

Abstract
Mutations in GBA1, which encode for the protein glucocerebrosidase (GCase), are the most common genetic risk factor for Parkinson’s disease and Dementia with Lewy Bodies. In addition, growing evidence now suggests that loss of GCase activity is also involved in onset of all forms of Parkinson’s disease, Dementia with Lewy bodies, and other dementias, such as progranulin-linked frontal temporal dementia. As a result, there is significant interest in developing GCase-targeted therapies that have the potential to stop or slow progression of these diseases. Despite this interest in GCase as a therapeutic target, there is significant inconsistency in the methodology for measuring GCase enzymatic activity in disease modeling systems and patient populations, which could hinder progress in developing GCase therapies. In this review, we discuss the different strategies that have been developed to assess GCase activity and highlight specific strengths and weaknesses of these approaches as well as the gaps that remain. We also discuss the current and potential role of these different methodologies in preclinical and clinical development of GCase targeted therapies.
Keywords: Glucocerebrosidase, GCase, GCase Enzyme Activity, Parkinson’s Disease
Introduction
Glucosylceramidases are a family of enzymes encoded by the genes GBA1, GBA2 and GBA3 that play an important role in maintaining cellular homeostasis via the metabolism of glucosylceramide to ceramide and glucose. Glucosylceramidase beta, better known as glucocerebrosidase (GCase), is encoded by GBA1, ubiquitously expressed and predominantly localized in the lysosome1. Glucosylceramidase beta 2, encoded by GBA2, is also ubiquitously expressed but localized in the cytoplasm2. Consequently, the enzymes encoded by GBA1 and GBA2 are also often referred to as lysosomal and non-lysosomal glucosylceramidase respectively. For the purpose of this review however, the enzymes encoded by GBA1 and GBA2 are referred to as GCase and GBA2 respectively. These two enzymes show little sequence homology to each other. However, they still share overlapping substrate specificity, with GCase and GBA2 metabolizing substrates at a different pH due to the different lysosomal/cytoplasmic intracellular locations. Glucosylceramidase beta 3, encoded by GBA3, is also cytoplasmic, but with an expression restricted to the liver and with seemingly much less affinity to metabolize glucosylceramide3. Due to its role in human disease, the majority of studies to date have focused on lysosomal GCase, encoded by GBA1. GCase is synthesized in the endoplasmic reticulum (ER) and contains 497 amino acids, including a signal peptide that is cleaved off to produce the mature protein. In the ER, GCase acquires 4 N-linked glycans4 and is complexed with lysosomal integral membrane protein-2 (LIMP-2), which is encoded by the SCARB2 gene. The LIMP-2-GCase complex is transported to the Golgi where additional glycosylation occurs. Once in the acidic late endosome/lysosomal compartments, the complex dissociates and GCase then interacts with Saposin C, which is a protein co-factor for GCase activity. In addition to metabolism of glucosylceramide, lysosomal GCase can also hydrolyze glucosylsphingosine, although this occurs at a much slower rate.
Homozygous or compound heterozygous GBA1 mutations lead to development of the lysosomal storage disorder, Gaucher disease (GD). More than 400 mutations in GBA1 have been associated with this disease5, including point mutations, splice-site mutations, deletions, insertions, and aberrant recombination that result in either disrupted translation, misfolding, impaired trafficking, reduced enzyme stability, reduced enzymatic efficiency or a combination of these defects. Different GBA1 mutation types may underlie the development of the different types of Gaucher disease (type-1, type-2, or type-3), which differ in severity and the manifestation of clinical symptoms. Regardless of the mutation type however, the end result is a significant impairment in GCase enzyme function in the lysosome resulting in the progressive accumulation of glucosylceramide, particularly in cells of the mononuclear phagocyte system. These cells are transformed into Gaucher cells which have a distinct enlarged lipid-laden macrophage phenotype6. Additionally, accumulating glucosylceramide in the lysosome can be converted to glucosylsphingosine by the lysosomal enzyme acid ceramidase.7 Glucosylsphingosine is more hydrophilic than glycosylceramide which is thought to allow its escape from the lysosome7 and contribute to toxicity in GD8. In severe GD, glucosylceramide also accumulates in the CNS, predominantly in perivascular macrophages9, but also in neurons10–12, which is thought to promote neuroinflammation observed in GD10.
