

Abstract
Analysis of 772 complete SARS-CoV-2 genomes from early in the Boston area epidemic revealed numerous introductions of the virus, a small number of which led to most cases. The data revealed two superspreading events. One, in a skilled nursing facility, led to rapid transmission and significant mortality in this vulnerable population but little broader spread, while other introductions into the facility had little effect. The second, at an international business conference, produced sustained community transmission and was exported, resulting in extensive regional, national, and international spread. The two events also differed significantly in the genetic variation they generated, suggesting varying transmission dynamics in superspreading events. Our results show how genomic epidemiology can help understand the link between individual clusters and wider community spread.
SARS-CoV-2 has now caused over fifty million infections and over one million reported deaths (1) in one of the worst public health crises of the past century. Cases are currently surging to unprecedented levels in the United States, reaching over 180,000 cases reported daily during November 2020. Massive ongoing transmission globally underscores that most countries have not found effective ways to control spread of the virus; better understanding of transmission dynamics could contribute to more targeted and effective responses to the pandemic. Reports of COVID-19 transmission have featured clusters of cases linked to gatherings, including ones in workplaces (2) and churches (3), and especially in close living environments such as care homes (4) and homeless shelters (5). These clusters are thought to often involve superspreading (6, 7), in which one individual infects many others (defined here as more than eight secondary cases; see Materials and Methods), yet the contribution of these events to regional and national transmission is not well understood. Instead, the evidence indicating that case clusters and superspreading events are major drivers of transmission has been based largely on time-series data showing an increase in cases following them (8), which has limited ability to determine the contribution of any event to overall transmission. Contact tracing from such events can be similarly uninformative, as it is resource intensive, invasive, and often limited in scope. Likewise, without genetic data about the viruses involved, it is often not possible to distinguish superspreading events from other forms of locally intense transmission, or from cases that occur in close proximity by chance. Yet understanding the role of superspreading events in transmission is critical for prioritizing public health interventions. To further that understanding, we used genomic epidemiology to investigate the introduction and spread of SARS-CoV-2 in the Boston, Massachusetts (MA) area, which was severely affected in the first wave of the pandemic. These data allowed us to study early outbreak dynamics and to examine the role of importations and superspreading events in fueling epidemic spread.
Genomic epidemiology of Boston superspreading events
The first known case in the Boston area was confirmed on February 1, 2020 (9); case counts rapidly increased through March and peaked in the third week in April. We performed viral genome sequencing and phylogenetic analysis of SARS-CoV-2-positive nasopharyngeal (NP) samples collected between March 4th and May 9th, 2020 by the Massachusetts Department of Public Health (MADPH) and Massachusetts General Hospital (MGH). Our dataset includes nearly all confirmed early cases of the epidemic (Fig. 1, A and B); samples from many of the highest-prevalence communities in the Boston area across the first wave (Fig. 1C), including Chelsea, Revere, and Everett (Fig. 1C and fig. S1); and samples from putative superspreading events involving an international conference and congregate living environments, specifically among residents and staff at a skilled nursing facility and in homeless shelters. As seen elsewhere, close-quarters living facilities like these have been disproportionately affected by COVID-19 in MA, accounting for 22% of confirmed cases and 64% of reported deaths through August 1, 2020 (10).
Fig. 1. Epidemiology of SARS-CoV-2 in Massachusetts and of sequenced viral genomes.
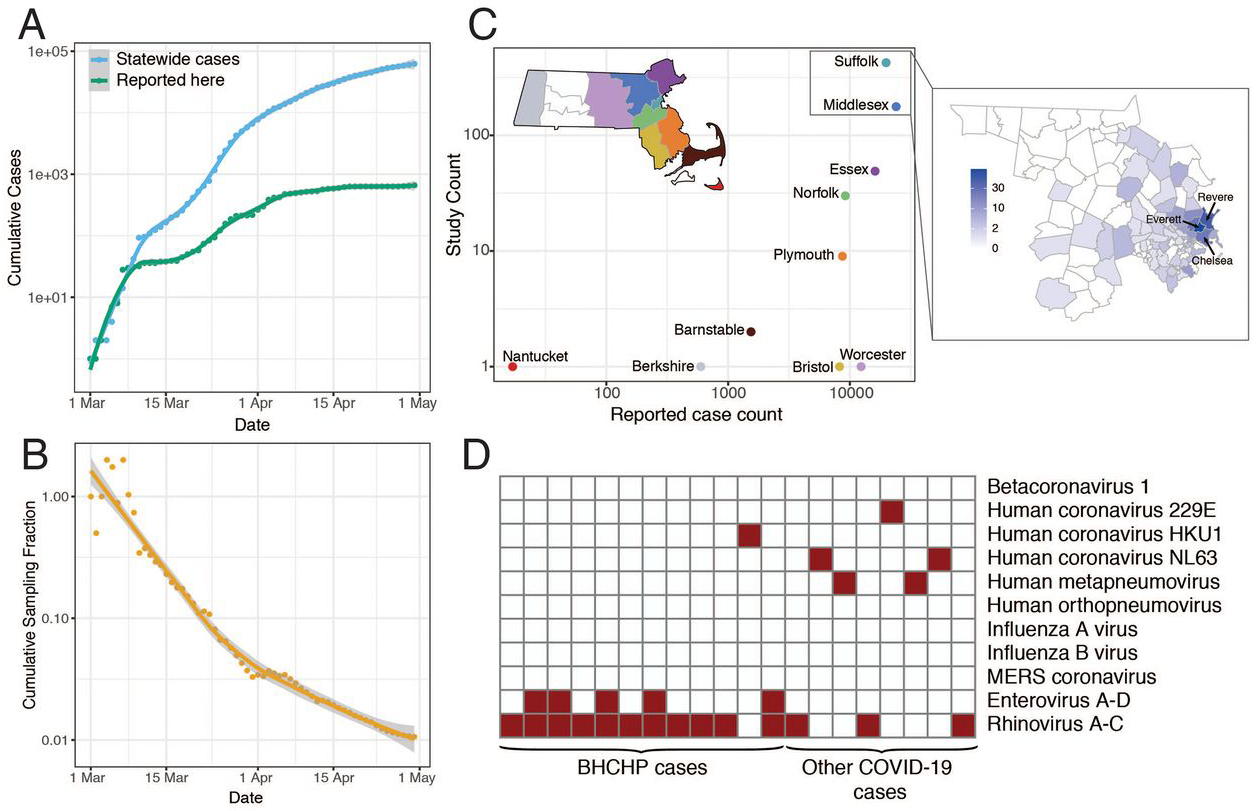
(A) Cumulative confirmed and presumed cases reported state-wide in MA (10) from March 1 through May 1, 2020, and the number of these cases that successfully yielded complete genomes with >98% coverage (green) in this study. (B) Cumulative proportion of all MA confirmed positive cases with complete genome sequences from unique individuals that are part of this dataset over time. (C) Total number of cases compared to cases in this study by MA county. Points are colored by state as shown in the state map. Suffolk and Middlesex counties are shown in detail to the right with counts from this study shown by zip code. (D) Detection of common respiratory viruses from metagenomic sequencing data. Samples with >10 reads mapped to at least 1 of these viruses using Kraken2 are shown in red. Enterovirus and Rhinovirus species have been grouped owing to the difficulty in discriminating at the sequence level.
