the reverse was not true as the SMARCB1 C-term alpha helix was unable to outcompete SSX from binding the nucleosomes, implicating stronger affinity of SSX compared to SMARCB1 C-term for nucleosomes. This result, coupled with the positioning of SS18 at the very N-terminus SMARCA4 subunit within the core module of BAF complexes (defined by CX-MS, Extended Data Fig. 7c and recent BAF complex structural insights 25–27and assessment of SMARCB1 levels across SS18-SSX mutant conditions (Extended Data Fig. 7d) suggests the mechanism of degradation of SMARCB1 observed in SS cell lines 14,15,28 is likely explained by the dominant, higher affinity SSX binding to the nucleosome acidic patch and the resulting configurational changes within the BAF core module.
Finally, to evaluate whether SS18-SSX-containing BAF complexes that are tethered to the nucleosome acidic patch via SSX in place of the BAF core module SMARCB1 C-terminal acidic patch binding region 25 are competent in remodeling, we performed chromatin remodeling assays using restriction enzyme accessibility assays (REAA) on endogenous BAF complexes containing either SS18 WT or SS18-SSX as well as assay for transposase-accessible chromatin using sequencing (ATAC-seq) in both CRL7250 fibroblasts and SS cell lines. Remodeling efficiency and ATPase activity of SS18-SSX-bound BAF complexes was slightly lower than that of WT SS18-bound complexes (Fig. 3h, Extended Data Fig. 7e–f), however, this reduced activity was sufficient to enable DNA accessibility over SS18-SSX target sites genome-wide (Fig. 3i, Extended Data Fig. 7g).
Taken together, these data resolve SSX as a nucleosome acidic patch binding ligand fused to SS18, a subunit bound to the BAF complex ATPase subunit, SMARCA4 at its N-terminal region within the core structural module 25,26., that dominantly competes for acidic patch binding with BAF core module subunit SMARCB1, resulting in its partial destabilization and degradation. These oncogenic SS18-SSX-containing complexes are still proficient in chromatin remodeling and catalytic activity, resulting in the aberrant activation of normally repressed chromatin regions.
SSX exhibits preference for H2AK119Ub-marked nucleosomes via its conserved C-terminal acidic region
Previously, we found that SS18-SSX-bound BAF complexes localize to polycomb-repressed regions 14. The engagement between the conserved SSX basic region and the nucleosome acidic patch is not, in itself, sufficient to explain why SS18-SSX complexes are preferentially recruited to repressed chromatin. We therefore reasoned that the SSX-nucleosome acidic patch interaction might be augmented in some manner by the presence of specific histone repressive marks. To explore this possibility, we performed CRISPR-Cas9-based screening of genes encoding proteins that are responsible for decorating and maintaining repressive chromatin. These studies were performed in the SS cell line, SYO-1, as well as in a cell line that is an SS histologic mimic lacking the SS18-SSX fusion, SW982 (Fig. 4a). Notably, we found that PRC1 subunits (specifically, RING1A and RING1B, as well as PCGF5 and PCGF3 components of PRC1.3 and PRC1.5 complexes) were selectively enriched as synthetic lethal dependencies in SS cell lines SYO1 as well as other SS cell lines including Yamato and SCS241 (Fig. 4a, Extended Data Fig. 8a,b). Importantly, all SS cell lines profiled exhibited significant dependency on SS18 and SSX (and hence the SS18-SSX fusion), relative to all other cell lines profiled (Extended Data Fig. 8c,d).
Figure 4. SSX preferentially binds H2A K119Ub-marked nucleosomes to promote BAF complex targeting to polycomb-repressed loci.
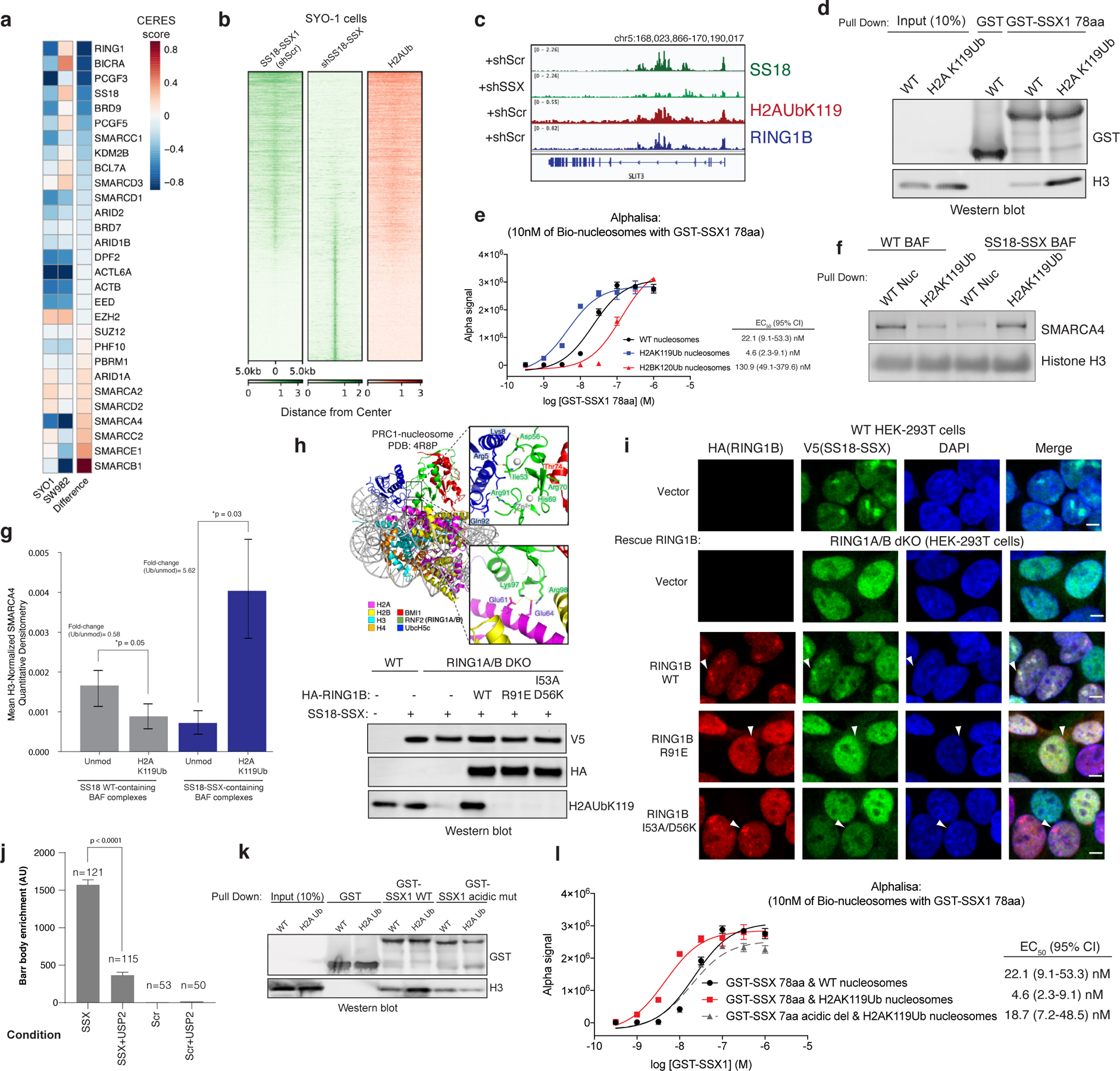
a, CERES dependency scores from CRISPR-Cas9 fitness screens (Project Achilles). Difference is the score calculated between SYO1 cells and SW982 cells.
b, SS18 localization in SYO-1 cells in control KD or shSS18-SSX conditions, aligned with H2AUb119 ChIP-seq.
c, Example tracks at the SLIT3 locus demonstrating co-localization of SS18-SSX BAF complexes, H2AK119Ub and RING1B.
d, GST-SSX pull down assays using either unmodified or H2AK119Ub nucleosomes.
e, Alphalisa performed with GST-SSX and 10nM biotinylated unmodified, H2AK119Ub or H2BK120Ub nucleosomes. Error bars= mean ± s.d. of n=4 experiments with EC50 and 95% CI values calculated by nonlinear regression curve fit.
f, Pull-down assays using HA-SS18- or HA-SS18-SSX-bound BAF complexes incubated with either unmodified or H2AK119Ub-modified nucleosomes. SMARCA4 and H3 immunoblots are shown.
g, Mean H3-normalized quantitative densitometry for SMARCA4 across SS18 and SS18-SSX-containing BAF complexes incubated with either unmodified or H2AK119Ub modified nucleosomes. Error bars indicate mean ± s.d. of n=5 independent experiments. P-value (SS18, unmodified vs. H2AK119Ub)= 0.05; P-value (SS18-SSX, unmodified vs. H2AK119Ub)= 0.03.
h, Representation of PRC1 complex-nucleosome structure (PDB: 4R8P), indicating mutated regions. Immunoblot of mutations which inhibit H2AK119Ub deposition.
i, Immunofluorescence for RING1B (red), V5 SS18-SSX (green), with DAPI nuclear stain in WT and RING1A-RING1B dKO 293T cells with rescued conditions as indicated.
j, Barr body localization for each condition, mean barr body fluorescence intensity (a.u., arbitrary units) is plotted. Peptides were incubated after mock or USP2 treatment. Error bars indicate mean ± s.d of n= 121 and 115 cells examined for SSX and SSX+USP2 conditions, respectively. P-value (SSX vs. SSX+ USP2) <0.0001.
k, Pull-down experiments performed using either GST-SSX or GST-SSXdel7aa (acidic C-term) with unmodified or H2AK119Ub nucleosomes, as indicated.
l, Alphalisa performed with GST-SSX or GST-SSXdel7aa and 10nM unmodified or H2AK119Ub nucleosomes. Error bars represent mean ± s.d. of n=4 experiments with EC50 and 95% CI values calculated by nonlinear regression curve fit. Data points shared between Fig. 4e. Uncropped gel images for panels d, f, h, k are presented in Supplementary Figure 1. Raw binding data for panels e,g,i,k are presented in Source Data.
Given that the key histone modification placed by PRC1 is the H2A K119Ub mark, we sought to determine whether SSX exhibited any preferential binding to nucleosomes decorated with this modification. Notably, we found that in SS cell lines, H2AK119Ub directly co-localized with sites of SS18-SSX BAF complex occupancy (Fig. 4b–c, Extended Data Fig. 8e). This was consistent with our IF observations suggesting substantial co-localization at Barr bodies (Fig. 2c, Extended Data Figs. 3a,b,e, 4c, 6a). Indeed, pulldown experiments and AlphaLisa binding assays performed with GST-SSX 78aa protein revealed higher affinity to H2AK119Ub -decorated nucleosomes relative to unmodified nucleosomes or H2BK120Ub nucleosomes (4.8-fold difference (Ub/unmod) by AlphaLisa) (Fig. 4d–e). Incubation of SSX 78aa with mammalian mononucleosomes also captured the higher molecular weight H2AUb species (Extended Data Fig. 8f), as did SS18-SSX-bound BAF complexes (Fig. 1a–b, Supplementary 1). Importantly, endogenously purified SS18-SSX-bound BAF complexes enriched for binding of recombinant H2A K119Ub-modified nucleosomes over unmodified nucleosomes by 5.62-fold (p-value=0.03) (Fig. 4f,g, Extended Data Fig. 8g–h), consistent with pull-down and fold changes detected by Alphalisa binding studies performed with GST-SSX 78aa protein above (Fig. 4d,e) and consistent with the finding that SS18-SSX fusion target sites directly overlay H2AK119Ub sites genome-wide in SS cell lines (Fig. 4b). Finally, we performed a screen for SSX binding to a range of differentially-marked recombinant mononucleosomes as well as mammalian (pooled) nucleosomes, and again, identified that GST-SSX 78aa preferentially bound to H2A K119Ub and mammalian nucleosomes over unmodified nucleosomes or nucleosomes with other histone marks (Extended Data Fig. 8i–k).
To understand the role of H2A K119Ub in SSX-BAF localization, we next double deleted the core, catalytic subunits of the PRC1 complex, RING1A and RING1B, using CRISPR-Cas9 in HEK-293T cells (RING1A, RING1B-dKO HEK-293T cells) and expressed SS18-SSX (Extended Data Fig. 9a). Following immunofluorescence, we observed complete loss of SS18-SSX localization to Barr bodies as compared to RING1A,RING1B WT cells (Fig. 4g–h). To address whether the catalytic activity of PRC1 rather than PRC1 complex formation is required for SS18-SSX Barr body recruitment, we performed structure-guided mutagenesis to selectively disrupt the ubiquitin ligase activity of PRC1 and hence block its placement of H2A K119Ub (Fig. 4g). We designed a series of point mutations in RING1B to disrupt acidic patch recognition (R98A), zinc binding (H69Y, R70C) and the E2 binding interface (R91A and I53A/D56K 29) (Fig 4g–h, Extended Data Fig. 9b–c). Rescue of WT RING1B in RING1A-RING1B-dKO cells was able to completely rescue SSX localization. However, restoration of RING1B mutant variants affected SS18-SSX localization in a manner directly proportional to the degree to which these RING1B mutations impacted H2A K119Ub deposition. Significantly for this study, RING1A ligase-deficient R91E and I53A/D56K were able to form polycomb foci but were unable to recruit SS18-SSX, further highlighting the importance of the H2A K119Ub mark placement. As controls, R98A, and combined H69Y and R70C mutants had similar loss-of- function effects on SS18-SSX localization 30,31 (Fig. 4g–h, Extended Data Fig. 9b–c). The widely-used I53A mutant 32–34 only partially attenuated H2A ubiquitination, and therefore had little effect on SSX targeting. As further support for a role for H2A K119Ub in SSX recruitment, we used a peptide hybridization assay performed on IMR90 cells pretreated with the deubiquitinating enzyme, USP2. USP2-mediated removal of H2A K119Ub disrupted SSX peptide hybridization to Barr bodies specifically and without affecting its overall nuclear staining pattern, consistent with the general ability of SSX to bind unmodified nucleosomes via its acidic patch binding region. (Fig. 4i, Extended Data Fig. 9d). EZH2 inhibitor treatment performed in WT HEK-293T or RING1A,RING1B dKO HEK-293T cells further highlighted the requirement for H2AKUb119 placement (and hence PRC1) rather than H3K27me3 and PRC2 activity (Extended Data Fig. 9e–i). Somewhat surprisingly, given the clear role for H2AK119Ub in recruiting SSX to chromatin, we did not observe direct binding between SSX and free ubiquitin, as assessed by a Ub-agarose pull down assay (Extended Data Fig. 9g), however, it is conceivable that SSX only engages Ub in the context of H2AK119Ub nucleosomes, as seen with other readers such as Dot1L 35–37), or alternatively, that it might recognize specific features of the nucleosome core itself that are sterically or allosterically affected by the presence of the ubiquitylation mark.