Subsequent clinical and genetic sequencing analysis revealed that heterozygous mutations in GBA1 are a major risk factor for the neurodegenerative diseases Parkinson’s disease (PD) and Dementia with Lewy bodies (DLB), with predicted frequencies of 7–12% in patient populations of both PD and DLB13–16. In a key early study, reduction in lysosomal GCase activity resulted in accumulation of glucosylceramide that stabilized toxic alpha synuclein oligomers. This study also found that accumulation of alpha synuclein interferes with ER to Golgi trafficking of GCase leading to formation of a positive feedback loop that, after a threshold, leads to self-propagating disease regardless of whether there is a mutation in GBA117. Subsequent studies have also demonstrated a reduction in wild-type GCase activity in patient blood samples18, CSF19, and post-mortem brain tissue20–22 highlighting a potential role for GCase in the pathogenesis of sporadic and familial forms of PD. Studies in iPSC-derived DA neurons from Patients with PD showed that either alpha-synuclein or oxidized dopamine could lower wild-type GCase activity in genetic or idiopathic forms of PD17, 23. Another recent study also described a reduction in GCase activity in idiopathic PD fibroblast driven by reduced LIMP2 expression24. Collectively these studies highlight decreases in GCase activity as an important contributor of PD pathogenesis and provide rationale for further studying the upstream regulators of GCase activity to develop additional novel strategies to target this protein in PD.
In GD, visceral symptoms are markedly improved by enzyme replacement therapy through chronic intravenous administration, which results in enzyme uptake by affected macrophages. However, the inability of the infused recombinant enzyme to pass through the blood-brain barrier prevents this approach from affecting neurological manifestation of GCase deficiency observed in PD or DLB. As a result, various strategies have been developed to restore or replace GCase activity in the brain for PD and neuronopathic GD. Small molecule therapeutics currently under development include molecular chaperones and positive allosteric modulators (Table 1). The goal of molecular chaperones is to assist in folding of mutant GCase in the ER, thereby improving trafficking from the ER to the lysosome and/or increasing the stability of the resulting lysosomal enzyme to improve protein longevity and accumulation of active protein in the lysosome. The goal of positive allosteric modulators is to pharmacologically increase the enzymatic efficiency of wild-type (WT) lysosomal GCase to compensate for activity lost by a heterozygous mutation. Other therapies that are in development or being tested include gene therapy to express wild-type GCase, linking recombinant GCase to a protein shuttle to enable active transport of enzyme into the brain, and CRISPR based approaches to correct mutations in the GBA1 gene (Table 1).
Table 1.
Current/Proposed therapeutic strategies targeting GCase
| Therapeutic Strategy | Example | Phase in drug development | Summary of Results | GCase Activity Measurement Technique |
|---|---|---|---|---|
| Molecular Chaperone | Ambroxol | Phase II Completed | Decreased CSF GCase activity and increased protein levels | 4-MU1 in vitro |
| Activator | BIA 28–6156/LTI-291 | Phase I Completed | Effects on GCase activity not publicly disclosed | N/A |
| Gene therapy | PR001 | Phase I/II Ongoing | CSF GCase activity increased from undetectable to within normal range | N/A |
| Transport Vehicle Modified Recombinant GCase | ETV:GBA2 | Preclinical Research Ongoing | No current publications | N/A |
4-MU (4-methylumbelliferyl-β-D-glucopyranoside)
ETV (Enzyme Transport Vehicle)
With different treatment modalities being tested preclinically and clinically, robust assays are required to measure the levels and activity of GCase so the effect of GBA-targeted therapies can be accurately assessed. These assays could also play a critical role in patient inclusion criteria for clinical trials. GCase activity can vary widely in the patient population, even in patients with GCase mutations. Therefore, the ability to identify patients with low GCase activity may be a way to select patients that are more likely to respond to GCase targeted therapy. This selection could increase the likelihood of success of new therapeutics and also ensure that future therapies are targeted to relevant patient populations. . Despite the considerable advances in assay technologies, there is significant inconsistency in the methodology for measuring GCase activity in disease modeling systems and patient populations. Hence, there is a critical need for uniform recognition of the strengths and weaknesses of these various approaches. Such an understanding is crucial for further development of strategies to measure target engagement of novel therapeutics for GCase.
Here we discuss different approaches that have been used to assess GCase activity, as well as potential roles of these measurements in the development/evaluation of new therapeutics (Table 2). The specific approaches discussed were selected because they are the most widely used in the field and most relevant to preclinical development. It is important to note that while each of these assays provide information on the function of GCase, they only serve as artificial proxies of the cellular function of GCase, which is the metabolic turnover of glycosylceramide and glucosylsphingosine in the lysosome. Therefore, the ultimate effect of GCase targeted therapies should be reliably measured through lipidomic based analyses. This has been done in PBMCs, serum and CSF, although for serum and CSF measurements it is unclear how accurately these levels reflect what is occurring in the lysosome.