We generated 778 complete SARS-CoV-2 assemblies (>98% complete) from 772 individuals, and an additional 72 partial genomes (>80% complete), using Illumina-based unbiased metagenomic short-read sequencing, followed by reference-guided assembly using viral-ngs 2.0.21 software (11) with the Wuhan-Hu-1 sequence (NC_045512.2) as the reference (Materials and Methods). Genome recovery and coverage were strongly correlated with viral abundance (fig. S2) and clinical diagnostic test results (fig. S3). Genomes were separated from one another by a median of 6 single nucleotide polymorphisms (SNPs) (interquartile range 4–9 SNPs; range 0–85 SNPs) (fig. S4, A and B). As expected during rapid population expansion, most alleles were rare, as assessed by a strongly negative Tajima’s D statistic throughout the genome (fig. S4C). In 20 samples (1.4% of sequenced cases) we identified the presence of at least one other common respiratory pathogen (Fig. 1D) via sequencing and confirmed it with a second assay (fig. S5). Co-infections were more commonly detected in residents and staff of homeless shelters (12/314) than in the other cases in the study (8/1117) (P = 0.0002, Fisher’s exact test).
We constructed a phylogenetic tree from this SARS-CoV-2 dataset alone, and additional trees from these data combined with repeated subsampling (Fig. 2A) from the Global Initiative on Sharing All Influenza Data (GISAID) (Materials and Methods). These trees form the basis of our analysis of the Boston area epidemic. The presence of a temporal signal in our dataset (fig. S6) means that a molecular clock can be fitted to infer the timing of ancestral branching based on the SARS-CoV-2 genomes.
Fig. 2. Introductions of SARS-CoV-2 into Massachusetts.
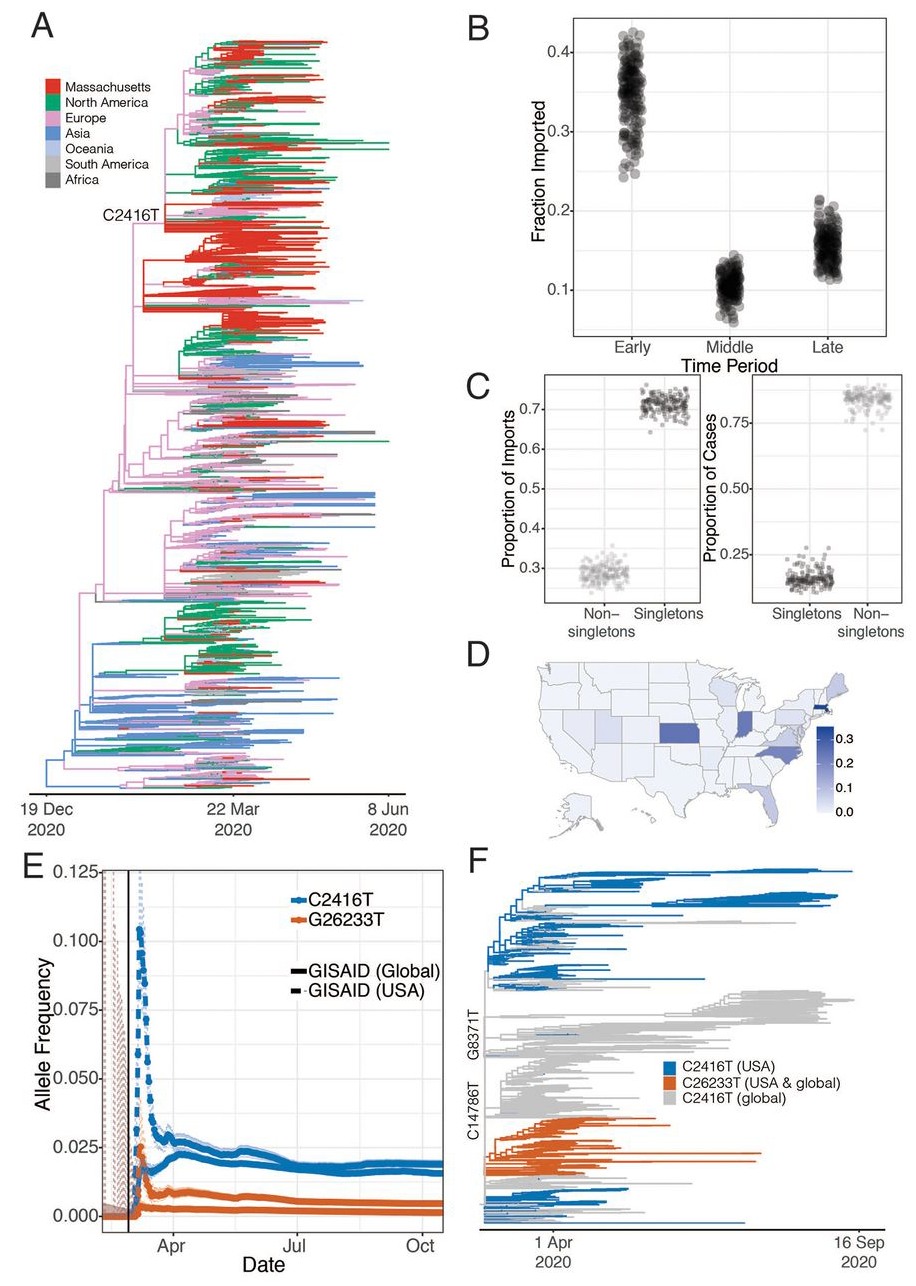
(A) Time tree of 772 MA genomes and a global set of 4,011 high-quality genomes from GISAID. To view an interactive version of this tree and for more information on specific sub-groupings within the MA dataset see auspice.broadinstitute.org. (B) Proportion of genomes that were inferred as imported (ancestral state as not from MA) in the early (prior to March 28, 2020), middle (March 28 – April 14, 2020) and late (after April 15, 2020) time periods of the MA epidemic. (C) The proportion of importation events and cases that were associated with singleton introductions (importation events associated with a single case in MA) into the Boston area over sub-sampled trees. (D) Allele frequency of the C2416T mutation by state. (E) Allele frequency of the C2416T and C26233T alleles in 159,043 GISAID samples reported through October 17, 2020. The vertical black line denotes the end of the business conference on February 27th. (F) Time tree of all sequences containing the C2416T variant collected before September 30th 2020
We identified putative introductions into the Boston area by carrying out ancestral state reconstruction for these phylogenetic trees (Materials and Methods). In total, we identified more than 122 [95% CI 122 – 161, median 143] putative introductions into the Boston area through May 9, stemming from sources on four continents (Table 1 and fig. S7, A and B). We characterize these introductions as putative because detailed ancestral reconstruction is limited by gaps in the global record of available genomes (12), and because the time scale of migration (hours to days) may exceed the rate of viral evolution (~1 new substitution every 13 days). Most of these inferred introductions occurred early in the pandemic, in March and early April, primarily from elsewhere in North America and from Europe (Table 1 and Fig. 2B). We observed close phylogenetic relatedness between genomes from the Boston area and genome sequences from elsewhere in the northeastern and eastern USA (fig. S8), consistent with frequent domestic travel that continued even after international routes were largely closed. The fraction of cases that were imported decreased over time (Fig. 2B), with the steepest decline during March (Fig. S9), likely reflecting the expansion of existing local clades as the outbreak accelerated and travel restrictions were implemented. By April 2020, the vast majority of cases (median 90.7%, 89.2 – 91.9%, 95% CI) resulted from local populations, rather than from new importations (Table 1, Fig. 2B, and fig. S9).
Table 1. Estimate of SARS-CoV-2 introductions into Massachusetts.