Finally, given that the conserved C-terminal acidic region of SSX did not disrupt SSX-nucleosome binding (Fig. 2f) but did affect SS18-SSX-specific BAF complex targeting and resultant gene expression and proliferation (Fig. 2g–i) in a manner comparable to loss of the basic region (acidic patch binding region), we sought to determine whether this region mediates the preference of SSX for H2AK119Ub-decorated nucleosomes. Excitingly, we found that mutation of the C-terminal acidic region of SSX to alanines (i.e. DPEEDDE→AAAAAAA) relieved the preference of SSX for H2AK119Ub nucleosomes, while not altering general SSX binding to nucleosomes (Fig. 4j–k). These data collectively indicate that the conserved C-terminal acidic amino acids are required to drive the preference of SSX for H2A K119Ub nucleosomes and hence SS18-SSX-bound BAF complex targeting to repressive regions genome-wide, as observed in cells.
Discussion
Here we identified an unexpected set of functionally critical properties of the fusion oncoprotein, SS18-SSX, the oncogenic driver of human synovial sarcoma (Fig. 5). We report an unusual case in which an additional nucleosome acidic patch binding domain is fused to a subunit of a major chromatin remodeling complex, the mammalian SWI/SNF (BAF) complex, conferring oncogenic properties to a tumor suppressor complex. Although several SNF2 helicase-based chromatin remodeling complexes are increasingly recognized to require the H2A-H2B nucleosome binding hub, we found that the minimal, conserved SSX 34 aa region dominantly binds the acidic patch of nucleosomes, and that SSX (or SS18-SSX-bound complexes) exhibit preferential binding to H2A K119Ub-modified nucleosomes, altering the interaction between the nucleosome-SMARCB1 C-terminal alpha helix interaction found in WT BAF complexes 25,26, and resulting in higher affinity nucleosome-binding properties augmented by specific repressive histone mark preferences 25. These data, coupled with recent structural elucidation of yeast and human SWI/SNF complexes27, provide strong support for SS18-SSX-mediated displacement of SMARCB1 from the acidic patch and its destabilization at the nucleosome-proximal region of the core (base) module of BAF complexes. A high-resolution, 3D structure of human BAF complexes containing SS18, as well as SS18-SSX is required to define the full repertoire of structural changes to nucleosome-bound BAF complexes upon incorporation of SS18-SSX. This is particularly true given that the SS18 subunit is metazoan-specific and hence is not found in yeast complexes, nor has it been resolved in structural efforts to date.
Figure 5. Model for SS18-SSX-bound BAF complex nucleosome engagement.
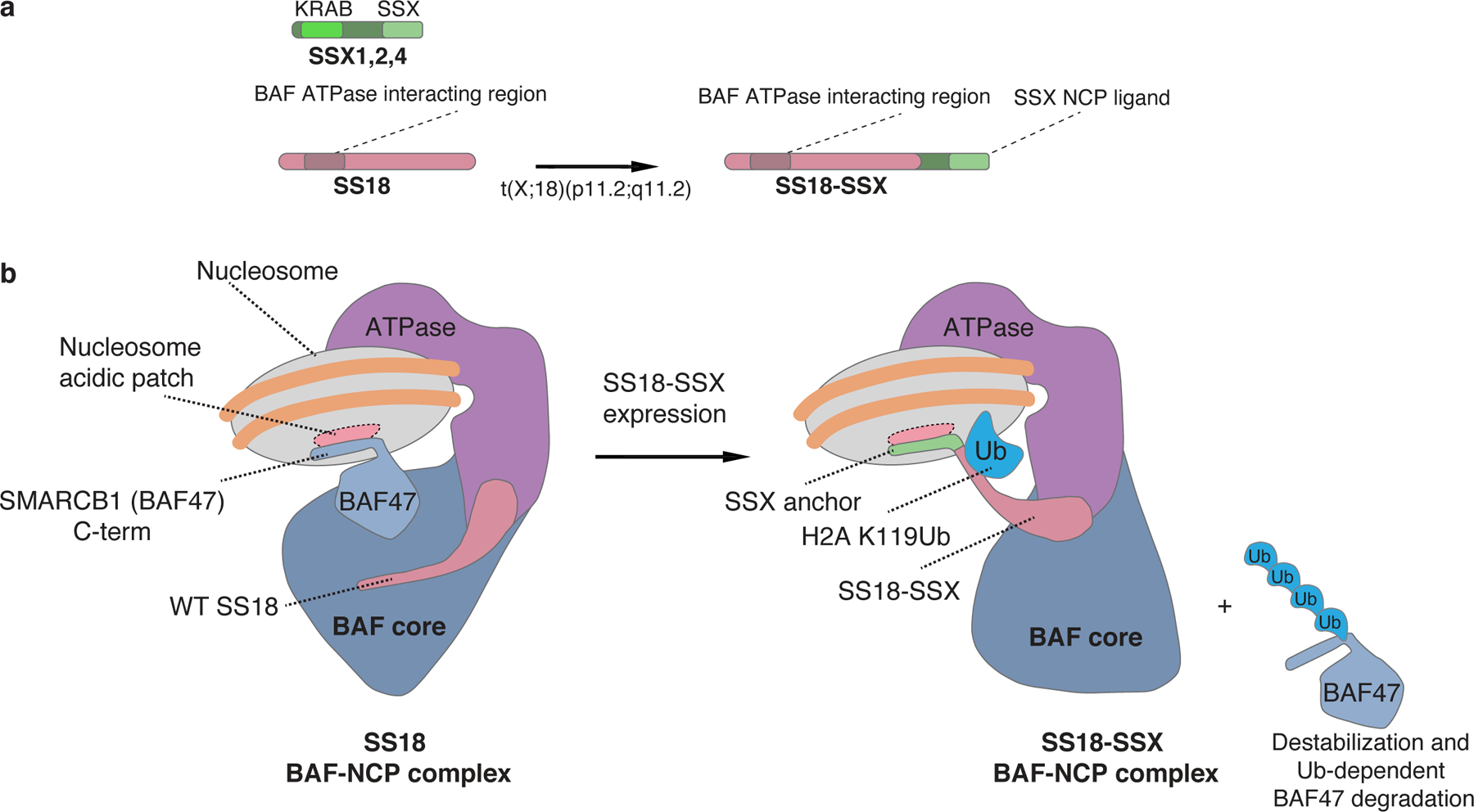
a, Schematic of SS18 WT and the SS18-SSX fusion oncoprotein.
b, Model for BAF complex engagement on nucleosomes in WT and SS18-SSX fusion oncoprotein states. In WT complexes, the core module of BAF complexes engages the nucleosome acidic patch via the SMARCB1 C-terminal alpha helical domain (aa 351–385). Upon expression of SS18-SSX, the SSX alpha helical basic region (RLRERK) dominantly engages the acidic patch, displacing SMARCB1, leading to its degradation, and changing the orientation of the BAF core module (21,27) on the nucleosome. This SS18-SSX-specific conformation of BAF complexes exhibits strong preference for H2AUbK119-decorated nucleosomes, conferring their preference for polycomb chromatin regions.
The expression of full length SSX is normally restricted to testes where it likely plays a role in sperm development, potentially involving polycomb-driven XY-body repression through engagement of H2A K119Ub-decorated sex chromosomes 38. Remarkably, this normal function of SSX as a binder of the nucleosome acidic patch and “reader” of this repressive state is leveraged in synovial sarcoma to alter BAF chromatin remodeling complex localization and gene expression patterns. Normally in testes, full-length SSX may function as a ligand for nucleosomes in this H2A K119Ub repressive state to promote further transcriptional repression through its N-terminal KRAB domain 39. In the case of SS, the KRAB domain is replaced with essentially the whole ATPase module of the BAF chromatin remodeling complex via fusion to SS18. This unfortunate scenario leads to gain of altered repressive chromatin reading properties of BAF complexes, loss of normal BAF complex -nucleosome acidic patch engagement, tight affinity and longer residency times at normally polycomb- repressed regions, and the activation of genes found in these regions (Figure 5).
We therefore characterized the SSX 78 aa tail, particularly the conserved 34aa C-terminus (Fig. 2d) as a ligand of the nucleosome acidic patch that confers preferential affinity of SS18-SSX-bound complexes for H2AK119Ub-marked nucleosomes. Our data suggests two non-mutually exclusive explanations for this reading preference: H2A K119Ub modification influences nucleosome structure by further exposing the acidic patch binding site; alternatively, SSX directly engages ubiquitin in the nucleosomal context. While our present data indicate that SSX does not bind directly to free (bead-bound) ubiquitin (Extended Data Fig. 9g), this does not rule out the possibility of direct ubiquitin engagement by the acidic C-terminal region of SSX when SS18-SSX-bound complexes are docked on nucleosomes. In a similar manner, DOT1Lwill not bind free ubiquitin but will be poised to interact with H2BUbK120 during substrate engagement 35–37. Determining the underlying basis of this binding preference requires structural characterization of SS18-SSX-bound human BAF complexes or at least high-resolution structures of SS18-SSX on H2A K119Ub-marked nucleosomes.. Nonetheless, the present results suggest that SSX acts as a nucleosome-specific binding ligand for the acidic patch on H2A K119Ub-decorated nucleosomes, and that this property underlies the chromatin localization, gene expression, and synthetic lethal profiles of this tumor type.
The biochemical and structural properties of the SSX fusion partner elucidated here underpin the dependency of SS on PRC1 complex activity that has been detected in fitness screening efforts and in our own structure-guided mutagenesis studies. In contrast to other reports 20, we do not find direct binding to PRC1 by the SS18-SSX fusion (or by SSX specifically), nor a selective dependency on KDM2B; rather, we find that SS18-SSX-bound complexes bind preferentially to H2AK119Ub-marked nucleosomes, and hence require PRC1 complex-mediated placement of the H2AUbK119 mark. Further, we do not detect peptides corresponding to PRC1 or PRC2, rather our mass spectroscopy data reveal highly abundant peptides corresponding to histones and ubiquitin itself, and enrichment of peptides corresponding to the ATPase module subunits of BAF complexes (SMARCA4, BCL7A, beta-actin, ACTL6A) to which SS18 is tethered. The increased abundance of SS18-SSX-containing BAF complexes over PRC1-decorated sites, and hence the frequency of molecules co-localized on chromatin, may help reconcile these previous suggestions.
In summary, we have identified an unanticipated nucleosome acidic patch-binding function of SSX, an oncoprotein fusion partner lacking canonical DNA-binding or chromatin-reader domains, that alters behavior of BAF chromatin remodeling complexes and activates oncogenic programs in a cancer-specific manner. Our results suggest that inhibition of the SSX- or SS18-SSX-bound BAF complex- H2A K119Ub nucleosome interactions using small molecules or peptides may prove a viable therapeutic strategy for synovial sarcoma.
Materials and Methods
Cell Lines and Cell Culture
The two synovial sarcoma cell lines, Aska and SYO1, were generous gifts from Kazuyuki Itoh, Norifumi Naka, and Satoshi Takenaka (Osaka University, Japan) and Akira Kawai (National Cancer Center Hospital, Japan), respectively. The CRL7250 human fibroblast cell line was obtained from Drs. Berkeley Gryder and Javed Khan (National Cancer Institute, Bethesda, MD). The HEK293T cell line was purchased ATCC (CRL-3216). Each cell line was cultured using standard protocols in DMEM medium (Gibco) supplemented with 10–20% fetal bovine serum, 1% Glutamax (Gibco), 1% Sodium Pyruvate (Gibco) and 1% Penicillin-Streptomycin (Gibco) and grown in a humidified incubator at 37°C with 5% CO2. All cell lines were routinely tested for mycoplasma contamination, confirmed to be negative prior to all experiments, and were authenticated prior to use.
Stable Gene Expression and shRNA Knockdown Constructs
Constitutive expression of SS18 wild-type (SS18), SS18-SSX1 (SS18-SSX1) and SS18-SSX1 mutations with HA or V5 N-terminus tag was obtained using an EF1alpha-driven expression vector (modified from Clonetech, dual Promoter EF-1a-MCS-PGK-Puro or EF-1a-MCS-PGK-Blast) expressed in cells by lentiviral infection and selected with puromycin (2 μg/mL) or blasticidin (10 µg/mL). Constitutive expression of shRNA hairpins targeting the 3’UTR region of SSX of the SS18-SSX fusion (5’-CAGTCACTGACAGTTAATAAA-3’) or a scramble non-targeting control (5’- CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3’) was obtained using lentiviral infection of the pLKO.1 vector with puromycin (2 μg/mL) selection.