Table 2.
Summary of commonly used strategies for assessing lysosomal GCase.
| Assay | Substrate Examples | Measures | Best Applications | Application for therapeutic development | Disadvantages |
|---|---|---|---|---|---|
| Recombinant protein In vitro activity | 4-MUG1, ResGlu2, BODIPY Glucosylceramide | GCase activity of recombinant protein | Analyzing direct effects of different environments/compounds on GCase enzyme kinetics | High throughput screening for GCase activators. Confirming lack of inhibitory activity for chaperones. | Does not account for variation in endogenous lysosomal factors that can affect activity |
| Cell Lysate in vitro activity | 4-MUG, ResGlu BODIPY Glucosylceramide | Total GCase protein that includes lysosomal and non-lysosomal GCase | Analyzing total GCase protein, the effect of GCase mutations and covalent modification on GCase activity | Proof of concept studies for GCase chaperones and gene therapies | Is not able to correct for difference in GCase levels, which affect measured activity |
| Patient Biofluid in vitro activity | 4-MUG, ResGlu | Total GCase protein | Activity measurement in serum and CSF | Evaluation of target engagement, Patient selection | Function of GCase in serum and CSF and correlation with tissue activity is unknown |
| Western Blotting | Antibody | Total GCase protein, ER GCase, post-ER GCase | Quantifying ER retention of GCase and post-ER GCase | Proof of concept studies for GCase chaperones and gene therapies | Does not report on enzyme activity |
| Inhibody | MDW333, MDW941 | Lysosomal GCase protein | Quantifying lysosomal GCase protein, Analyzing GCase protein by microscopy | Proof of concept studies for GCase chaperones and gene therapies | Quantifies levels of active protein not enzyme activity |
| In situ GCase activity – cell culture | PFB-FDGlu3 | In situ lysosomal GCase activity | Analyzing lysosomal GCase activity while accounting for endogenous factors | Screening, proof of concept studies for GCase chaperons, gene therapies and activators | Measurement will be affected by differences in substrate uptake |
| In situ GCase activity – PBMC | PFB-FDGlu | In situ lysosomal GCase activity | Analyzing lysosomal GCase activity while accounting for endogenous factors | Verify target engagement of chaperones and activators, patient selection | Measurement will be affected by differences in substrate uptake |
| Dry Blood Spot assay | C12 Glucosylceramide | Total GCase protein that includes lysosomal and non-lysosomal GCase | Analyzing total GCase protein, the effect of GCase mutations, covalent modification on GCase activity | Patient selection, target engagement of GCase chaperones | Requires specialized sample preparation and equipment. Does not account for variation in endogenous lysosomal factors |
4-MU (4-methylumbelliferyl-β-D-glucopyranoside)
ResGlu (Reresorufin-β-D-glucopyranoside)
PFB-FDGlu (5-(Pentafluorobenzoylamino) Fluorescein Di-beta-D-Glucopyranoside)
In vitro GCase Activity using Fluorescent Substrates
The most commonly used method to evaluate GCase activity is the use of artificial fluorescent substrates combined with in vitro systems. This technique uses either recombinant GCase protein or protein extracted from cellular model systems including patient fibroblasts and iPSCs as well as animal or patient tissues or biofluids. The protein is then diluted in an acidic enzyme assay buffer to mimic the low pH of the lysosome. A critical component of the assay system is the addition of a lipid or detergent to maintain the enzyme in an active confirmation. This is necessary as delipidate GCase is essentially inactive25. There is significant variation in the lipid/detergent used in literature. The most commonly used is the bile salt taurocholate, however, neutral detergents or the acidic phospholipid, phosphatidylserine, are also common. To monitor enzymatic activity, several fluorescent probes have been developed. These include the blue fluorogenic substrate 4-methylumbelliferyl-β-D-glucopyranoside (4-MUG)26 or the red fluorogenic substrate resorufin-β-D-glucopyranoside (Res-Glu)27. While the lower pH in the reaction buffer is selective for lysosomal GCase, it is common to simultaneously treat samples with a selective GCase inhibitor such as conduritol B epoxide (CBE), or isofagomine, to determine the background signal in the system and remove any contribution of substrate hydrolysis by GBA2 which can also hydrolyze 4-MUG, although far less efficiently at lower pH. Alternatively, a GBA2 selective inhibitor such as NB-DNJ could be used to isolate GCase specific activity28. There are a number of factors that are essential to consider when setting up an in vitro GCase assay. The most important is to ensure that the enzyme kinetics are linear at the time of fluorescence measurement. Dilution of GCase into an assay buffer has been shown to reduce the stability of the enzyme. This is particularly important when assessing activity of mutant enzymes which are less stable than the WT enzyme. Linear kinetics is essential for accurate comparison of GCase activity and should be optimized prior to quantification. Another important consideration is the lysis buffer used to generate the cell/tissue lysates because GCase activity is very sensitive to the presence of detergent, the specific detergent used in the cell or tissue lysis buffer can significantly affect the apparent activity of GCase.