Results of ancestral trait inference using a binary model (MA vs non-MA) and regional model (regional geographic categories) are shown. 95% confidence intervals are shown in parentheses and derived from subsampling the database of global strains (Materials and methods).
| Region | Before 28 March | 28 March to 15 April | After 15 April |
|---|---|---|---|
| Binary model | |||
| Non-MA | 76 (61 to 86) | 40 (33 to 46) | 28 (23 to 33) |
| Regional Model | |||
| Africa | 0 (0 to 1) | 0 (0 to 1) | 0 (0 to 1) |
| Asia | 2 (1 to 4) | 0 (0 to 1) | 1 (0 to 2) |
| Europe | 11 (7 to 16) | 6 (3 to 9) | 2 (0 to 3) |
| North America | 56 (43 to 66) | 29 (22 to 34) | 22 (17 to 28) |
| Oceania | 0 (0 to 0) | 0 (0 to 0) | 0 (0 to 1) |
| South America | 1 (1 to 1) | 0 (0 to 0) | 0 (0 to 0) |
The majority of cases in our dataset are associated with a minority of importation events: only 29% (26–32%, 95% CI) of importations involved more than one case, but those 29% accounted for 85% (78–88%) of the cases in our dataset (Fig. 2C and fig. S9C). As expected, early importation events resulted in large clades (fig. S9, B and C)—likely due to a combination of longer time to expand and unchecked spread before public health measures were implemented. Several clades established early in the Boston area showed continued community transmission throughout the study period (Table 2 and Fig. 3A), with the lineage containing C2416T, associated with a superspreading event early in the epidemic (described below), being the largest. The C2416T lineage was likely the first of these clades imported into Boston (median estimated time to the most recent common ancestor (tMRCA), February 14, 2020; 95% highest posterior density (HPD) February 4 – 20, 2020) (Fig. 3B). The other four major lineages (G3892T, G105T, G28899T, and C20099T) appeared to enter the region between March and early April 2020. These major lineages, including the superspreading event-associated viruses, circulated widely in the Boston area (fig. S10). This included the communities of Chelsea, Revere, and Everett, which were among the most deeply affected in the state (fig. S11). Consistent with a larger global trend (13, 14), we observed a rise in frequency of viruses harboring the D614G amino acid polymorphism in the Spike protein, conferred by a SNP at nucleotide 23,403 in the Wuhan reference strain, which rose to near-fixation in our dataset by the end of the study period (Fig. 3C) and is present in all of the dominant lineages.
Table 2.
Major Boston-area lineages identified by lineage-defining mutation.
| Lineage | Root | C20099T | G3892T | C2416T | G105T | G28899T |
|---|---|---|---|---|---|---|
| Number of genomes | 772 | 21 | 77 | 288 | 98 | 34 |
| Epidemiology | BHCHP | SNF | Conference, BHCHP | BHCHP | ||
| Amino acid substitution | ORF1b: A2211V; NSP15: A160V | ORF1a: E1209D; NSP3: E391D | N: R56I, ORF14: E56* | |||
| Median tMRCA (95% HPD) | 15 December 2019 (20 November 2019 to 4 January 2020) | 4 April 2020 (30 March 2020 to 8 April 2020) | 19 March 2020 (13 March 2020 to 23 March 2020) | 14 February 2020 (4 February 2020 to 20 February 2020) | 10 March 2020 (1 March 2020 to 16 March 2020) | 15 March 2020 (4 March 2020 to 21 March 2020) |
Fig. 3. SARS-CoV-2 spread in the Boston area.
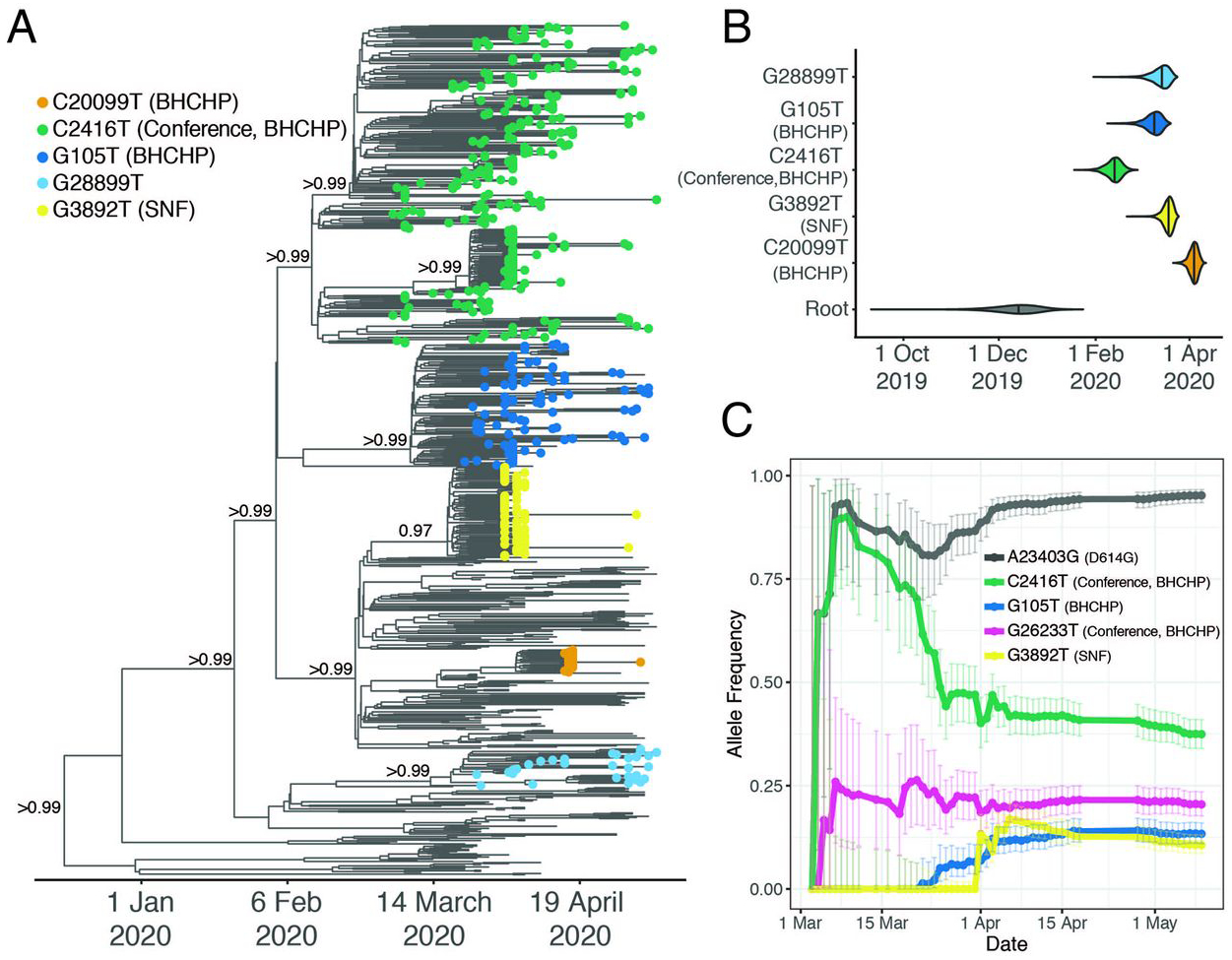
(A) Time-measured maximum clade credibility tree of 772 MA genomes with tips labeled by clade. Nodes with posterior support > 0.8 are labeled. (B) Violin plots of tMRCA for the major Boston-area clades. (C) Estimated allele frequency in sequenced genomes over time for major Boston-area clades. We use the following abbreviations; Boston Healthcare for the Homeless Program (BHCHP); Skilled Nursing Facility (SNF); large international business conference (Conference).
Based on tMRCA estimates for the major Boston-area clades, we do not find evidence of cryptic transmission in the region before mid-February, and none of the importation events we inferred (Table 1) occurred prior to known cases. However, Since testing for SARS-CoV-2 in MA was restricted to a narrow definition prior to established community spread (15), we cannot rule out the possibility that isolated importation events and small outbreaks may have escaped detection with the current resolution of sampling.