Lentivirus Generation and Harvesting
Lentivirus production was obtained from PEI (Polysciences) transfection of HEK293T LentiX cells (Clontech) with co-transfection of the packaging vectors pspax2 and pMD2.G along with the gene delivery vector. Viral supernatants were collected 72 hours after transfection, underwent ultracentrifugation at 20,000 rpm for 2.5 hr at 4°C to concentrate, and then virus pellets were resuspended in PBS. For infection, the viral pellets were added to cells in a drop wise manner in the presence of polybrene (10 μg/mL). After 48 hours, the media containing the lentivirus was replaced and infected cells were selected by addition of puromycin (2 μg/mL) or blasticidin (10 µg/mL).
Western Blot Analysis
Detection of proteins by western blot (WB) analysis was achieved using standard protocols with primary antibodies (Supplementary Table 4). Samples were separated on 4–12% Bis-Tris SDS PAGE gel (Invitrogen) and transferred to PVDF membrane. The membranes were then blocked in 5% milk and incubated with primary antibody in PBST over night at 4°C. Following incubation with the primary antibody, membranes were washed 3X in PBST, incubated with IRDye (LI-COR Biosciences) secondary antibodies for 3 hours, washed 3X in PBST with a final PBS wash, and then visualized by the LI-COR Odyssey Imaging System (LI-COR Biosciences).
Cell Lysate Collection
Whole cell extractions (WCE) were obtained by washing harvested cell pellets with PBS pH 7.4, resuspending in whole cell lysis buffer (PBS pH 7.4 and 1% SDS) and then heating for 3 minutes at 95°C. Lysates were sonicated until fully liquid. Nuclear extractions (NE) were obtained by suspending the harvested cells in Buffer 0 (50 mM Tris pH 7.5, 0.1% NP-40, 1 mM EDTA, 1 mM MgCl2 with protease inhibitor cocktail, 1 mM DTT and 1 mM phenylmethylsulfonyl fluoride (PMSF)), centrifuging at 5,000 rpm for 5 minutes at 4°C, and discarding the supernatant. The pellet (nuclei) were resuspended in EB300 (50 mM Tris pH 7.5, 0.1% NP-40, 1 mM EDTA, 1 mM MgCl2, 300 mM NaCl with protease inhibitor cocktail, 1 mM DTT and 1 mM phenylmethylsulfonyl fluoride (PMSF)), vortexed, incubated on ice, centrifuged at 15,000 rpm for 10 minutes at 4°C and supernatant containing the nuclear extract collected.
Nuclear Extraction
Nuclear extracts for 293T V5-SS18WT and V5-SS18-SSX1 cells were prepared as previously described 14. Specifically, cells were scraped from plates, washed with cold PBS, pelleted at 3,000 rpm for 5 min at 4C, and resuspended in Buffer A hypotonic buffer (50 mM Hepes, pH 7.6, 25 mM KCl, 10% Glycerol, 0.1% NP-40, 0.05 mM EDTA, 5 mM MgCl2 supplemented with protease inhibitor cocktail, and 1 mM phenylmethylsulfonyl fluoride (PMSF)). Lysates were pelleted at 3,000 rpm for 5 min at 4C. Supernatants were discarded, and nuclei were resuspended in Buffer C high salt buffer (10 mM Hepes, pH 7.6, 100 mM KCl, 10% Glycerol, 0.5 mM EDTA, 3 mM MgCl2 supplemented with protease inhibitor and 1 mM PMSF). Lysates were incubated at 4C at constant rotation. Lysates were then pelleted at 40,000 x rpm for 1 hour at 4C. Supernatants were collected, and mixed with (NH4)2SO4 at 300mg/ml for 30 min. Samples were pelleted at 15,000 rpm for 30 minutes and supernatant was discarded. Protein concentrations were quantified via bicinchonic acid (BCA) assay (Pierce). Finally, samples were supplemented with 1 mM DTT.
Co-Immunoprecipitations
Nuclear extracts were quantified by Bradford assay and 150–200 μg of protein was incubated with 2 μg of antibody in Buffer EB300 (50 mM Tris pH 7.5, 0.1% NP-40, 1 mM EDTA, 1 mM MgCl2, 150 mM NaCl with protease inhibitor cocktail, 1 mM DTT and 1 mM phenylmethylsulfonyl fluoride (PMSF)) overnight at 4°C. Each sample was then incubated with Protein G Dynabeads (Thermo Scientific) for 2–3 hours. Beads were washed three times with Buffer EB300 followed by elution with 20 μL of elution buffer (NuPage LDS buffer (2X) (Life Technologies) containing 100 mM DTT and water).
Cell Proliferation Assay
To measure cell proliferation following lentiviral infection, 2.5×104 cells per well were seeded in 12-well plates following 48-hour exposure to lentivirus and 5-day selection with puromycin or blasticidin, with Day 7 denoting the day cells were plated after infection and selection. The cell viability in three wells was then measured using a Vi-CELL Cell Counter (Beckman, Brea, CA) every 72 hours.
Differential Salt Extraction
Following collection of 5.0×107 cells, cells were resuspended in elution 0 buffer (50 mM Tris-HCl pH 7.5, 1 mM EDTA, 0.1% NP40 with protease inhibitor cocktail and 1 mM PMSF), incubated on ice for 5 minutes, and pelleted by centrifugation. The supernatant was collected (0 mM fraction), and the cell pellet was resuspended in elution 150 buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl) 1 mM EDTA, 0.1% NP40 with protease inhibitor cocktail and 1 mM PMSF) and vortexed. This process was repeated sequentially with elution 300 buffer, elution 500 buffer, and elution 1000 buffer that contained increasing concentrations of NaCl in order to obtain 0, 150, 300, 500, and 1,000 mM NaCl soluble fractions. Each of these soluble fractions, along with a total sample (5×106 cells in elution buffer) and the chromatin pellet (non-soluble material remaining following extraction with 1000 mM NaCl) fractions, was denatured in SDS to a final concentration of 1%, protein quantified by Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), and analyzed (1.5 μg of protein) by immunoblot.
Purification of mSWI/SNF (BAF) Complexes
Stable HEK293T cell lines expressing by lentiviral infection HA-SS18 WT or HA-SS18-SSX1 were grown in 150mm dishes. Complexes were purified using methods previously described with a few modifications21. Confluent plates were scraped to remove cells and cells were washed with PBS. Cell suspension was spun down by centrifugation at 3000 rpm for 5 minutes at 4°C and pellets were resuspended in hypotonic buffer (10mM Tris HCl pH 7.5, 10mM KCl, 1.5 mM MgCL2, 1mM DTT, 1mM PMSF) and incubated on ice for 5 minutes. Following incubation, cell suspension was spun down by centrifugation at 5000 rpm for 5 minutes at 4°C, and pellets were resuspended in 5X volume of fresh hypotonic buffer (with protease inhibitor cocktail) and then cells were homogenized using a Dounce homogenizer (glass). Cell suspension was layered onto hypotonic buffer sucrose cushion made with 30% sucrose w/v, spun down by centrifugation at 5000 rpm for 1 hour at 4°C followed by removal of the cytosol-containing layer. The nuclei containing pellets were resuspended in high salt buffer (50mM Tris HCl pH 7.5, 300mM KCl, 1mM MgCL2, 1mM EDTA, 1mM, 1% NP40, 1mM DTT, 1mM PMSF and protease inhibitor cocktail) and then the homogenate rotated for 1 hour at 4°C. Homogenates were then spun down by centrifugation at 20,000 rpm for 1 hour at 4°C in a SW32Ti rotor (Beckman Coulter). The soluble proteins, consisting of the nuclear extract (NE) fraction, was separated from the insoluble chromatin pellet, consisting of the chromatin (CHR) fraction. The chromatin pellet was further solubilized by treatment with Benzonase (Sigma Aldrich) for 30 minutes and subsequently additional KCl was added to final concentration of 700 mM (50mM Tris HCl pH 7.5, 700mM KCl, 1mM MgCL2, 1mM EDTA, 1mM, 1% NP40, 1mM DTT, 1mM PMSF and protease inhibitor cocktail), and sonicated 3 times for 30 seconds with 5 minute intervals. The solubilized chromatin fraction was then spun down by centrifugation at 20,000 rpm for 1 hour at 4°C in a SW32Ti rotor (Beckman Coulter) and supernatant collected. The collected nuclear extract and chromatin fractions were filtered with a 0.45um filter and rotated overnight at 4°C with HA magnetic resin. HA beads were washed in high salt buffer and eluted with 1 mg/mL of HA peptide for 4 times at durations of 1.5 hour each. Eluted proteins were then subjected to density gradient centrifugation or dialysis.
Colloidal blue and Silver Stain
HA-SS18 WT and HA-SS18-SSX1 mSWI/SNF complexes were purified via HA-epitope dependent complex purification. Importantly, for Figure 1A, the same number of cells were used for both HA-SS18 WT and HA-SS18-SSX expressing cells, and nuclear material from both cell lines was split into NE and CHR fractions, representing an equal total amount of complexes in the nucleus. Hence, equal input/output loading by volume was achieved. Samples were run on a 4–12% Bis-Tris SDS PAGE gel, stained using Colloidal blue kit or SilverQuest Silver Staining Kit (Invitrogen), and imaged using LI-COR Odyssey Imaging System (LI-COR Biosciences) or Epson-Perfection V600 Photo scanner, respectively.
Density Sedimentation Gradients
Purified protein complexes were added to the top of a linear, 11ml 10%–30% glycerol gradients containing 25mM HEPES pH 7.9, 0.1 mM EDTA, 12.5 mM MgCl2, 100 mM KCl with 1 mM DTT and protease inhibitors. Gradient tubes were placed into SW41 rotor (Beckman Coulter) and spun by centrifugation at 40000 rpm for 16 hours at 4°C. Fractions of 550 µL volume were collected sequentially from the top of the gradient. 100 µL of each fraction was concentrated with 10 µL of Strataclean beads (Agilent Technologies, 400714) loaded and run on a SDS-PAGE gel, and then analyzed by SYPRO Ruby Protein Gel Stain (Thermo Fisher Scientific) and scanned using Typhoon FLA 9500 scanner.
Mass Spectrometry Proteomics Analysis of Purified Complexes
Equal amounts of purified HA-SS18 WT and HA-SS18-SSX1 complexes were loaded onto SDS-PAGE gels from both the nuclear extract (NE) and chromatin (CHR) fractions. Samples were migrated into the gel for a length of 2cm, gels were stained with colloidal blue stain and protein bands were excised for protein detection by mass spectrometry. The samples were then prepared and data analyzed by the Taplin Biological Mass Spectrometry Facility directed by Dr. Steven Gygi (Harvard Medical School).
Protein and Peptide Pull Downs
Recombinant purified proteins with affinity tags (MBP or GST) or biotinylated peptides were purified using magnetic beads (Maltose, glutathione or streptavidin respectably ) by incubation in EB150 buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl) 1 mM EDTA, 0.1% NP40 with protease inhibitor cocktail and 1 mM PMSF at 4°C overnight. The flow through was removed, the immobilized bait was incubated with 1–2 µg of purified mammalian mono-nucleosomes from HEK293T cells, recombinant mono-nucleosomes (EpiCypher, 16–0006), recombinant H2AK119Ub mono-nucleosomes (EpiCypher, 16–0020) or recombinant protein for 3 hours at 4°C, the beads were washed 3X with EB150 buffer and then eluted in 2X LDS with 200mM DTT with heating at 95°C for 5 minutes. The pull downs were then visualized by immunoblot analysis or colloidal blue staining.
Peptide Competition Experiments
The peptide competition experiments were set up in a similar manner as the peptide pull down experiments with the following exceptions: SSX1 (aa 55–78) or SMARCB1-CC (aa 351–385) biotin-labeled peptides at 10 μM in EB150 were bound to Streptavidin Dynabeads (Pierce Streptavidin Magnetic Beads, Thermo Scientific) in parallel to 1–2 μg of mononucleosomes incubated with LANA, SSX (aa 155–188) or SMARCB1-CC (aa 351–385) peptide (KE Biochem) at varying concentrations ranging from 0–30 μM overnight at 4°C. Beads were washed 3 times in EB150, and resuspended with the mononucleosomes-LANA peptide solutions. The suspension was rotated for 3–5 hours at 4°C. The beads were washed 5 times in EB150, and eluted in Sample Buffer (2X LDS with 200 mM DTT) to load onto 10–20% Tricine gels.
Quantitative Targeted Mass Spectrometry
Mammalian mono-nucleosomes purified from MBP-SSX1 78aa pull downs along with representative input samples were prepared and analyzed by the targeted mass spectrometry pipeline described previously 40. Briefly, samples were prepared by histone extraction by acid precipitation followed by protein digestion from incubation with trypsin. To these prepared samples, synthesized isotopically labeled peptides of histone tails with numerous modifications were added at a known quantity. Each sample was then separated using a Proxeon EASY-nLC 1000 UHPLC system (Thermo Scientific) and detected with a Q Exactive mass spectrometer (Thermo Scientific). The fold change in abundance of each histone peptide from the input sample compared to the pull down was calculated from the light:heavy ratio in detected peak size.