As discussed, significant variation exists in the exact conditions used for in vitro GCase activity assay. Instead of delving into the different buffer systems, pH, and detergents used in published in vitro GCase assays, it is important to determine what the buffering system accomplishes. In any in vitro enzyme assay, the biochemical activity of the enzyme is measured outside a biological system. As a result, this assay does not take into account in situ factors like variations lysosomal pH, natural allosteric regulators, the presence of co-factors like saposin C, or the current state of GCase in the ER or the lysosome. In an in vitro assay, the activity measurement is proportional with the total GCase protein in the sample. This limitation is highlighted by the observation that in rare instances of Gaucher-like disease caused by loss of saposin C, the activity of GCase is normal when measured by an in vitro GCase assay29. Therefore, in this assay design, the only major factor that could influence observed reaction rate is the presence of mutations that affect enzyme function or the presence of covalent post-translational modifications23. This is why in vitro GCase assays are diagnostic in GD and may help to identify GBA1 mutation carriers.
Advantages/Disadvantages
The use of in vitro GCase activity assays has had a significant impact on GCase research and therapeutic development. These assays have been used to diagnose GD and evaluate GCase activity derived from the tissue of patients with PD. Because of the robust assay signal, these approaches have been successfully used in high throughput screening30. Additionally, when evaluating the efficacy of molecular chaperones or gene therapy, the resulting increase in GCase protein can be detected using these strategies. Lastly, a major advantage of this approach is that is allows for the absolute quantification of GCase enzyme activity. In cell/tissue lysates this is expressed as nmol/mg protein/hour and in patient fluids this is expressed as μMol/L/Hr. In theory this enables comparison across different studies in the literature, however this is only possible if identical assay conditions are used, which is rarely the case.
A major limitation of this assay is that it does not account for endogenous factors that could influence GCase activity. These include mutations in lysosomal enzymes, chemical agents that cause lysosomal dysfunction, or agents that increase lysosomal pH which can lead to the accumulation and enlargement of lysosomes. The effects of these endogenous factors may display as normal or increased levels of GCase activity when using in vitro assays but may significantly alter the in situ GCase activity. An additional concern with this approach is that it does not exclude GCase located in the ER. As we have seen with certain GCase mutations and overexpression of GCase, there is considerable GCase retained in the ER that could also be included using such in vitro analyses (Figure 1). In vitro GCase assays are also not useful to assess GCase activation in cellular systems treated with putative positive allosteric modulators, as any modulator is likely to be significantly diluted upon cellular lysis and addition of reaction buffer. Lastly, differences in structure and affinity of the artificial substrates to mutant GCase may not reflect the affinity of the natural substrate31. In recombinant systems, this has been overcome by the use of natural substrate with mass spectrometry31 or using BODIPY labeling with HPLC32.
FIG 1. ER-retention of GCase in neurons overexpressing GBA1 and in fibroblasts from patients with Gaucher disease.