Spread of SARS-CoV-2 at an international business conference
Sustained local transmission of SARS-CoV-2 in the Boston area was first detected in early March, and with it case clusters began to appear. The first large cluster was recognized in the context of an international business conference held in Boston from February 26 – 27 (8). Public health investigation with contact tracing identified approximately 100 cases associated with this conference (16), raising suspicion that a superspreading event had occurred there. We sequenced SARS-CoV-2 genomes from 28 of these cases. These genomes indeed showed the signature of superspreading: they form a tight phylogenetic cluster of highly similar viruses within a narrow time window.
All 28 conference-associated genomes were collected between March 5th and 11th and form a well-supported monophyletic cluster (posterior probability > 0.99) (Fig. 3A and fig. S12) marked by the presence of the SNP C2416T (Fig. 3A). The parent lineage of C2416T, defined by G25563T, was widely distributed in Europe in January and February 2020. The C2416T variant can serve as a marker for tracking the spread of SARS-CoV-2 from the conference, within MA and the USA: it is first reported in the USA in patients associated with the conference and there is no evidence that it had entered the country independent of its appearance there. In our dataset, all C2416T-containing viruses collected prior to March 10th were sampled from individuals with conference exposure, and it was not seen in other publicly available genome data from cases anywhere in the US prior to March 7th, when it appeared in cases that were also likely associated with the conference (17). Prior to that, it is seen in the global GISAID database only in 2 French patients, ages 87 and 88, on February 29, 2020 (Fig. 2E). The estimated tMRCA for C2416T-containing genomes is February 14 (95% HPD February 4 – February 20). Taken together, this strongly suggests low-level community transmission of C2416T in Europe in February 2020 before the allele came to Boston via a single introduction, which was then amplified by superspreading at the conference.
We also identified a second variant, G26233T, with a strong conference association. Evidence suggests G26233T emerged during (or theoretically, immediately after) the conference, as it was first seen in 7 of 28 individuals with known conference exposure, including in one sample at intermediate frequency (26%). It is not seen elsewhere in any public genome databases prior to cases associated with the conference (Figs. 2E and 3C). The presence of these two genetic signatures-C2416T in all conference-associated genomes in our dataset, and G26233T in a subset of them-with little or no evidence of transmission prior to the conference, provide markers to track the onward spread of SARS-CoV-2 from the event (Fig. 2F).
The conference-associated lineage was the most common one in our dataset, with C2416T representing 35% (261/744) and C2416T/G26233T representing 20% (151/744) of genomes (excluding those known to be directly associated with the conference). SARS-CoV-2 containing the C2416T allele spread extensively in the Boston area (Fig. 3C and fig. S10A), accounting for between 30% and 46% of genomes from the four counties that make up the Boston area; by the end of the study period, these four counties had reported 51,718 cases. The allele was already at high frequency, in fact, by the time it became clear that an epidemic was underway in the region (fig. S13B), establishing the conditions for extensive spread within Massachusetts and elsewhere.
C2416T began to appear in multiple other US states in early March and increased rapidly in frequency (Fig. 2D and figs. S14 and S15). The effect of this spread was long-lasting. By November 1 2020, viruses containing C2416T could be found in 29 states (fig. S15), and this lineage contributed 1.9% (675/35,566) of all US SARS-CoV-2 genomes in GISAID. States with the largest numbers of cases included ones with known travel by or reported epidemiological links to conference participants returning from the meeting, including Florida, (125/1552 genomes contain C2416T), North Carolina (18) (20/94 genomes), and Indiana (19) (10/42 genomes) (fig. S15A).
Two additional lines of evidence suggest that the conference superspreading event in Boston contributed substantially to the spread of C2416T outside Massachusetts. First, the C2416T/G26233T sublineage, which arose in the context of the conference, was exported from Boston to at least 18 US states as well as to other countries, including Australia, Sweden, and Slovakia (Fig. 2, D and F, and fig. S14A), with evidence of community spread in many places (fig. S15, C, D, and K). Second, there is evidence from other non-conference associated C2416T sublineages that additional importations from Europe were not major contributors to C2416T prevalence in the US. Two sub-lineages (C2416T/G8371T and C2416T/G20578T) appear frequently among European SARS-CoV-2 genomes in GISAID (295 genomes and 312 genomes, respectively), but are extremely rare among genomes from the US (4 and 1 genomes, respectively) (fig. S14, B and C). This evidence, along with the epidemiological data connecting multiple conference-linked cases to other US states (18–21), suggests that most C2416T viruses in the US likely derive from this initial introduction.
Genome data reveal that the impact of the conference was far larger than the approximately 100 cases directly associated with the event. Using state-reported case counts, we estimate that by the end of the study period, approximately 50,000 diagnosed cases [44,000 – 56,000] in the US resulted from conference-associated viruses; of these, 46% [40.4 – 51.8%] were in Massachusetts. Through November 1, 2020, we estimate that a total of 245,000 [205,000 – 300,000] cases marked by C2416T, and 88,000 [56,000 – 139,000] cases marked by G26233T, were linked to the conference in the United States. While Massachusetts accounted for most early spread related to the conference, Florida accounted for the greatest proportion of cases overall (29.2% [22.8 – 36.0%], S15G).
While we have attempted to adjust for geography (by using state-level data) and time period as potential confounders, we note that the accuracy of these estimates is limited by the available data: 1) GISAID is not a random sample of the US epidemic, leading to unknown biases in the estimates; 2) existing state-level data are too sparse for detailed spatiotemporal modeling; 3) we have omitted states with ten or fewer available genomes, leading to possible underestimation; 4) diagnosed cases substantially underestimate true incidence (22), and 5) the estimates do not account for subsequent transmission of the virus (e.g., 4 million new infections in the US in November 2020). While these estimates are provisional, they convey the likely scope of regional, national, and international spread resulting from a single superspreading event early in the pandemic.
Spread of SARS-CoV-2 In a skilled nursing facility
We investigated a second large cluster of cases, this time at a skilled nursing facility (SNF) in the Boston area, that also proved to involve a superspreading event. The cluster was discovered accidentally: screening of residents prior to a planned relocation in early April revealed widespread infection, and ultimately 85% (82/97) of the residents and 37% (36/97) of the staff (23) tested positive for SARS-CoV-2, even though none were known to be symptomatic when screening began. From these individuals we assembled 83 SARS-CoV-2 genomes, 75 of which were found to comprise a single cluster, part of the G3892T lineage described above (Fig. 3A). There was very little genetic variation within the cluster and 59 of the genomes were identical (Fig. 4A), suggestive of a superspreading event. The estimated tMRCA for the cluster of March 20 (Fig. 3B, 95% HPD: March 13 – March 24, 2020), along with the high proportion (30/45) of residents who tested negative on April 1, 2020 but were found to be positive 5 days later (23), suggests rapid spread within the facility in late March and early April 2020. Like other outbreaks reported from nursing facilities, the mortality rate was high. While spread outside the facility appeared rare, as only 1% (2/194) of samples in our dataset after April 15, 2020 harbored G3892T, twenty-four residents who tested positive for SARS-CoV-2 died within two weeks of testing.
Fig. 4. SARS-CoV-2 superspreading events.