Detection of Nucleosome Acidic Patch Interactions by Photocrosslinking
Details of the design and preparation of diazirine containing nucleosomes for photo-crosslinking studies will be described elsewhere 41. Briefly, diazirine-containing recombinant nucleosomes (0.5 uM) were incubated with biotinylated SSX peptides (12.5 uM) in binding buffer (20 mM HEPES, pH 7.9, 4 mM Tris, pH 7.5, 150 mM KCl, 10 mM MgCl2, 10% glycerol, and 0.02% (v/v) IGEPAL CA-630) at 30 °C for 30 mins, and cooled on ice for 5 mins. The reaction mixtures were then irradiated at 365 nm for 10 minutes. Reactions were then analyzed by western blotting employing IRDye® 800CW streptavidin on a LI-COR Odyssey Infrared Imager. Additional details are found in Dao et al., 2019 41.
Immunofluorescence
Immunofluorescent images were obtained as previously described 42. Following lentiviral infection and/or drug treatment, cells were prepared by fixation in 3% PFA-PBS and then were permeabilized with PBS 0.1% NP40. Following incubation with primary antibodies, the Anti-rabbit Alexa Fluor® 594 and Anti-mouse Alexa Fluor® 488 (Life Technologies) secondary antibodies were used for visualization. Staining with 4’,6-diamidino-2-phenylindole (DAPI) was used to visualize nuclei. Images were acquired using Zeiss Axio Imager Z2 microscope and images were processed using ImageJ program (NIH).
Fluorescent Recovery After Photobleaching (FRAP)
The FRAP experiments were carried out in the same manner as previously described 43. Briefly, HEK293T cells expressing GFP-SS18 WT or GFP-SS18-SSX1 by lentiviral infection or Aska cells co-expressing BRG1-Halo fusion with pLKO.1 shScramble control or shSSX were imaged to measure the mean fluorescence intensity of a defined nuclear region pre and post-photobleaching at 5 second intervals. The relative fluorescence intensity (RFI) for each image was calculated by normalizing the maximal difference in fluorescence intensity post-bleaching to 1. The t1/2 values and mobile fractions were determined using the software Prism (GraphPad Software) from n=27–30 cells in each condition over two biological replicates.
Chromatin Immunoprecipitation (ChIP)
For chromatin immunoprecipitation (ChIP) experiments, prepared cells were harvested following 48 hours of lentiviral infection and 5 day selection (unless otherwise indicate) with puromycin or blasticidin. Capture of chromatin bound proteins was performed using standard protocols (Millipore, Billerica, MA). Briefly, cells were cross-linked with 1% formaldehyde for 10 minutes at 37°C, reaction was quenched by addition of 125 mM glycine for 5 min and then 5 (for synovial sarcoma cell lines) or 10 (for fibroblast cell lines) million fixed cells were used per experiment. Chromatin was fragmented by sonication with a Covaris E220 and the solubilized chromatin was incubated with a primary antibody overnight at 4°C to form antibody-chromatin complexes. These complexes incubated with Protein G-Dynabeads (Thermo Scientific) for 3 hours at 4°C, beads washed 3X and eluted. The samples then underwent crosslink reversal, treatment with RNase A (Roche), and treatment with proteinase K (Thermo Scientific) followed by DNA capture with AMP Pure beads (Beckman Coulter).
RNA Isolation from Cell Lines
Cells (1×106) were collected following 48 hours of lentiviral infection and 5 days (7 days post-infection) of selection with puromycin or blasticidin for extraction of RNA for RNA-seq experiments. Samples for RNA-seq were prepared in biological duplicate (collected using independent production of lentivirus, infection, selection, and cell culture). Total RNA was collected using the RNeasy Mini Kit (Qiagen) following homogenization of cell lysates using the QIAshredder (Qiagen).
Library Preparation and Sequencing for RNA and ChIP Samples
Library preparations for next-generation sequencing of RNA-seq samples were performed using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs) to purify mRNA from 1 µg of total RNA isolated from cells. Next, the this isolated mRNA was used with the NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina (New England BioLabs ) to generate DNA. The DNA from these prepared RNA samples as well as the ChIP-seq samples were then prepared for sequencing using the NEBNext® Ultra™ II (New England BioLabs ) to amplify and barcode each sample. The fragments sizes were determined using a D1000 ScreenTape system (Agilent) and the DNA quantified by Kapa Library Quantification Kit Illumina Platforms (Kapa Biosystems). The samples were then diluted and loaded on a buffer cartridge for 75bp single end sequencing on the NextSeq 500 system (Illumina).
CRISPR–Cas9 and shRNA synthetic lethal screening data analyses
CRISPR-Cas9 datasets (Avana-19Q3) were obtained from the Project Achilles Data Portal (https://depmap.org/portal/achilles/). Fitness (CERES) scores were extracted for each cell line and hierarchical clustering was performed using complete linkage and correlation as a distance measure. Heatmaps were generated using pheatmap in RStudio. DRIVE data is publicly available and can be downloaded from the Novartis DRIVE Data Portal (https://oncologynibr.shinyapps.io/drive/). Waterfall plots were generated using ggplot2 in RStudio.
Purification of Mammalian Mononucleosomes
Mammalian mononucleosomes were purified from HEK293T cells transfected with pCDNA3 Flag-H2A as previously described 44. Cells were scraped from plates, washed with cold PBS, and centrifuged at 5,000 rpm for 5 min at 4°C. Pellets were resuspended in hypotonic buffer (EB0: 50 mM Tris HCl, pH 7.5, 1mM EDTA, 1mM MgCl2, 0.1% NP40 supplemented with 1 mM DTT, 1 mM PMSF, and protease inhibitor cocktail and incubated for 5 min on ice. The suspension was centrifuged at 5,000 rpm for 5 min at 4°C, and pellets were resuspended in 5 volumes of high salt buffer (EB420: 50 mM Tris HCl, pH 7.5, 420mM NaCl, 1 mM MgCl2, 0.1% NP40 with supplemented with 1 mM DTT and 1 mM PMSF containing protease inhibitor cocktail. Homogenate incubated on rotator for 1 hour at 4°C. The supernatant was then centrifuged at 20,000 rpm (30,000 x g) for 1 hour at 4°C using a SW32Ti rotor. Supernatant was then discarded and chromatin pellet was washed in MNAse buffer (20 mM Tris-HCl pH 7.5, 100 mM KCl, 2 mM MgCl2, 1 mM CaCl2, 0.3 M sucrose, 0.1% NP-40, and protease inhibitor cocktail) 3 times. Following MNase treatment (3 U/mL for 30 min at room temperature, Sigma-Aldrich), the reaction was quenched with 5 mM of EGTA and 5 mM of EDTA. The samples were then centrifuged at 20,000 x g for 1 hour at 4°C to obtain the soluble chromatin fraction. This fraction was then incubated with M2 anti-Flag magnetic beads (Sigma) over night. Beads were washed with EB300 (EB300: 50 mM Tris HCl, pH 7.5, 300mM NaCl, 1 mM MgCl2, 0.1% NP40 with supplemented with 1 mM DTT and 1 mM PMSF containing protease inhibitor cocktail). And eluted with EB300 containing 0.2mg/ml of Flag peptide 3 times for 1.5 hours at 4°C. Elution fractions were loaded onto 10–30% glycerol gradient 21 and fractions containing mononucleosomes were isolated and concentrated using ultra concentrators (Amicon, EMD Millipore).
Restriction Enzyme Accessibility Assay (REAA) Nucleosome Remodeling Assay
SMARCA4 (BRG1) levels of the ammonium sulfate nuclear extracts were normalized via BCA protein quantification and Silver Stain analyses for HA-SS18 and HA-SS18-SSX conditions. Protein was diluted for final reaction concentration of 150 ug/mL in REAA buffer (20mMHEPES, pH 8.0, 50mMKCl, 5mMMgCl2) containing 0.1 mg/mL BSA, 1 mMDTT, 20 nM nucleosomes (EpiDyne Nucleosome Remodeling Assay Substrate ST601-GATC1, EpiCypher). The REAA mixture was incubated at 37C for 10 min, and reaction was initiated using 1–2 mM ATP (Ultrapure ATP, Promega) and 0.005 U/mL DpnII Restriction Enzyme (New England Biolabs). The REAA reaction mixture was quenched with 20–24 mM EDTA and placed on ice. Proteinase K (Ambion) was added at (100 mg/mL) for 30–60 min, followed by either AMPure bead DNA purification and D1000 HS DNA ScreenTape Analysis (Agilent) or mixing with GelPilot Loading Dye (QIAGEN) and loading onto 8% TBE gel (Novex 8%TBE Gels, Thermo Fisher). TBE gels were stained with either SYBR-Safe (Invitrogen) or Syto-60 Red Fluorescent Nucleic Acid Stain (Invitrogen), followed by imaging with UV light on an Alpha Innotech AlphaImager 2200 and/or with 652 nm light excitation on a Li-Cor Odyssey CLx imaging system (LI-COR).
ATPase assays
ATPase consumption assays were performed using the ADP-Glo Kinase Assay kit (Promega) according to manufacturer’s instructions. The same conditions as the REAA nucleosome remodeling assay described above were used. Following incubation with desired substrates for 40 min at 37C, 1X volume of ADP-Glo Reagent was used to quench the reaction and incubated at RT for 40 min. 2X volume of the Kinase Detection Reagent was then added and incubated at RT for 1 h. Luminescence readout was recorded. Substrates used for this assay measuring nucleosome-bound ATPase activity were purified recombinant mononucleosome (EpiDyne Nucleosome Remodeling Assay Substrate ST601-GATC1, EpiCypher, Cat#16–4101). Nuclear extract material was used at 150ug for each ARID1A-IP using ARID1A antibody (Cell Signaling, Cat# 12354S).
Preparation of Peptides
Custom peptide sequences were prepared using standard synthesis techniques from KE Biochem. The peptides were confirmed to have >95% purity by HPLC and obtained as a white to off-white lyophilized powder. The powder was re-suspended in DMSO (Sigma) for use in experiments.
Expression and Purification of Recombinant Proteins
DNA constructs of human SSX1 aa111–188 and related mutates in pGEX-6P2 expression vector were transformed in E. coli BL21 (DE3) cells and overexpressed in TB medium in the presence of 100 μg/ml of ampicillin. Cells were grown at 37°C to an OD600 of 0.6, cooled to 17°C, induced with 500 μM isopropyl-1-thio-D-galactopyranoside (IPTG), incubated overnight at 17°C, collected by centrifugation, and stored at −80°C. Cell pellets were resuspended in buffer A (25 mM HEPES, pH 7.5, 200 mM NaCl, 5% glycerol, and 0.5 mM TCEP) supplemented with 1mM PMSF, lysed in a Microfluidizer (Microfluidics) and centrifuged at 16,000 x g for 45 min. Glutathione sepharose beads (GE healthcare) were incubated with lysate supernatant for 90 min to captured GST-tagged proteins and washed with buffer A. Beads with bound protein were transferred to an FPLC-compatible column and the bound protein was washed with high salt buffer (buffer A containing 1M NaCl) followed by elution with buffer A supplemented with 15mM glutathione (Sigma). Eluted protein fractions were collected, concentrated and purified by size exclusion chromatography using a Superdex 75 10/300 column (GE healthcare) equilibrated with buffer A. Eluted protein was incubated with GST-3C protease at 4°C overnight. Cleaved samples were incubated with a second round of glutathione beads to remove GST-3C and free GST, and desired protein product contained within the flow-through fractions was further purified by ion-exchange chromatography using mono-Q column (GE healthcare). Fractions containing the cleaved protein product were pooled, concentrated and stored at −80°C.
Peptide hybridization assay
IMR90 fibroblast were grown on coverslips, washed with PBS and fixed using 100% ice cold methanol for 3 minutes. Coverslips were then washed with IF wash buffer (PBS 0.1% NP40 1mM Sodium azide) 3 times. Selected groups were treated with 200 ng/ml of recombinant USP2 catalytic domain (Boston biochem) for 1 hour. Coverslips were then washed 3 times with IF wash buffer and incubated with 2uM of biotinylated peptides. Coverslips were subsequently washed 3 times with IF wash buffer and fixed in 3% PFA-PBS for 20 minutes. The rest of the procedure followed accordingly to standard IF protocol. In brief, following incubation with primary antibodies, the Anti-rabit Alexa Fluor® 594 and Streptavidin Alexa Fluor® 488 (Life Technologies) secondary antibodies/reagent were used for visualization of primary antibodies or biotinylated peptides. Staining with 4’,6-diamidino-2-phenylindole (DAPI) was used to visualize nuclei. Images were acquired using Zeiss Axio Imager Z2 microscope and images were processed using ImageJ program (NIH).
Statistics
All graphical representations of data and statistical analyses were performed using either Mann-Whitney U tests or two-tailed Student’s t-test. Error bars representing standard deviations (s.d.), the number of events (n), number of biological replicates (n) and p-values are indicated in the figure legends.
Extended Data
Extended Data Figure 1. SS18-SSX-containing BAF complexes exhibit high-affinity interactions with histones and longer residency times on chromatin.