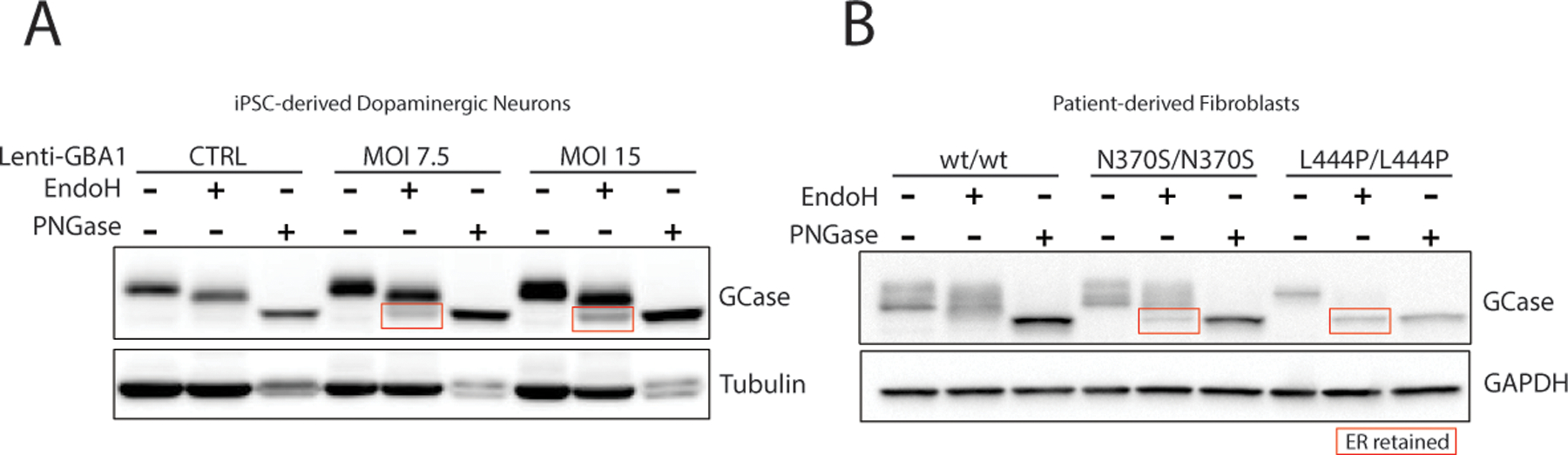
(A) Western blot analysis of lysates treated with Endo H, PNGase F or untreated from patient-derived dopaminergic neurons after lentiviral-mediated over expression of GBA1 for 2 weeks at multiplicity of infection (MOI) of 7.5 or 15 and control neurons treated with lentivirus expressing GFP. (B) Western blot analysis of lysates treated with Endo H, PNGase F or untreated from fibroblasts for control or patients with Gaucher disease type I (N370S/N370S) or type II (L444/L444P).
Use in therapeutic development
The in vitro GCase activity assay has played an important part in determining the role of GCase in the onset of PD. The assay has been used to show reduced GCase activity as a result of GBA1 mutations in patient-derived brain tissue. The assay has also been adapted to measure GCase activity in serum and, more recently, was optimized for measuring GCase activity in the CSF33. As discussed above, in vitro measurements of GCase activity reflect the amount of GCase protein in the sample. As such, this assay is well suited to measure the effects of molecular chaperones. Systemic administration of molecular chaperones would lead to increased GCase in all cell types including blood cells. Such treatments may also lead to increased GCase protein in serum, although the mechanism through which GCase is released into the serum is unclear. Similarly, increased GCase activity in CSF has recently been reported upon administration of Ambroxol, a GCase molecular chaperone34. While CNS administration of gene therapy would limit peripheral measurements, similar effects on GCase in the CSF could be expected from this approach. This was observed in recent data published by Prevail Therapeutics which demonstrated significantly increased GCase activity in the CSF in a patient following treatment with PR00135, although the patient in this report was homozygous for GBA1 mutations which does not reflect most GBA1-PD patients.
Measurement of GCase protein by SDS-PAGE
Western blot is the most widely used analytical technique to assess specific proteins in a cell or tissue homogenate. A number of commercial antibodies to GCase have been developed with varying degrees of success. One recent analysis of several antibodies used murine neural cells deficient in GCase, which invalidated a surprising number of commercially available antibodies36. This study serves as a key resource for researchers investigating GCase using western blot techniques and highlights the importance of proper antibody validation.
The glycosylation of GCase creates an additional challenge for western blot detection of GCase. Early pulse chase studies revealed that GCase is initially glycosylated in the ER by N-linked high-mannose-type oligosaccharides on 4 of it 5 putative sites4. When fully glycosylated, this species runs at an apparent molecular weight of 64 kDa and can be completely deglycosylated by Endo H treatment. As GCase is transported towards the lysosome, further maturation of these oligosaccharides occurs in the Golgi apparatus yielding a species with an apparent molecular weight of 69 kDa. The half-life for this conversion in patient-derived fibroblasts was found to be 3 hours37. After an additional 48 hours, the glycosylation can be further modified to a species with an apparent molecular weight of 59 kDa, presumably through modification by lysosomal exoglycosidases. Therefore, both the 59 and 69 kDa species represent post-golgi GCase protein as they are largely resistant to endo H treatment. Treatment with PNGase F, which removes all N-linked glycosylations, results in species that have the same apparent molecular weight, indicating that the shift in molecular weights is not due to proteolytic processing of GCase37.