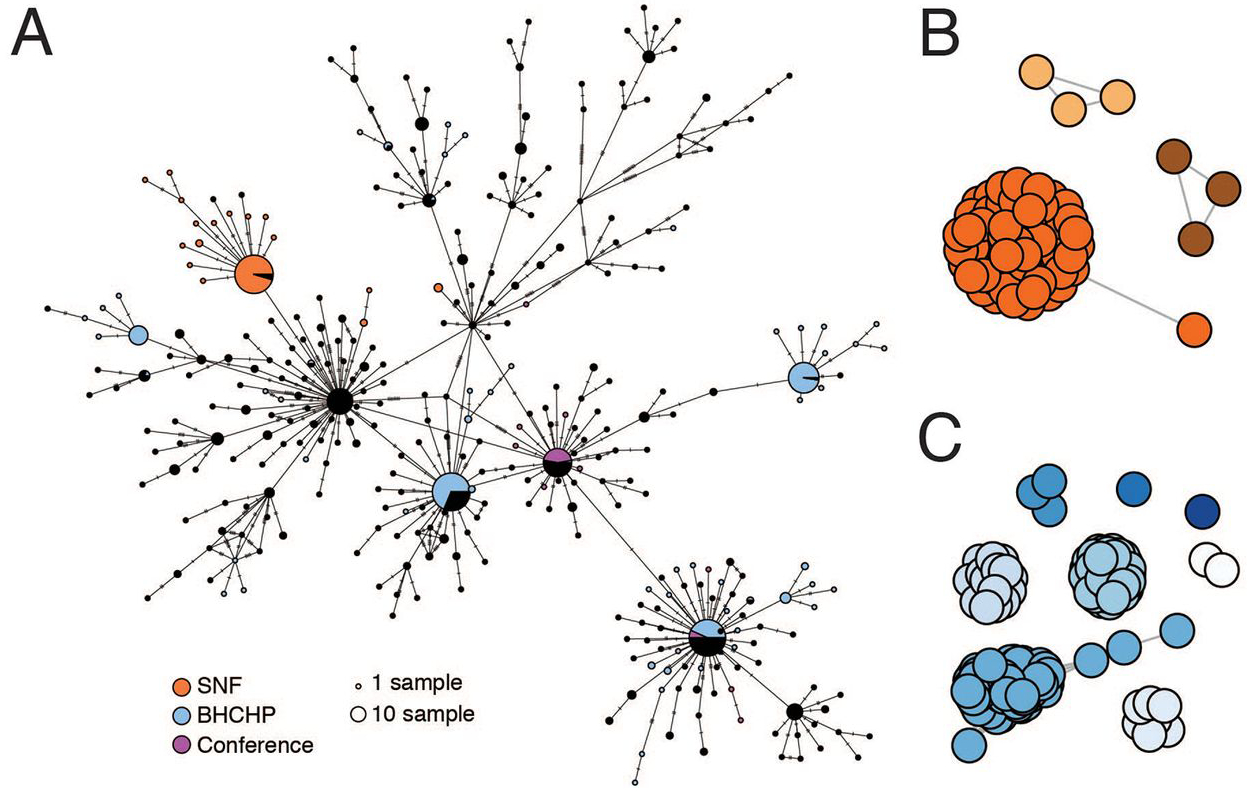
(A) Minimal spanning network showing genetic similarity of SARS-CoV-2 genomes in the MA dataset, with genomes from major known superspreading events highlighted. (B and C) Gene graphs showing clusters of highly similar sequences among viral genomes from the SNF (B) and BHCHP (C) cohorts. Sequences are clustered when they are separated by < 4 SNPs, and the lengths of lines between points reflect genetic distance.
In addition to the major cluster, another 1–2 small clusters can be seen among the patients and staff in the SNF (95% HPD 2–3 total importations, Fig. 4, A and B, and fig. S16). The different outcome of the introductions—one leading to massive spread within the facility (90% of sampled genomes) and the other(s) to little spread (10% of sampled genomes)—illustrates how superspreading can dramatically impact the transmission dynamics of SARS-CoV-2, and how under the right circumstances it can amplify the effect of any given introduction and associated lineage. Notably, these introductions occurred despite infection control policies—including a restriction on visitors (24), universal masking for all staff, masking for all residents when leaving their rooms, and vigilance with hand hygiene—in place for at least two weeks before the first detected infection (23).
Upon examination, we concluded that the genetic diversity in the main SNF cluster was strikingly low even under the assumption of recent transmission from a single source. The 18 mutations seen in the cluster are significantly fewer than expected based on the conference cluster (P = 0.019), which occurred over a similarly short time window, and much lower than the ~32 mutations expected under a simple model of SARS-CoV-2 substitution (P = 0.009, Materials and Methods). This discrepancy might have resulted from low diversity in the SNF index patient, but it may also hint that heterogeneous mechanics of superspreading were at work in the two events. For example, if more virions than usual were transmitted from the SNF index patient to each secondary case — such as through unusually close or prolonged contact, or the initial case having a very high viral load at the time — then we would expect that the resulting infections would more often have the same consensus genome as the index case.
Cluster investigations in other close contact settings
We studied several additional case clusters with the goal of providing viral genomic data to support public health investigations. These included potential transmission in homeless shelters and within a hospital. First, we analyzed the introduction and spread of SARS-CoV-2 among guests and staff at homeless shelters affiliated with the Boston Health Care for the Homeless Program (BHCHP). We produced 193 complete genomes from 314 samples collected in March and April 2020, including those collected during universal screening at Boston’s largest homeless shelter (5). Based on the position of these 193 SARS-CoV-2 genomes from BHCHP in the overall Boston-area tree (Fig. 3A), we identified at least 14 introductions into the BHCHP community (95% HPD 14–18). Of these, 4 resulted in clusters consistent with superspreading, each containing 20 or more highly similar viral genomes (Fig. 4, A and C, and fig. S16B). Two of the clusters descended from the conference-associated C2416T lineage, including one that contained C2416T/G26233T. In total, 54% (105/193) of the genomes in this cohort contained C2416T, of which half (54/105) also contained G26233T, demonstrating that BHCHP guests and staff were affected by community transmission resulting from amplification and spread of conference-associated SARS-CoV-2.
The other two case clusters occurred at Massachusetts General Hospital, where the Infection Control Unit sought genomic data to inform their investigations of possible nosocomial outbreaks. In the first cluster, two patients in the same hospital ward tested positive for SARS-CoV-2 during their hospital stay, after testing negative at the time of admission. In the second, unrelated cluster, four patients who received care in a specialty ward were diagnosed with SARS-CoV-2 infections over a period of several days. For each cluster, complete genomes (2 of 2 from the first cluster and 4 of 4 from the second cluster) were genetically very distinct, a pattern inconsistent with having been infected from the same source during hospitalization (fig. S17). Although we cannot exclude the possibility of nosocomial transmission per se because independent introductions from multiple asymptomatic staff could theoretically have occurred, this demonstrated that the individuals in each cluster were not part of the same transmission chain.
Conclusions
Genomic analysis of the first wave of the COVID-19 outbreak in the Boston area provides powerful evidence of the importance of superspreading events in shaping the course of this pandemic. In this study we show that importation events occurred very frequently—we identified over 120 independent introductions during the three-month study period—and that they varied enormously in terms of their subsequent impact on local transmission. Consistent with an over-dispersed offspring distribution for SARS-CoV-2 (25), in our dataset, a small minority of importations accounted for the majority of observed cases. At least some of this variation in clade sizes results from superspreading events amplifying some lineages and not others. This can be seen in microcosm in one of the two superspreading events we studied in detail: SARS-CoV-2 was introduced at least twice into the skilled nursing facility; one introduction led to widespread transmission and numerous deaths, while the other 1–2 introductions led to a total of six cases.
The other superspreading event, which occurred at an international business conference early in the local epidemic, had a much greater impact on community transmission. Because SARS-CoV-2 viruses circulating at the conference happened to be marked by distinct genomic signatures, we were able to track its downstream effects far beyond the superspreading event itself, tracing the descendants of the virus as they made a large contribution to the local outbreak in the Boston area and as they spread throughout the US and the world, likely causing hundreds of thousands of cases. The different genetic diversity seen in the two events raises the possibility that superspreading encompasses varied transmission dynamics.