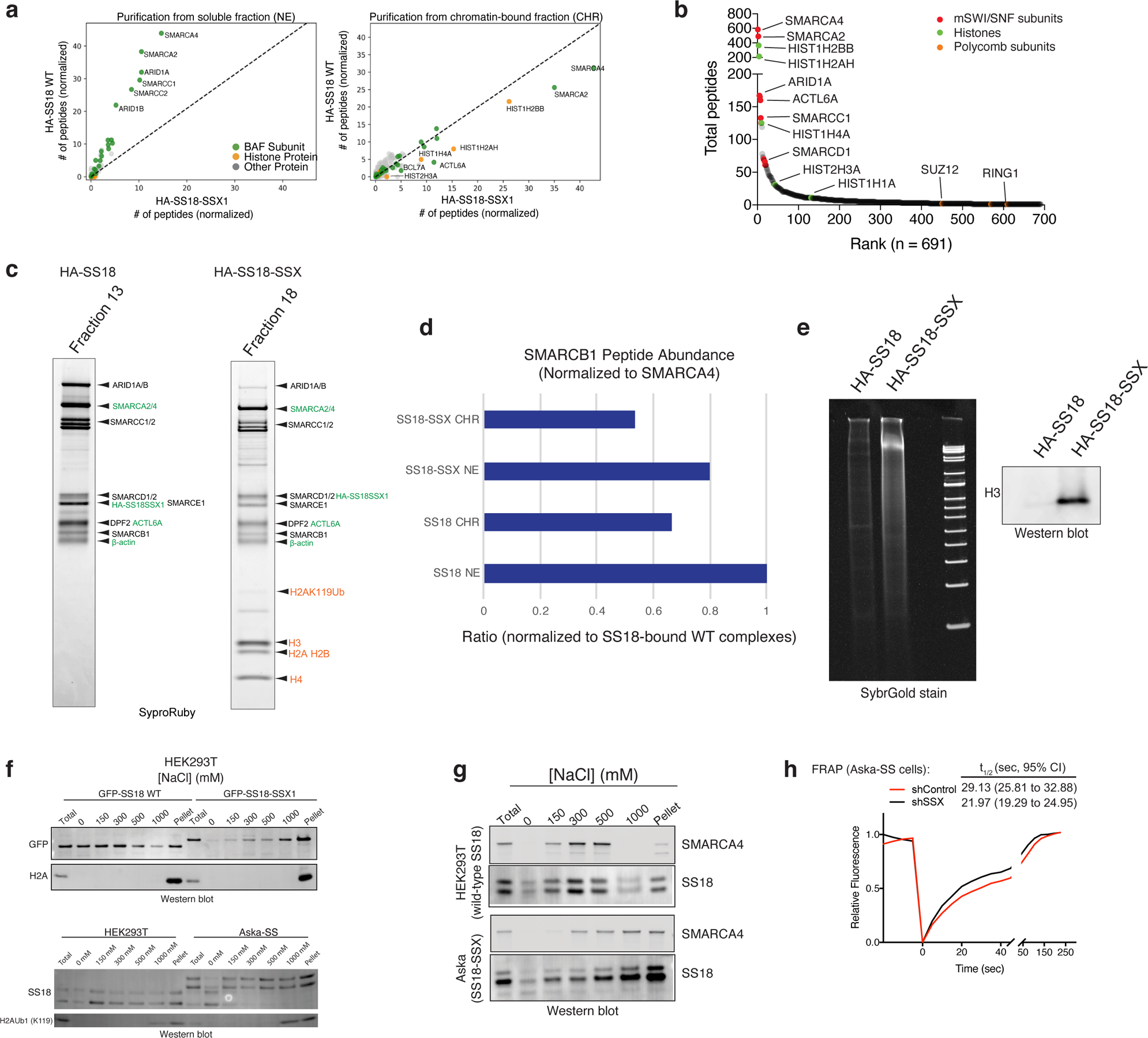
a, MS spectral counts for BAF complex subunits and histone proteins from HA-SS18 WT and HA-SS18-SSX purifications from soluble nuclear extract NE and CHR fractions from Fig. 1a. Total number of peptides (number of peptides normalized to bait, SS18, are shown.
b, Ranked peptides captured in HA-SS18-SSX purification (chromatin-bound fraction). Red, mSWI/SNF complex subunits. Green, histones. Orange, members of PRC1 and PRC2 complexes, shown for comparison. See also Extended Data Table 1.
c, SyproRuby staining indicating identified proteins from Fig.1c in Fraction 13 (HA-SS18 WT) and Fraction 18 (HA-SS18-SSX).
d, SMARCB1 peptide abundance (normalized to SMARCA4) and relative to SS18 WT-bound complexes (soluble NE fraction). NE, nuclear extract; CHR, chromatin-bound fraction.
e, (left) Cyber-gold staining of complexes purified from untreated (no benzonase) nuclear extracts isolated via ammonium sulfate extraction. (right) H3 immunoblot reveals prominent histone binding in HA-SS18-SSX-bound complexes but not in HA-SS18 WT-bound complexes.
f, (Top) Immunoblot for GFP and H2A performed on HEK-293T cells infected with either GFP-SS18 WT or GFP-SS18-SSX following differential salt extraction (0–1000 mM NaCl).(Bottom) Immunoblot for SS18 and H2A K119Ub performed on HEK-293T cells (naive) and Aska-SS cells following differential salt extraction (0–1000 mM NaCl) experiments.
g, Immunoblot for SMARCA4 and SS18 performed HEK-293T cells or Aska SS cells (SS18-SSX+) following differential salt extraction (0–1000 mM NaCl).
h, FRAP experiments performed in the Aska SS cell line modified to express BRG1 (SMARCA4)-Halo. Aska-SS cells were treated with either shControl shRNA hairpin or shSSX (targeting the SS18-SSX fusion). Recovery t1/2 times (seconds) with 95% CIs are shown, n=20 cells.
Extended Data Figure 2. The SSX 78aa protein binds mononucleosomes, with preference for nucleosomes decorated with repressive histone modifications.
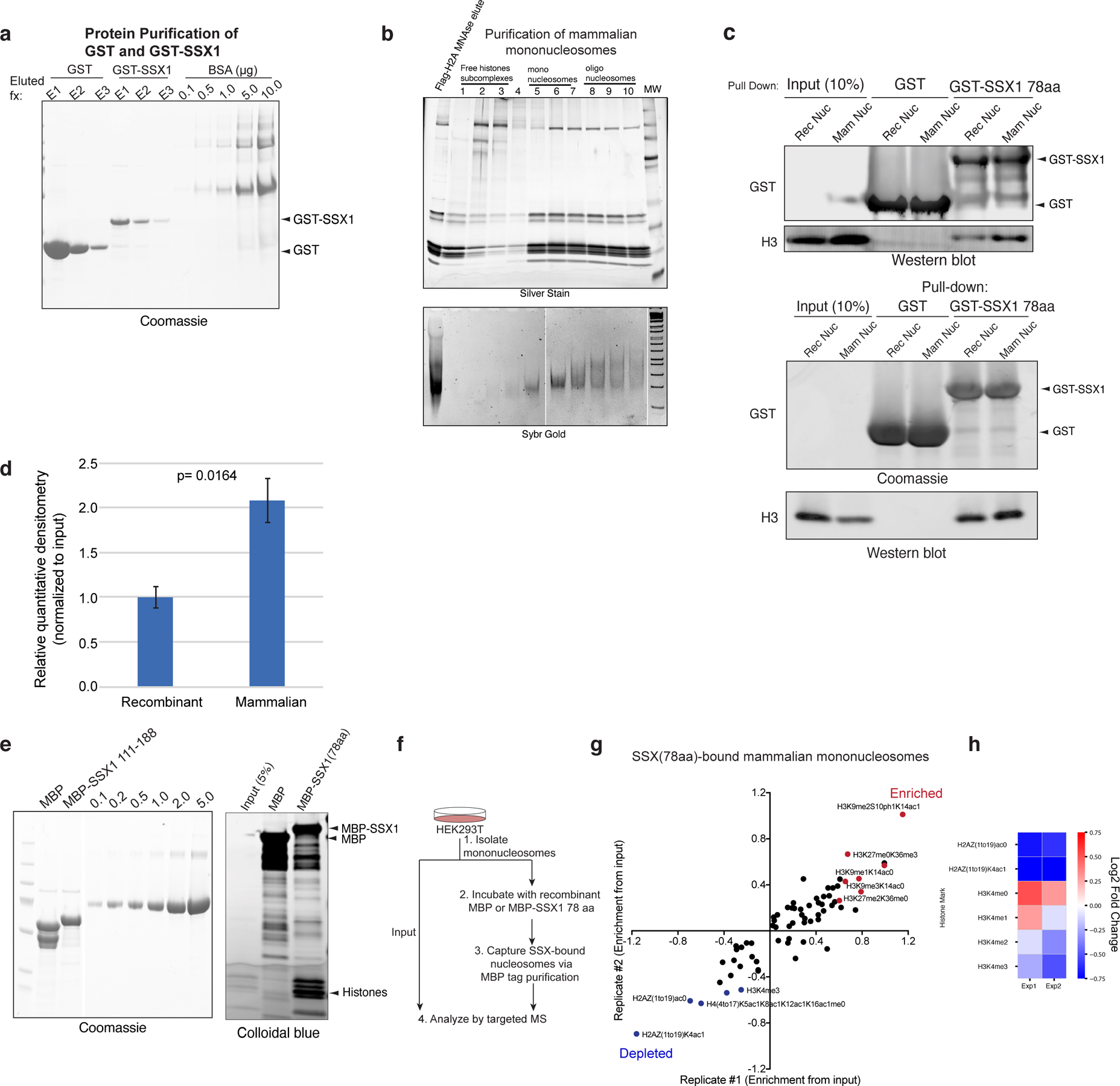
a, Coomassie-stained gel of recombinantly-purified GST, GST-SSX (78aa) proteins, run next to BSA protein as control.
b, Purification of mammalian mononucleosomes from HEK-293T cells using MNase digestion.
c, Incubation of GST or GST-SSX (78aa) with either recombinant or mammalian mononucleosomes, resolved by immunoblot for GST and histone H3 or Coomassie and histone H3. Two representative experiments are shown.
d, Quantitative densitometry of histone H3 normalized to input reflecting GST-SSX 78aa preferential binding to mammalian mononucleosomes (prepared via MNase digestion in HEK-293T cells) versus recombinant, unmodified nucleosomes. Bars represent averages of n=3 independent experiments, error bars represent standard deviation; p-value= 0.0164.
e, Purification of MBP and MBP-SSX (78aa) proteins for targeted, quantitative histone mass-spectrometry.
f, Schematic for targeted MS experiments.
g, Enrichment of SSX-bound histone peptides, over input. Enriched and depleted proteins are shown in red and blue, respectively. Quantitative histone mass spectrometry performed on MBP-SSX1 (versus MBP control) incubated with pooled mononucleosomes isolated from HEK-293T cells via MNase digestion. Experiment performed in n=2 replicates. See also Extended Data Table 2.
h, Heatmap reflecting enrichment or depletion of selected histone marks, including H2AZ and H3K4 methylation states. Scale= log2FC
Extended Data Figure 3. Nucleosome binding and nuclear localization properties of SS18-SSX and SSX variants.
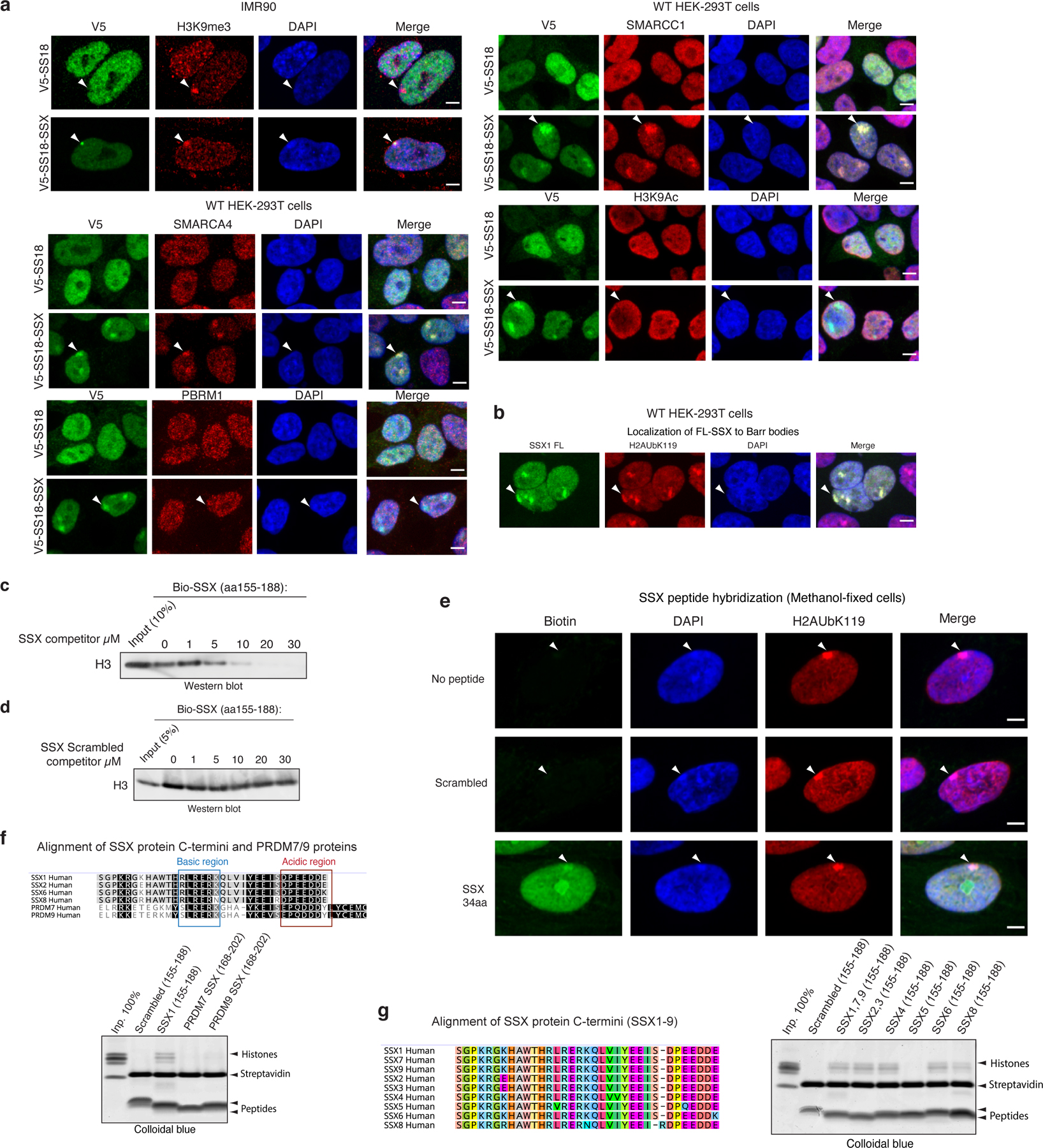
a, Immunofluorescence imaging performed on IMR90 fibroblasts and HEK-293T cells infected with either V5-SS18-SSX or V5-SS18. Visualized in red for H3K9me3, SMARCA4, PBRM1, SMARCC1, H3K9Ac across experiments. DAPI is shown as nuclear stain and merged images are provided with scale bars, Scale bar, 5μm.