Not all case clusters were the result of superspreading. Both hospital clusters consisted of unrelated cases that happened to occur in close proximity to one another. Cases associated with the homeless shelters likely resulted from a mix of superspreading events and more general transmission, although we lack the detailed epidemiological data to explore their history in depth. Where we were able to study superspreading events in detail, in the SNF and the conference, it was not because they were unique in size or character, but because circumstances allowed close study. For both, we had dense sampling during a narrow time window of a clearly demarcated exposed population, aided by good data on prevailing genetic diversity to provide context.
Our findings highlight the close relationships between seemingly disconnected groups and populations: viruses from international business travel seeded major outbreaks among individuals experiencing homelessness, spread throughout the Boston area including to other higher risk communities, and were exported to other domestic and international sites. It also illustrates the role of chance in the trajectory of an epidemic: a single introduction had an outsize effect on subsequent transmission because it was amplified by superspreading in a highly mobile population very early in the outbreak, before many public health precautions were put in place, and when its effects would be further amplified by exponential growth and subsequent superspreading events (e.g., among the homeless). By contrast, other early introductions led to very little onward transmission, and the superspreading event in the SNF, while devastating to the residents, had little large-scale effect because it occurred later and in a more isolated population. While superspreading events among medically vulnerable populations, such as nursing home residents, have a larger immediate impact on mortality, our findings raise the possibility that—paradoxically—the implications may be greater, when measured as a cost to society, for superspreading events that involve younger, healthier and more mobile populations because of the increased risk of subsequent transmission. With the possibility of vaccines that protect against disease but not infection, this consideration may be increasingly important. In summary, this study provides clear evidence that superspreading events may profoundly alter the course of an epidemic and implies that prevention, detection, and mitigation of such events should be a priority for public health efforts.
Materials and Methods
Sample and data collection
This study was approved by the Partners Institutional Review Board under protocol 2019P003305 and MDPH IRB 00000701. We obtained samples and selected metadata from the MGH Microbiology Laboratory and MADPH under a waiver of consent for viral genome sequencing. All samples were nasopharyngeal (NP) swabs that tested positive for SARS-CoV-2 by RT-qPCR. Epidemiological data on exposure and geography were obtained from medical record review (MGH) or collected by the DPH laboratory in the process of clinical testing. Samples included individuals with known exposures to suspected superspreading events and individuals where no possible exposures were known. We compared known information about these cases to publicly available daily and weekly data on cases of SARS-CoV-2 in MA for the period January 1 – August 1 (https://www.mass.gov/info-details/covid-19-response-reporting).
Viral sequencing and analysis
Total RNA was extracted from inactivated NP swabs and presence of virus was confirmed using an RT-qPCR assay detecting the N1 gene of the virus. Metagenomic sequencing libraries were prepared as previously described (26)). Briefly, following DNase treatment to remove residual DNA and depletion of human rRNA, cDNA was synthesized using random hexamer priming. Illumina sequencing libraries were prepared from cDNA and sequenced with 100-nucleotide paired-end reads.
We conducted all analyses using viral-ngs 2.0.21 on the Terra platform (app.terra.bio). All of the workflows named below are publicly available via the Dockstore Tool Registry Service (dockstore.org/organizations/BroadInstitute/collections/pgs). Code is also archived at doi:10.5281/zenodo.4306358 and doi:10.5281/zenodo.4306362. Briefly, samples were demultiplexed (demux_only workflow), filtered for known sequencing contaminants and SARS-CoV-2 genomes were assembled using a reference-based assembly approach (assemble_refbased) with the reference genome NC_045512.2. Following a stringent quality control and filtering, we identified a final set of 772 high-quality assemblies from unique individuals that was used for all subsequent analyses and deposited in GenBank and GISAID. We used R (27), Bioconductor (28), ggplot2, tidyverse (29), and ggtree (30) to clean and plot data and trees, and choroplethr to draw maps.
To detect the presence of 20 common respiratory viruses in sequenced samples, we used Kraken2 (31) implemented in the classify_single and merge_metagenomics workflows. A virus was determined to be present if more than 10 reads mapped to that species. Wherever possible, these co-infections were confirmed using the BioFire FilmAssay Respiratory Panel.
We constructed phylogenetic maximum likelihood (ML) and time trees with associated visualizations using the Augur pipeline (augur_with_assemblies) and SARS-CoV-2-specific procedures taken from github.com/nextstrain/ncov for our 772 genomes and a representative background set of 4,011 subsampled from the GISAID database on 15 June, 2020. We separately constructed ML trees from trimmed alignments to estimate root-to-tip distances and obtain branch support for ML phylogenies. To estimate coalescence dates of major lineages we constructed Bayesian time-trees using BEAST 2.6.2 with a general time reversible substitution model with 4 rate categories drawn from a gamma distribution (GTR4G), a strict clock, coalescent exponential tree prior, a uniform [-inf, inf] prior for the clock rate, a 1/x [-inf, inf] prior for the coalescent exponential population size; and a laplace [-inf, inf] prior for the growth rate.
Ancestral state reconstruction
We used 3 orthogonal approaches to reconstruct the ancestral location of unsampled nodes: 1) a ML approach using the augur pipeline, 2) a maximum parsimony approach using the Narushima and Hanazawa method as implemented in the MPR function of the ape package in R, and 3) a bayesian approach using BEAST1.10.4. In each case, we use a binary classification of “MA” vs “non-MA” to identify nodes that represent a likely importation event into Massachusetts. For full details of each approach see the Supplementary Materials and Methods.
Analysis of superspreading events
To estimate the number of cases linked to the conference we estimated the proportion of genomes with C2416T and C2416T/G26233T per state by multiplying the observed proportion in genomes reported in GISAID through November 2nd 2020 by case counts reported in the New York Times COVID data repository (https://github.com/nytimes/covid-19-data). We summed across states using a Monte Carlo simulation (n = 10,000).
To show clustering within the SNF and BHCHP cases, we constructed a minimal spanning haplotype network from the trimmed ML alignment of 772 genomes using PopART v1.7 (32) with masking of regions where any sequence had ambiguous bases. Gene graphs were constructed using pairwise distance matrices computed on aligned SARS-CoV-2 genomes and clustered using the R package adegenet (33). Importations into the SNF and BHCHP populations were calculated using a bayesian approach similar to that described above (see Supplementary Materials and Methods for more details).
We define a superspreading event as the transmission from a single source to a large number of secondary infections, where the number is large enough that it would occur < 1% of the time in a simple Poisson model of transmission (34). For this study, using an Reff value of 3.0, we set the threshold at a minimum of nine transmissions. We compared the number of mutations among conference-associated and SNF-associated genomes with the expected number based on a generation time of 5.0 days (35) and a mean substitution rate of 1.04 × 10−3/bp/year (fig. S6C) and calculated a p-value based on the fraction of draws yielding fewer mutations than observed.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the microbiology lab staff and infection control personnel at MGH and DPH and all members of the COVID-19 emergency response efforts at MGH, BHCHP, and MADPH. We also thank Hayden C. Metsky, Aviv Regev, Avi I. Feller, and members of the CDC SPHERES consortium for valuable feedback and helpful discussions. This study was approved by the Partners Institutional Review Board under protocol 2019P003305 and MDPH IRB 00000701.
Funding: This work was sponsored by the National Institute of Allergy and Infectious Diseases (U19AI110818 to P.C.S; R37AI147868 and R01AI148784 to J.L.), the National Human Genome Research Institute (K99HG010669 to S.K.R.), the National Institute of General Medical Sciences of the National Institutes of Health (U54GM088558 W.P.H.), the Centers for Disease Control and Prevention (U01CK000490; MGH), the Bill and Melinda Gates Foundation (Broad Institute), and the US Food and Drug Administration (HHSF223201810172C), with in-kind support from Illumina, Inc., as well as support from the Doris Duke Charitable Foundation (J.E.L.), the Howard Hughes Medical Institute (P.C.S.), the Herchel Smith Fellowship (K.A.L.), and the Evergrande COVID-19 Response Fund Award from the Massachusetts Consortium on Pathogen Readiness (J.L). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests: J.E.L. has received consulting fees from Sherlock Biosciences. J.B. has been a consultant for T2 Biosystems, DiaSorin, and Roche Diagnostics. A.P. is a Venture Partner at Google Ventures. P.C.S. is a co-founder and shareholder of Sherlock Biosciences, and a Board member and shareholder of Danaher Corporation.
Data and materials availability: Sequences and genome assembly data are publicly available in the Broad Institute’s Terra platform in a featured workspace for COVID-19. Researchers can use this workspace to reproduce analyses described here or perform similar analyses on their own viral sequence data. Assembled genomes and raw metagenomic reads from this dataset have been deposited at NCBI’s GenBank and SRA databases under BioProject PRJNA622837 in accordance with NIAID’s Data Sharing policy and will soon be available to visualize on nextstrain.org/ncov. Experimental protocols are publicly available on Benchling and can be accessed here: https://benchling.com/sabetilab/f_/gaLGu5X9-sabeti_group_sars-cov-2_metagenomic_sequencing_protocols/.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/cgi/content/full/science.abe3261/DC1
Materials and Methods
Figs. S1 to S17
Tables S1 to S3
MDAR Reproducibility Checklist
REFERENCES AND NOTES
- 1.Center for Systems Science and Engineering (CSSE) at Johns Hopkins University, (JHU), COVID-19 Dashboard, (available at https://coronavirus.jhu.edu/map.html).
- 2.Waltenburg MA, Rose CE, Victoroff T, Butterfield M, Dillaha JA, Heinzerling A, Chuey M, Fierro M, Jervis RH, Fedak KM, Leapley A, Gabel JA, Feldpausch A, Dunne EM, Austin C, Pedati CS, Ahmed FS, Tubach S, Rhea C, Tonzel J, Krueger A, Crum DA, Vostok J, Moore MJ, Kempher H, Scheftel J, Turabelidze G, Stover D, Donahue M, Thomas D, Edge K, Gutierrez B, Berl E, McLafferty M, Kline KE, Martz N, Rajotte JC, Julian E, Diedhiou A, Radcliffe R, Clayton JL, Ortbahn D, Cummins J, Barbeau B, Carpenter S, Pringle JC, Murphy J, Darby B, Graff NR, Dostal TKH, Pray IW, Tillman C, Rose DA, Honein MA; CDC COVID-19 Emergency Response Team, Coronavirus Disease among Workers in Food Processing, Food Manufacturing, and Agriculture Workplaces. Emerg. Infect. Dis 27, (2020). 10.3201/eid2701.203821 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei WE, Li Z, Chiew CJ, Yong SE, Toh MP, Lee VJ, Presymptomatic Transmission of SARS-CoV-2 - Singapore, January 23-March 16, 2020. MMWR Morb. Mortal. Wkly. Rep 69, 411–415 (2020). doi: 10.15585/mmwr.mm6914e1 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMichael TM, Currie DW, Clark S, Pogosjans S, Kay M, Schwartz NG, Lewis J, Baer A, Kawakami V, Lukoff MD, Ferro J, Brostrom-Smith C, Rea TD, Sayre MR, Riedo FX, Russell D, Hiatt B, Montgomery P, Rao AK, Chow EJ, Tobolowsky F, Hughes MJ, Bardossy AC, Oakley LP, Jacobs JR, Stone ND, Reddy SC, Jernigan JA, Honein MA, Clark TA, Duchin JS; Public Health—Seattle and King County, EvergreenHealth, and CDC COVID-19 Investigation Team, Epidemiology of Covid-19 in a Long-Term Care Facility in King County, Washington. N. Engl. J. Med 382, 2005–2011 (2020). doi: 10.1056/NEJMoa2005412 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baggett TP, Keyes H, Sporn N, Gaeta JM, Prevalence of SARS-CoV-2 Infection in Residents of a Large Homeless Shelter in Boston. JAMA 323, 2191–2192 (2020). doi: 10.1001/jama.2020.6887 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamner L, Dubbel P, Capron I, Ross A, Jordan A, Lee J, Lynn J, Ball A, Narwal S, Russell S, Patrick D, Leibrand H, High SARS-CoV-2 Attack Rate Following Exposure at a Choir Practice - Skagit County, Washington, March 2020. MMWR Morb. Mortal. Wkly. Rep 69, 606–610 (2020). doi: 10.15585/mmwr.mm6919e6 Medline [DOI] [PubMed] [Google Scholar]
- 7.James A, Eagle L, Phillips C, Hedges DS, Bodenhamer C, Brown R, Wheeler JG, Kirking H, High COVID-19 Attack Rate Among Attendees at Events at a Church -Arkansas, March 2020. MMWR Morb. Mortal. Wkly. Rep 69, 632–635 (2020). doi: 10.15585/mmwr.mm6920e2 Medline [DOI] [PubMed] [Google Scholar]
- 8.Schuchat A; CDC COVID-19 Response Team, Public Health Response to the Initiation and Spread of Pandemic COVID-19 in the United States, February 24-April 21, 2020. MMWR Morb. Mortal. Wkly. Rep 69, 551–556 (2020). doi: 10.15585/mmwr.mm6918e2 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MA Department of Public Health, Man returning from Wuhan, China is first case of 2019 Novel Coronavirus confirmed in Massachusetts (2020), (available at https://www.mass.gov/news/man-returning-from-wuhan-china-is-first-case-of-2019-novel-coronavirus-confirmed-in).
- 10.COVID-19 Response Reporting, Massachusetts Department of Public Health (2020), (available at https://www.mass.gov/info-details/covid-19-response-reporting).
- 11.Park DJ, Dudas G, Wohl S, Goba A, Whitmer SLM, Andersen KG, Sealfon RS, Ladner JT, Kugelman JR, Matranga CB, Winnicki SM, Qu J, Gire SK, Gladden-Young A, Jalloh S, Nosamiefan D, Yozwiak NL, Moses LM, Jiang P-P, Lin AE, Schaffner SF, Bird B, Towner J, Mamoh M, Gbakie M, Kanneh L, Kargbo D, Massally JLB, Kamara FK, Konuwa E, Sellu J, Jalloh AA, Mustapha I, Foday M, Yillah M, Erickson BR, Sealy T, Blau D, Paddock C, Brault A, Amman B, Basile J, Bearden S, Belser J, Bergeron E, Campbell S, Chakrabarti A, Dodd K, Flint M, Gibbons A, Goodman C, Klena J, McMullan L, Morgan L, Russell B, Salzer J, Sanchez A, Wang D, Jungreis I, Tomkins-Tinch C, Kislyuk A, Lin MF, Chapman S, MacInnis B, Matthews A, Bochicchio J, Hensley LE, Kuhn JH, Nusbaum C, Schieffelin JS, Birren BW, Forget M, Nichol ST, Palacios GF, Ndiaye D, Happi C, Gevao SM, Vandi MA, Kargbo B, Holmes EC, Bedford T, Gnirke A, Ströher U, Rambaut A, Garry RF, Sabeti PC, Ebola Virus Epidemiology, Transmission, and Evolution during Seven Months in Sierra Leone. Cell 161,1516–1526 (2015). doi: 10.1016/j.cell.2015.06.007 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Worobey M, Pekar J, Larsen BB, Nelson MI, Hill V, Joy JB, Rambaut A, Suchard MA, Wertheim JO, Lemey P, The emergence of SARS-CoV-2 in Europe and North America. Science 370, 564–570 (2020). doi: 10.1126/science.abc8169 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yurkovetskiy L, Pascal KE, Tompkins-Tinch C, Nyalile T, Wang Y, Baum A, Diehl WE, Dauphin A, Carbone C, Veinotte K, Egri SB, Schaffner SF, Lemieux JE, Munro J, Sabeti PC, Kyratsous C, Shen K, Luban J, SARS-CoV-2 Spike protein variant D614G increases infectivity and retains sensitivity to antibodies that target the receptor binding domain. bioRxiv (2020), p. 2020.07.04.187757. [Google Scholar]
- 14.Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, de Silva TI, McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, Montefiori DC; Sheffield COVID-19 Genomics Group, Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 182, 812–827.e19 (2020). 10.1016/j.cell.2020.06.043 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MA Department of Public Health, Update and Interim Guidance on Outbreak of 2019 Novel Coronavirus (2019-nCoV) in Wuhan, China (2020), (available at https://www.mass.gov/clinical-advisory/update-and-interim-guidance-on-outbreak-of-2019-novel-coronavirus-2019-ncov-in).