b, IF-based localization of SSX1 FL (1–188aa) in fibroblasts. H2A K119Ub, DAPI counterstain, and merged images are shown. Scale bar, 5μm.
c, Peptide competition experiment using Biotinylated SSX peptide (aa 155–188) and unlabeled SSX (aa 155–188). Visualization for Histone H3 using immunoblot.
d, Peptide competition experiment using Biotinylated SSX peptide (aa 155–188) and Scrambled control SSX peptide (aa 155–188). Visualization for Histone H3 using immunoblot.
e, SSX peptide hybridization experiments performed on methanol-fixed cells. Streptavidin (SA) used for visualization of biotinylated SSX peptides, H2A K119Ub for Barr bodies. DAPI counterstain and merged images shown. Scale bar, 5μm.
f, Top, conservation analysis among SSX and PRDM 7/9 human protein regions. Bottom, peptide pull down experiments with recombinant nucleosomes performed with Scrambled control SSX1, SSX1, PRDM7, PRDM9. Visualization by colloidal blue staining.
g, Left, alignment of SSX proteins (SSX 1–9). Right, peptide pull down experiments with recombinant nucleosomes performed with aa 155–188 of SSX family members. Visualization by colloidal blue staining.
Extended Data Figure 4. Defining a minimal 34-aa SSX region responsible for chromatin engagement and oncogenic gene expression.
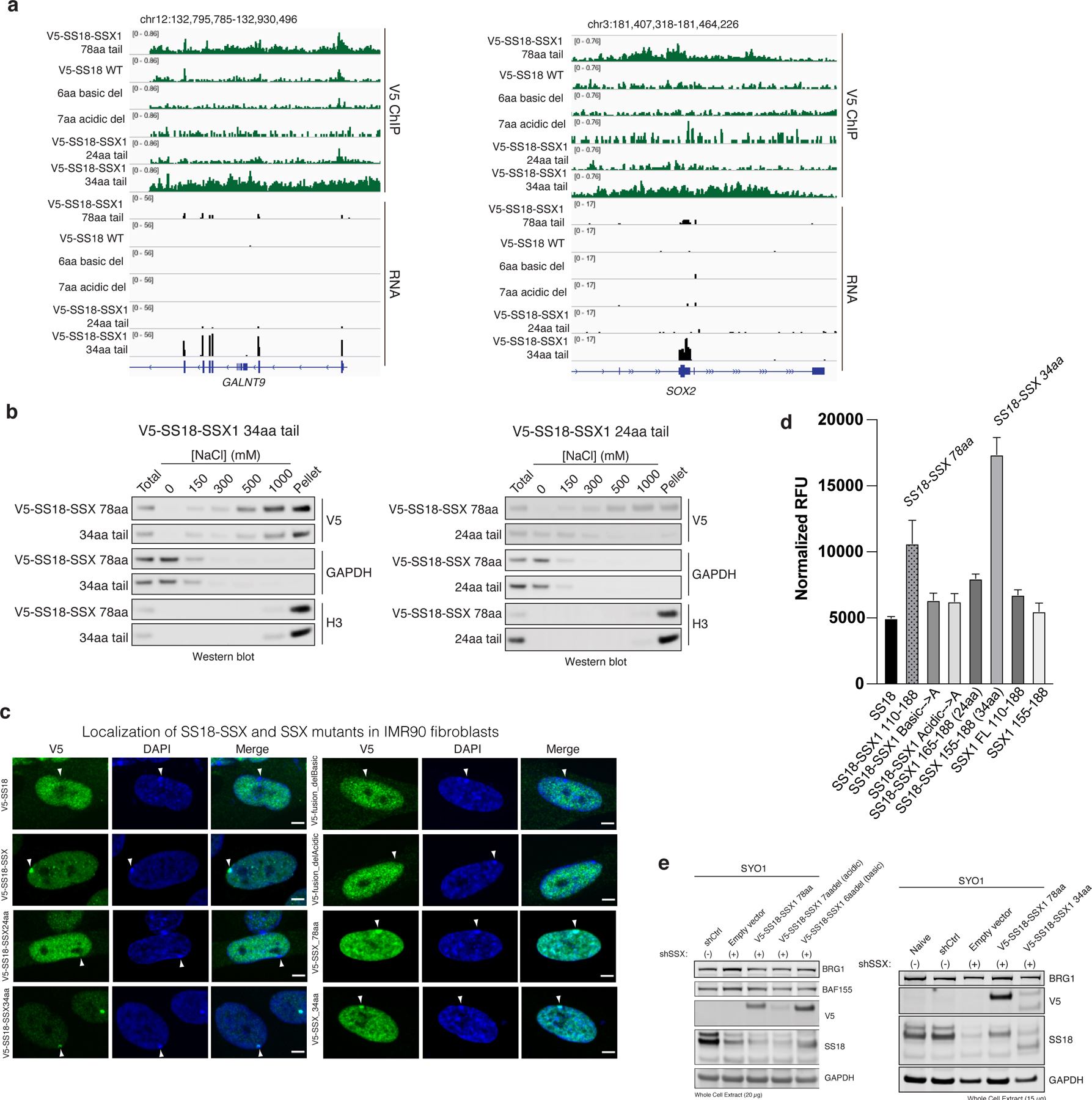
a, Additional representative V5 ChIP-seq and RNA-seq tracks, here shown at the GALNT9 and SOX2 loci.
b, Differential salt experiments ([0–1000mM NaCl]) performed on HEK-293T cells infected with either SS18-SSX 34aa versus SS18-SSX 24aa. Immunoblots for V5 as well as GAPDH and H3 (controls) are shown.
c, Immunofluorescence imaging of IMR90 fibroblasts infected with SS18 and SS18-SSX variants, as indicated, and stained for V5 (SS18-SSX or SSX variant) and DAPI; merged images are shown. Localization to H2AUb119-high sites (Barr bodies) is highlighted. Scale bar, 5μm.
d, Beta-gal senescence assay performed on IMR90 cells infected with WT SS18, SS18-SSX and SSX FL and 78aa variants, as indicated. Averaged normalized RFUs are shown (bar graph indicates mean RFU across n=3 independent experiments; error bars represent standard deviation.
e, SYO-1 synovial sarcoma cells were treated with either shCtrl (control hairpin) or shSSX (shRNA targeting SSX) to reduce levels of endogenous fusion, followed by rescue of SS18-SSX WT and mutant variants or empty vector control. Proliferation was evaluated over 16 days (see also Fig. 2i).
Extended Data Figure 5. The SSX basic region and SMARCB1 C-terminal alpha helical domain compete for nucleosome acidic patch binding.
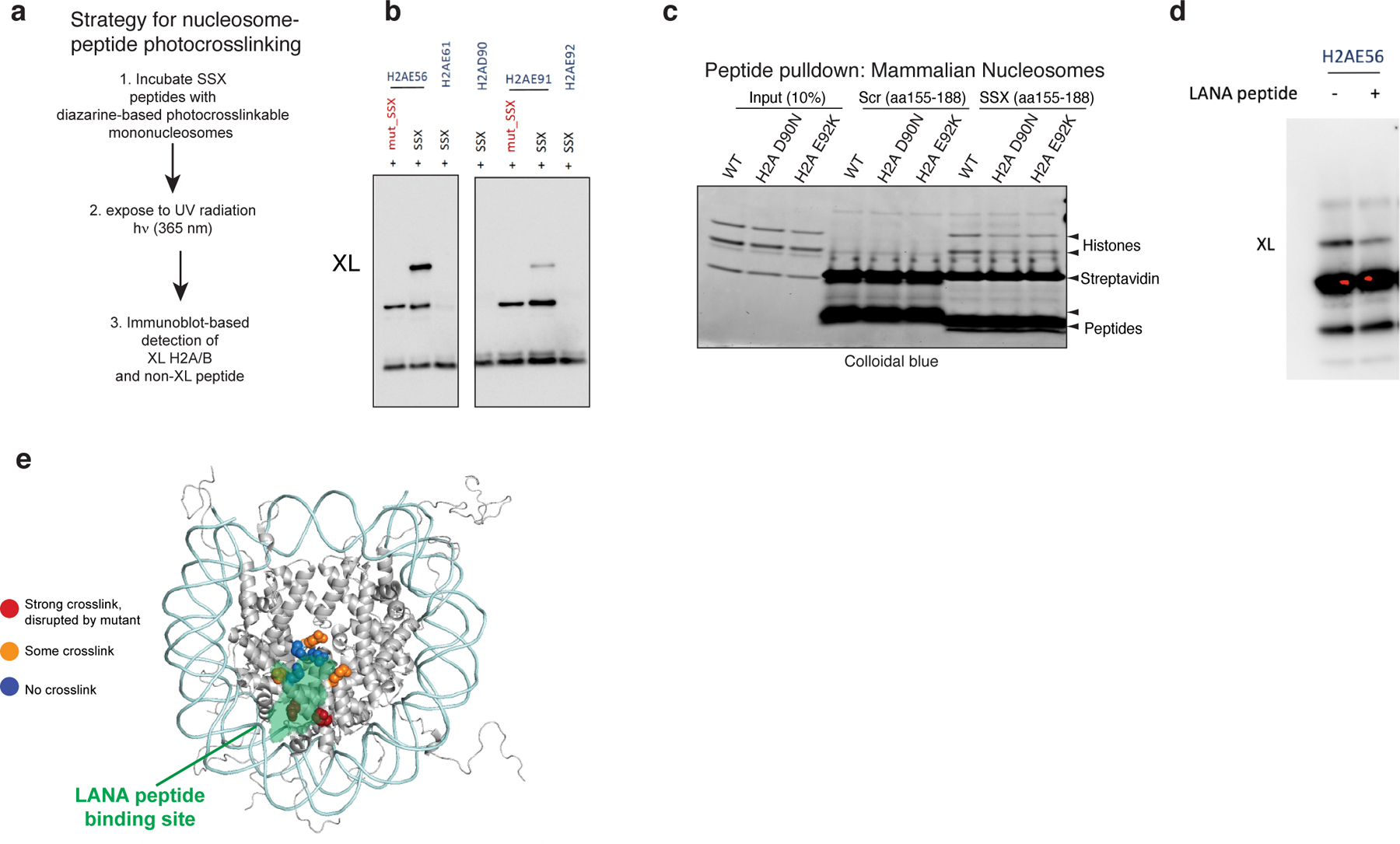
a, Schematic outlining strategy for nucleosome-peptide photocrosslinking.
b, Additional (replicate) photocrosslinking experiments performed with reactive diazirine probes localized throughout the nucleosome acidic patch region indicate strongest binding to H2A E56 and H2B E113 residues, weaker binding to H2A E91, and no binding to E61, E92, and D90 residues. Experimental conditions: 0.3uM mononucleosomes, 3uM SSX, 150mM KCl.
c, Pulldown experiments performed with either Scrambled or SSX 34aa peptides (biotinylated) incubated with mammalian mononucleosomes prepared from cells infected with WT H2A, or H2AD90N, H2A E92K mutant variants.
d, Photocrosslinking experiments performed with SSX 34aa peptide incubated with nucleosomes modified at the H2A E56 residue, with and without LANA peptide competition.
e, Model indicating docking of solved LANA peptide-nucleosome binding region and SSX peptide crosslinking sites in the nucleosome acidic patch. Interacting residues from photocrosslinking experiments are highlighted.
Extended Data Figure 6. Mutations in the basic region of SSX affect the targeting and function of SS18-SSX-containing BAF complexes.