- 16.MA Department of Public Health, Coronavirus Disease 2019 (COVID-19) Cases in MA, March 15 2020 (2020).
- 17.The CDC sequenced 19 MA genomes prior to March 8 2020.17/19 cases (89%) contained C2416T. The CDC MA genomes are not annotated with exposure information, but given official MADPH data reporting that 23/28 cases as of March 8 2020 were linked to the conference (19), and the five non-conference associated cases include the travel-associated cases from this time period (MA-1, DPH_00002, and DPH_00003) and one from the Berkshire county cluster (all of which lack C2416), it can be inferred that a minimum of 16/17 C2416T-containing samples sequenced by the CDC were conference-associated.
- 18.North Carolina Department of Health and Human Services, Five More People in North Carolina Test Positive for COVID-19 (2020), (available at https://www.ncdhhs.gov/news/press-releases/five-more-people-north-carolina-test-positive-covid-19).
- 19.Indiana State Department of Health, State Health Department Announces 2nd COVID-19 Case (2020), (available at https://calendar.in.gov/site/isdh/event/isdh-news-release-state-health-department-announces-2nd-covid-19-case/).
- 20.Tennesse Department of Health, TDH Releases Further Information Regarding COVID-19 Case, (2020), (available at https://www.tn.gov/health/news/2020/3/5/tdh-releases-further-information-regarding-covid-19-case.html).
- 21.Indiana State Department of Health, State Health Department Confirms 1st Case of COVID-19 in Hoosier with Recent Travel (2020).
- 22.Reese H, Iuliano AD, Patel NN, Garg S, Kim L, Silk BJ, Hall AJ, Fry A, Reed C, Estimated incidence of COVID-19 illness and hospitalization - United States, February-September, 2020. Clin. Infect Dis ciaa1780 (2020). doi: 10.1093/cid/ciaa1780 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldberg SA, Lennerz J, Klompas M, Mark E, Pierce VM, Thompson RW, Pu CT, Ritterhouse LL, Dighe A, Rosenberg ES, Grabowski DC, Presymptomatic Transmission of Severe Acute Respiratory Syndrome Coronavirus 2 Among Residents and Staff at a Skilled Nursing Facility: Results of Real-time Polymerase Chain Reaction and Serologic Testing. Clin. Infect. Dis 10.1093/cid/ciaa991 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bharel M, Order of the Commisioner of Public Health, (2020), (available at https://www.mass.gov/doc/march-15-2020-assisted-living-visitor-restrictions-order/download).
- 25.Endo A, Abbott S, Kucharski AJ, Funk S: Centre for the Mathematical Modelling of Infectious Diseases COVID-19 Working Group, Estimating the overdispersion in COVID-19 transmission using outbreak sizes outside China. Wellcome Open Res. 5, 67 (2020). doi: 10.12688/wellcomeopenres.15842.3 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matranga CB, Andersen KG, Winnicki S, Busby M, Gladden AD, Tewhey R, Stremlau M, Berlin A, Gire SK, England E, Moses LM, Mikkelsen TS, Odia I, Ehiane PE, Folarin O, Goba A, Kahn SH, Grant DS, Honko A, Hensley L, Happi C, Garry RF, Malboeuf CM, Birren BW, Gnirke A, Levin JZ, Sabeti PC, Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 15, 519 (2014). doi: 10.1186/s13059-014-0519-7 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ihaka R, Gentleman R, R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat 5, 299–314 (1996). [Google Scholar]
- 28.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, lacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J, Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004). doi: 10.1186/gb-2004-5-10-r80 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wickham H, Averick M, Bryan J, Chang W, McGowan LD, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen T, Miller E, Bache S, Müller K, Ooms J, Robinson D, Seidel D, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H, Welcome to the Tidyverse. J. Open Source Softw 4, 1686 (2019). doi: 10.21105/joss.01686 [DOI] [Google Scholar]
- 30.Yu G, Smith DK, Zhu H, Guan Y, Lam TT, ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol 8, 28–36 (2017). doi: 10.1111/2041-210X.12628 [DOI] [Google Scholar]
- 31.Wood DE, Lu J, Langmead B, Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019). doi: 10.1186/s13059-019-1891-0 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leigh JW, Bryant D, POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol 6, 1110–1116 (2015). doi: 10.1111/2041-210X.12410 [DOI] [Google Scholar]
- 33.Jombart T, Ahmed I, adegenet 1.3–1: New tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011). doi: 10.1093/bioinformatics/btr521 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lloyd-Smith JO, Schreiber SJ, Kopp PE, Getz WM, Superspreading and the effect of individual variation on disease emergence. Nature 438, 355–359 (2005). doi: 10.1038/nature04153 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferretti L, Wymant C, Kendall M, Zhao L, Nurtay A, Abeler-Dörner L, Parker M, Bonsall D, Fraser C, Quantifying SARS-CoV-2 transmission suggests epidemic control with digital contact tracing. Science 368, eabb6936 (2020). doi: 10.1126/science.abb6936 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matranga CB, Gladden-Young A, Qu J, Winnicki S, Nosamiefan D, Levin JZ, Sabeti PC, Unbiased Deep Sequencing of RNA Viruses from Clinical Samples, Unbiased Deep Sequencing of RNA Viruses from Clinical Samples. J. Vis. Exp (113): (2016). 10.3791/54117 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K, Standley DM, MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol 30, 772–780 (2013). doi: 10.1093/molbev/mst010 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guindon S, Dufayard JF, Hordijk W, Lefort V, Gascuel O, in Infection Genetics and Evolution (ELSEVIER SCIENCE; BV PO BOX 211, 1000 AE AMSTERDAM, NETHERLANDS, 2009), vol. 9, pp. 384–385. [Google Scholar]
- 39.Rambaut A, Lam TT, Max Carvalho L, Pybus OG, Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016). doi: 10.1093/ve/vew007 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ, IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phytogenies. Mol. Biol. Evol 32, 268–274 (2015). doi: 10.1093/molbev/msu300 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sagulenko P, Puller V, Neher RA, Tree Time: Maximum-likelihood phylodynamic analysis. Virus Evol. 4, vex042 (2018). doi: 10.1093/ve/vex042 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, Sagulenko P, Bedford T, Neher RA, Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018). doi: 10.1093/bioinformatics/bty407 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong E, Du H, Gardner L, An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis 20, 533–534 (2020). doi: 10.1016/S1473-3099(20)30120-1 Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.