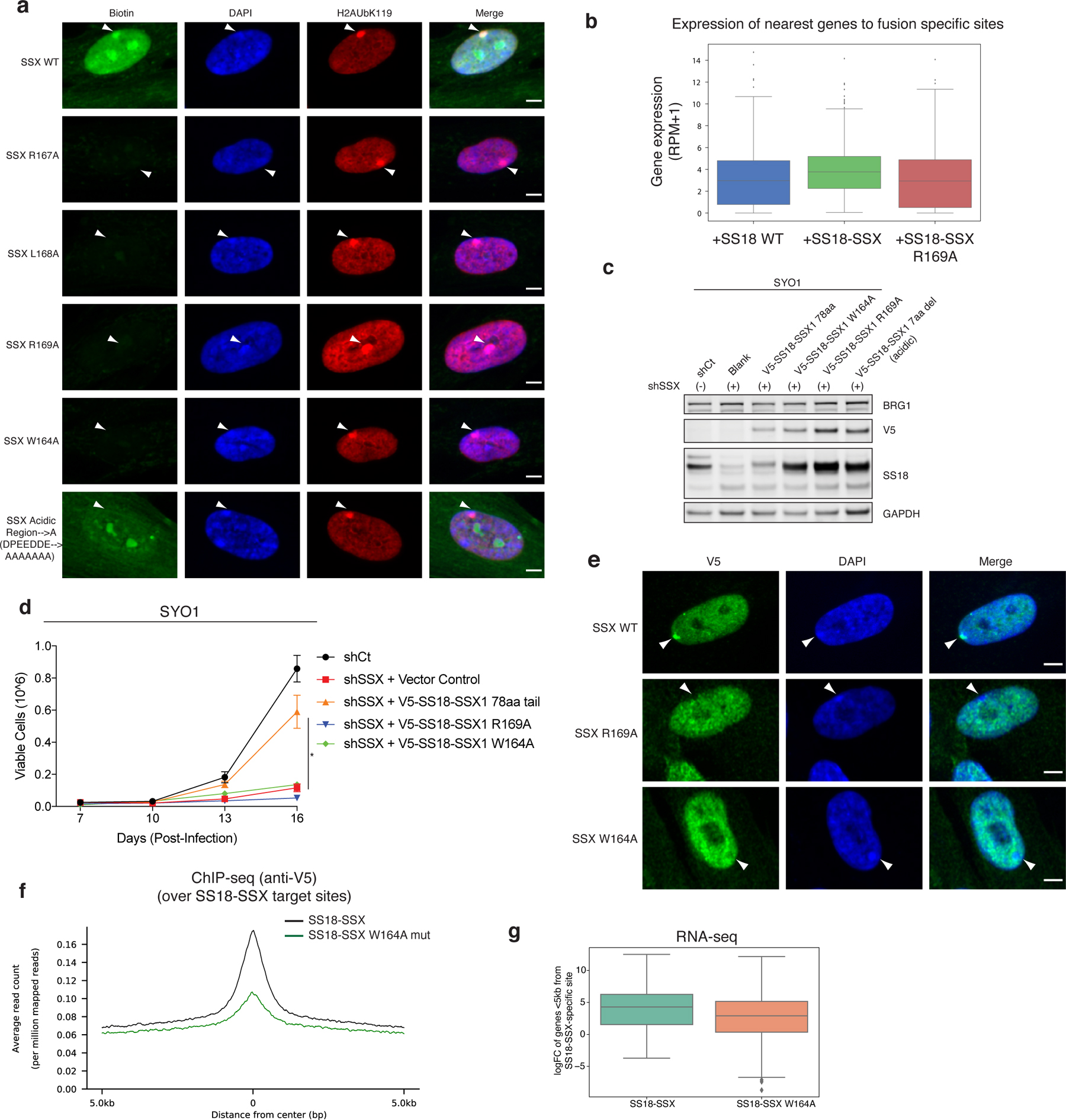
a, Peptide hybridization of IMR90 cells using SSX and mutant basic region mutant peptides. Arrows indicate positions of the Barr bodies. Scale bar 5μm.
b, Gene expression changes across each SS18 WT and SS18-SSX variant conditions from Fig. 3g.
c, Immunoblot performed on whole-cell extracts (RIPA extraction) from SYO1 cells treated with either shCtrl or shSSX and infected with either empty vector or SS18-SSX variants, used in proliferation experiments in Extended Data Fig. 6d.
d, Proliferative rescue experiments performed in SYO-1 SS cell line treated with shSSX and rescued with either vector control, SS18-SSX or SS18-SSX (R169A or W164A) variants. n=3 independent experiments performed, error bars represent standard deviation; *p<0.05.
e, Peptide hybridization of IMR90 cells using SSX and mutant basic region mutant (W164A and R169A) peptides. Arrows indicate positions of the Barr bodies. Scale bar 5μm.
f, ChIP-seq studies (anti-V5) performed in CRL7250 cells infected with either SS18-SSX or SS18-SSX W164A mutant, mapped as summary plot over SS18-SSX target sites.
g, RNA-seq (gene expression), box and whisker plots indicating average expression in SS18-SSX versus SS18-SSX W164A mutant conditions.
Extended Data Figure 7. Subunit composition, chromatin binding, and functional properties of SS18-SSX-bound BAF complexes.
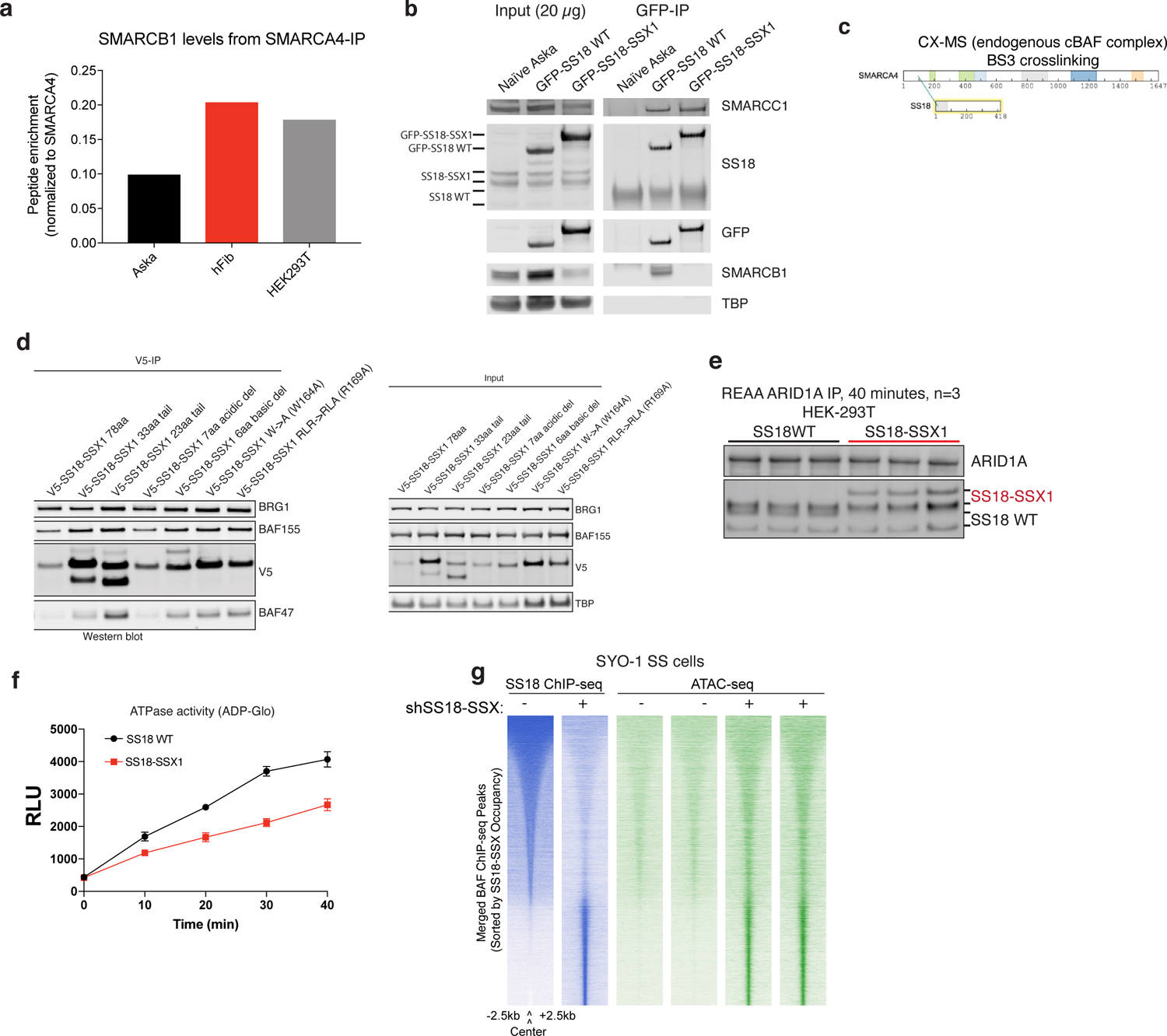
a, SMARCB1 peptide abundance calculated from MS experiments (anti-SMARCA4 (BRG1) IPs) performed in Aska-SS synovial sarcoma cells, human Fibroblasts, and HEK-293T cells. Peptide abundance normalized to SMARCA4 abundance.
b, Input and GFP IPs performed in Aska-SS cells infected with either GFP-SS18 or GFP-SS18-SSX. SMARCC1, SS18, GFP, SMARCB1, and TBP levels are shown.
c, SS18-SMARCA4 crosslinks detected in CX-MS experiments of intact, fully-formed canonical BAF complexes in Mashtalir et al., Cell 2018.
d, Immunoblot studies performed on CRL7250 cells infected with SS18-SSX variants indicated.
e, Immunoblot performed for ARID1A and SS18 on complexes captured via ARID1A , used for nucleosome remodeling and ATPase assays.
f, ATPase activity calculated by ADP-Glo for SS18 WT- and SS18-SSX-containing BAF complexes. t, 0–40min time course, n=3 experimental replicates at each time point; error bars represent standard deviation.
g, ATAC-seq studies performed in SYO-1 SS cells in shCtrl and shSSX conditions, mapped over SS18 ChIP-seq.
Extended Data Figure 8. SS18-SSX-bound BAF complexes preferentially bind H2A K119Ub-marked nucleosomes.
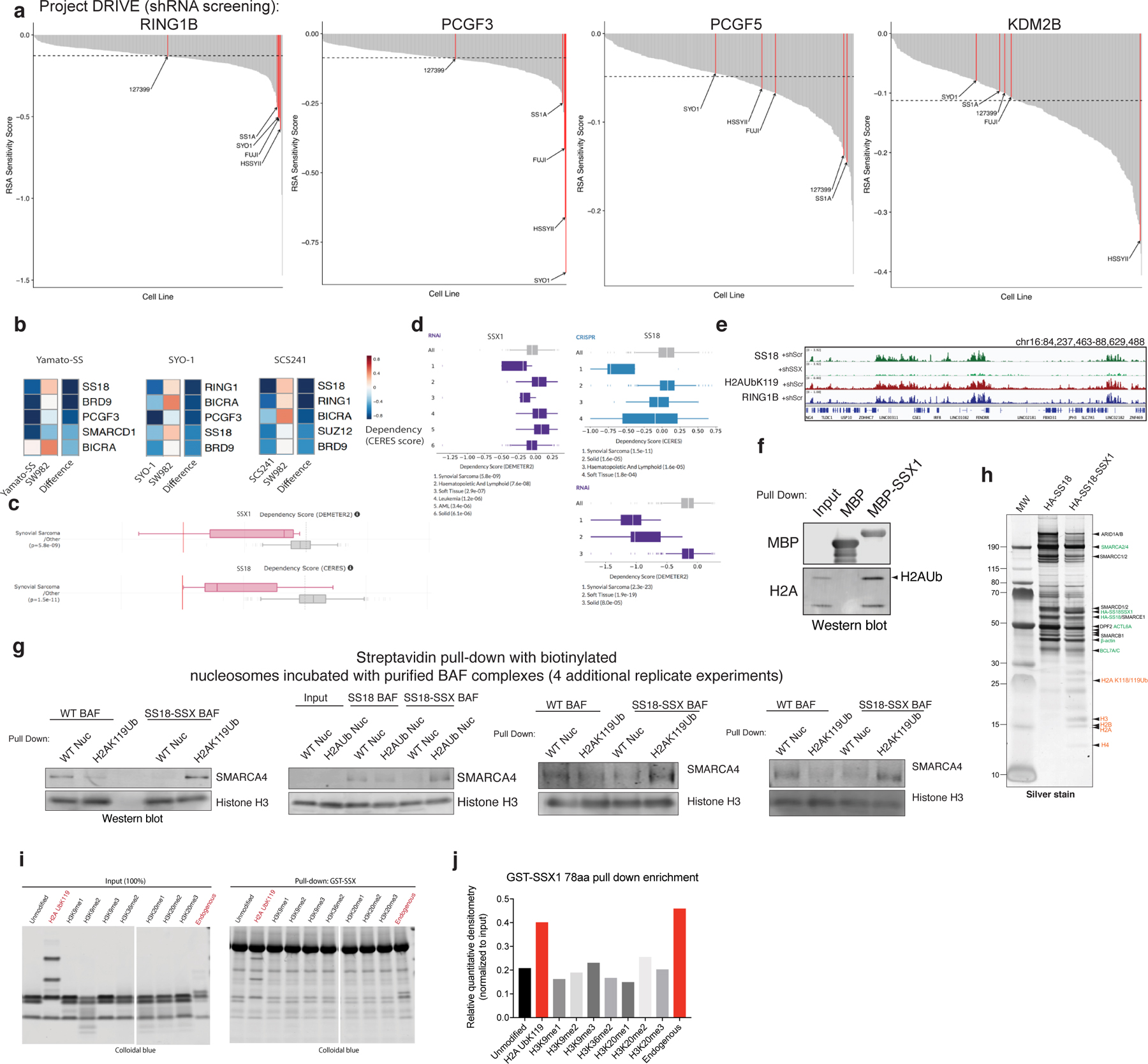
a, Waterfall dependency plots for RING1B, PCGF3, PCGF5 and KDM2B genes across n=387 cell lines in Project DRIVE Dataset (Novartis).
b, CERES dependency scores (fitness dropout) derived from genome-scale fitness screens performed using CRISPR-Cas9-based methods (Achilles, Broad Institute; https://depmap.org/portal/achilles/). Difference is the score calculated between SYO1, Yamato-SS, SCS241 (SS18-SSX+) cells and SW982 cells (negative for fusion, histologic mimic). Blue, enriched for dependency. mSWI/SNF, PRC1, PRC2 members are shown.
c, CERES and DEMETER Dependency scores for SSX1 and SS18 genes for CRISPR-Cas9 and RNAi datasets, respectively. Synovial sarcoma cell lines are indicated in pink; all other cell lines are represented in gray.
d, CERES and DEMETER Dependency scores for SSX1 and SS18 genes for CRISPR-Cas9 and RNAi datasets, respectively. Synovial sarcoma and soft tissue (SS cell lines) exhibit preferential dependency. (Project DRIVE; https://oncologynibr.shinyapps.io/drive/). SS cell lines containing the SS18-SSX fusion oncoprotein are highlighted in red.
e, H2A K119Ub and RING1B ChIP-seq tracks over selected loci, aligned with SS18 (BAF) localization in SYO-1 cells treated with shScramble or shSS18-SSX.
f, MBP-SSX1 (78aa) pull down experiments indicate capture of histones, and specifically, H2A K119Ub species.
g, Streptavidin-based pull-down experiments (n=4 independent replicate experiments) using endogenous, fully-assembled HA-SS18- or HA-SS18-SSX-bound BAF complexes incubated with biotinylated WT nucleosomes (unmodified) or H2A K119Ub-modified nucleosomes. SMARCA4 and Histone H3 immunoblots are shown.
h, Representative silver stain of the WT SS18 complexes and SS18-SSX fusion complexes isolated using ammonium sulfate nuclear extraction protocol. Identified proteins are labeled.
i, Pull down experiments performed using GST-SSX incubated with unmodified or a series of modified recombinant mononucleosomes, or endogenous mononucleosomes (mammalian, purified via MNase digestion from HEK-239T cells).
j, Quantitative densitometry performed on experiment in Extended Data Fig. 8i.
Extended Data Figure 9. SSX targeting requires PRC1 complex-mediated H2A K119Ub placement.
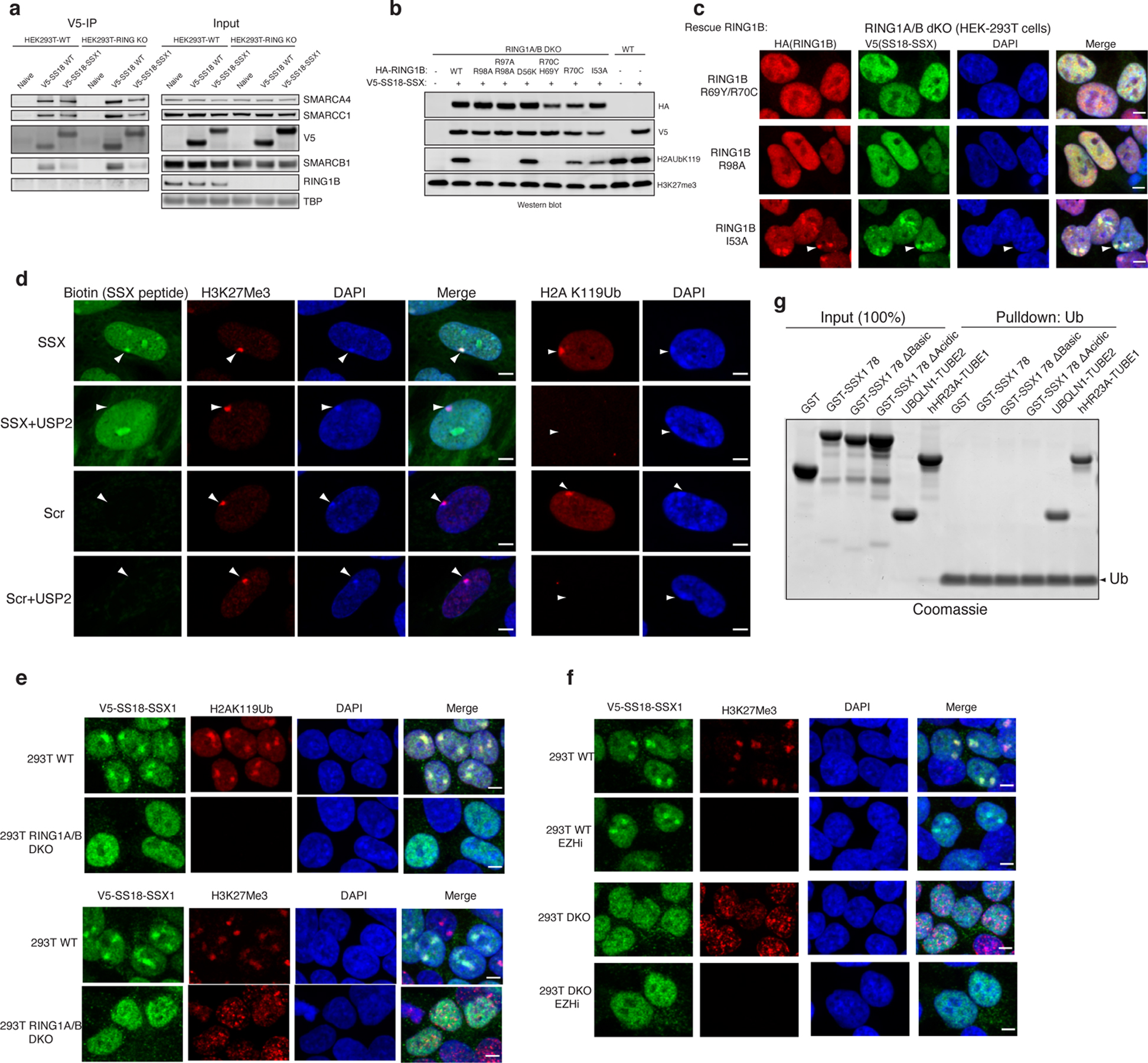
a, Immunoblots performed on V5 IP and Input protein levels in WT and RING1A/B double KO (dKO) HEK-293T cells.
b, Immunoblot of representative, structurally-guided RING1B mutations which inhibit H2AK119Ub deposition partially, fully, or not at all.
c, Immunofluorescence imaging for RING1B (red), V5 SS18-SSX (green), with DAPI nuclear stain, and merged images.
d, Peptide hybridization experiments. Representative Images of SSX labeling of Barr bodies (inactive X) identified for each condition using H3K27me3 staining. Peptides (SSX or Scrambled) were incubated methanol-fixed cells, untreated or treated with USP2 deubiquitinating enzyme.
e, V5-SS18-SSX, H2A K119Ub, and H3K27me3 IF studies performed in WT and RING1A/B dKO 293T cells.
f, DMSO control or EZH2 inhibitor treatment (to inhibit H3K27me3 placement) indicates no change to SS18-SSX foci localized to Barr bodies.
g, Incubation of GST-SSX WT, SSX mutant variants, or UBQLN1-TUBE2 or hHR23A-TUBE1 (pos controls) with Ub-coated beads.
Supplementary Material
Acknowledgements
We thank members of the Kadoch lab for critical feedback and advice. We thank members of the M.J.M. dissertation advisory committee, including Scott Armstrong, David Liu, and Matthew Meyerson, for their guidance and mentorship throughout the development of this project. M.J.M. was supported by the Harvard Medical School GSAS Fellowship Program. N.M. is supported by NIH K99/R00 K99CA237855. H.T.D. was funded by a postdoctoral fellowship from the Jane Coffin Childs Memorial fund. This work was supported in part by awards from the NIH DP2 New Innovator Award 1DP2CA195762–01(C.K.), the American Cancer Society Research Scholar Award RSG-14–051-01-DMC (C.K.), the Pew- Stewart Scholars in Cancer Research Grant (C.K.), Alex’s Lemonade Stand Foundation ‘A Award’ (C.K.) , NIH 1R01 CA237241–01 (C.K.), the NIH Cancer Moonshot FusOnc2 1U54 CA231638–01 (C.K.), NIH R37-GM086868 (T.W.M.), and NIH PO1- CA196539 (T.W.M.).
Footnotes
Competing Interests Statement
C.K. is the scientific founder, fiduciary Board of Directors member, Scientific Advisory Board member, consultant and shareholder of Foghorn Therapeutics, Inc. (Cambridge, MA).
Reporting Summary Statement
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data Availability Statement
All sequencing data has been deposited with the Gene Expression Omnibus with accession number GSE139055.(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139055) . Proteomic data have been deposited in PRIDE with accession number PXD018715. Uncropped gel and blot images are presented in Supplementary Figure 1.
References
- 1.Fujisawa T & Filippakopoulos P Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol 18, 246–262 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Hyun K, Jeon J, Park K & Kim J Writing, erasing and reading histone lysine methylations. Exp Mol Med 49, e324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiong X et al. Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat Chem Biol 12, 1111–1118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dann GP et al. ISWI chromatin remodellers sense nucleosome modifications to determine substrate preference. Nature 548, 607–611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levendosky RF & Bowman GD Asymmetry between the two acidic patches dictates the direction of nucleosome sliding by the ISWI chromatin remodeler. Elife 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dao HT, Dul BE, Dann GP, Liszczak GP & Muir TW A basic motif anchoring ISWI to nucleosome acidic patch regulates nucleosome spacing. Nat Chem Biol (2019). [DOI] [PMC free article] [PubMed]
- 7.Sandoval GJ et al. Binding of TMPRSS2-ERG to BAF Chromatin Remodeling Complexes Mediates Prostate Oncogenesis. Mol Cell 71, 554–566.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boulay G et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 171, 163–178.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadoch C et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45, 592–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valencia AM & Kadoch C Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat Cell Biol 21, 152–161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kadoch C & Crabtree GR Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 1, e1500447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwase S et al. Epigenetic Etiology of Intellectual Disability. J Neurosci 37, 10773–10782 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet 49, 289–295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McBride MJ et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 33, 1128–1141.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kadoch C & Crabtree GR Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark J et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet 7, 502–8 (1994). [DOI] [PubMed] [Google Scholar]
- 17.Crew AJ et al. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. Embo j 14, 2333–40 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Leeuw B, Balemans M, Olde Weghuis D & Geurts van Kessel A Identification of two alternative fusion genes, SYT-SSX1 and SYT-SSX2, in t(X;18)(p11.2;q11.2)-positive synovial sarcomas. Hum Mol Genet 4, 1097–9 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Smith HA & McNeel DG The SSX family of cancer-testis antigens as target proteins for tumor therapy. Clin Dev Immunol 2010, 150591 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banito A et al. The SS18-SSX Oncoprotein Hijacks KDM2B-PRC1.1 to Drive Synovial Sarcoma. Cancer Cell 33, 527–541.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mashtalir N et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1288.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakayama RT et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet 49, 1613–1623 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan J et al. The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity-independent genomic targeting. Nat Genet 51, 618–626 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbera AJ et al. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311, 856–61 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Valencia AM et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell 179, 1342–1356 e23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye Y et al. Structure of the RSC complex bound to the nucleosome. Science (2019). [DOI] [PMC free article] [PubMed]
- 27.He S et al. Structure of nucleosome-bound human BAF complex. Science 367, 875–881 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Kohashi K et al. Reduced expression of SMARCB1/INI1 protein in synovial sarcoma. Mod Pathol 23, 981–90 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Blackledge NP et al. PRC1 catalytic activity is central to Polycomb system function. bioRxiv, 667667 (2019). [DOI] [PMC free article] [PubMed]
- 30.Wang H et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–8 (2004). [DOI] [PubMed] [Google Scholar]
- 31.McGinty RK, Henrici RC & Tan S Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buchwald G et al. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J 25, 2465–74 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eskeland R et al. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol Cell 38, 452–64 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Illingworth RS et al. The E3 ubiquitin ligase activity of RING1B is not essential for early mouse development. Genes Dev 29, 1897–902 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson CJ et al. Structural Basis for Recognition of Ubiquitylated Nucleosome by Dot1L Methyltransferase. Cell Rep 26, 1681–1690 e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Worden EJ, Hoffmann NA, Hicks CW & Wolberger C Mechanism of Cross-talk between H2B Ubiquitination and H3 Methylation by Dot1L. Cell 176, 1490–1501 e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valencia-Sanchez MI et al. Structural Basis of Dot1L Stimulation by Histone H2B Lysine 120 Ubiquitination. Mol Cell 74, 1010–1019 e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baarends WM et al. Histone ubiquitination and chromatin remodeling in mouse spermatogenesis. Dev Biol 207, 322–33 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Huntley S et al. A comprehensive catalog of human KRAB-associated zinc finger genes: insights into the evolutionary history of a large family of transcriptional repressors. Genome Res 16, 669–77 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Creech AL et al. Building the Connectivity Map of epigenetics: chromatin profiling by quantitative targeted mass spectrometry. Methods 72, 57–64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dao HT, Dul BE, Dann GP, Liszczak GP & Muir TW A basic motif anchoring ISWI to nucleosome acidic patch regulates nucleosome spacing. Nat Chem Biol (2019). [DOI] [PMC free article] [PubMed]
- 42.Daou S et al. Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proc Natl Acad Sci U S A 108, 2747–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carvalho P, Gupta ML Jr., Hoyt MA & Pellman D Cell cycle control of kinesin-mediated transport of Bik1 (CLIP-170) regulates microtubule stability and dynein activation. Dev Cell 6, 815–29 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Mashtalir N et al. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell 54, 392–406 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data has been deposited with the Gene Expression Omnibus with accession number GSE139055.(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139055) . Proteomic data have been deposited in PRIDE with accession number PXD018715. Uncropped gel and blot images are presented in Supplementary Figure 1.