The presence of two apparent molecular weight GCase species in the lysosome has generated some confusion. The prevalence of one species over the other appears to vary depending on the cell line or tissue source that is analyzed. Some researchers have incorrectly indicated the lower 59 kDa molecular weight band as ER-retained GCase, which has led to the conclusion that in the absence of GBA1 mutations, a significant fraction of cellular GCase is basally retained in the ER. This is unlikely as the half-life of GCase in the ER is very short and is supported by evidence that knockdown of the GCase transporter, LIMP-2, which would theoretically cause all GCase protein to be retained in the ER, leads to an almost complete loss of GCase highlighting the speed at which ER-GCase is degraded. Unlike the WT enzyme, many of the mutations in GCase can lead to its retention in the ER, which can be identified by examining Endo H sensitivity (Figure 1). This has led to speculation that misfolding in the ER could promote ER-stress and modification of disease phenotypes. This has been observed in patient derived fibroblasts and animal models of GD38,39, however only preliminary studies have shown a connection in PD20. Further studies are required to establish whether ER-stress contributes to pathogenesis of GBA-PD. A major goal of the molecular chaperone strategy is to assist in proper folding of these ER-retained forms to allow for optimum transport from the ER. This strategy could be beneficial two-fold as it reduces the amount of misfolded protein in the ER and potentially increases the amount of GCase in the lysosome.
Advantages/Disadvantages
Evaluation of the level of ER-GCase using Endo H sensitivity can be an effective strategy to evaluate the potential of molecular chaperones to improve trafficking of mutant GCase to the lysosome. This approach could also serve to evaluate a concern associated with gene therapy which is that excessive GCase overexpression will overwhelm the ability of LIMP-2 to traffic the protein to the lysosome leading to an undesired consequence of GCase accumulation in the ER (Figure 1). A disadvantage of this approach is that there is considerable noise in western blotting technique making it challenging to accurately obtain quantification. This is especially challenging for GCase as glycosylation provides an additional variable that may affect affinity of the primary antibody to its GCase epitope. Therefore, treatment of all samples with PNGase F can be used to improve the reliability of total GCase quantification by western blot.
Use in therapeutic development
The cumbersome, low throughput and variable nature of the SDS-PAGE technique gives this measurement limited usability in translational approaches. However, the ability to measure ER-retained GCase makes this a critical method to evaluate therapeutic strategies in cellular and animal models. This technique could provide important proof of mechanism in cell and animal models for molecular chaperones that are designed to bind mutant GCase retained in the ER and enable trafficking to the lysosome. Additionally, a concern for the development of small molecules that bind GCase is that they could cause structural changes that affect the LIMP-2 binding site. These molecules would therefore affect the trafficking of GCase resulting in ER accumulation and potentially less lysosomal GCase. A similar concern exists for gene therapy strategies where increased expression of GCase may lead to ER-retention by overwhelming the capacity of LIMP-2 to traffic GCase to the lysosome. These concerns could be alleviated by examining ER-retained GCase and titrating the level of expression to ensure that ER-retention is mitigated.
Measurement of GCase using Inhibodies
Another approach that has been developed to visualize lysosomal GCase levels is the development of inhibodies40. This approach made use of epoxides like CBE and cyclophellitol which first bind non-covalently to GCase at the active site and then reacts with glutamate 340, forming a covalent bond that irreversibly inhibits the enzyme. Fluorescent boron dipyrromenthene (BODIPY) analogues were attached to cyclophellitol using a triazole linker which led to the generation of MDW333 and MDW94140. These fluorescent probes can be incubated with cultured cells where they react with lysosomal GCase and produce a clear lysosomal staining pattern in live cells that can be analyzed by microscopy (Figure 2a,b) or flow cytometry. Similarly, the probes can be injected intravenously in mice. After incubation, the level of GCase in tissue lysates can be examined using SDS-PAGE, although this utility is limited to peripheral tissue as the probe is not able to access GCase in the brain. More recently this limitation was overcome by directly applying probes to the CNS through i.c.v. administration41.
FIG 2. Decreased GCase levels in superior temporal gyrus (STG) from GBA1 mutation carriers and sporadic PD revealed by Fluorescent GCase Probe.
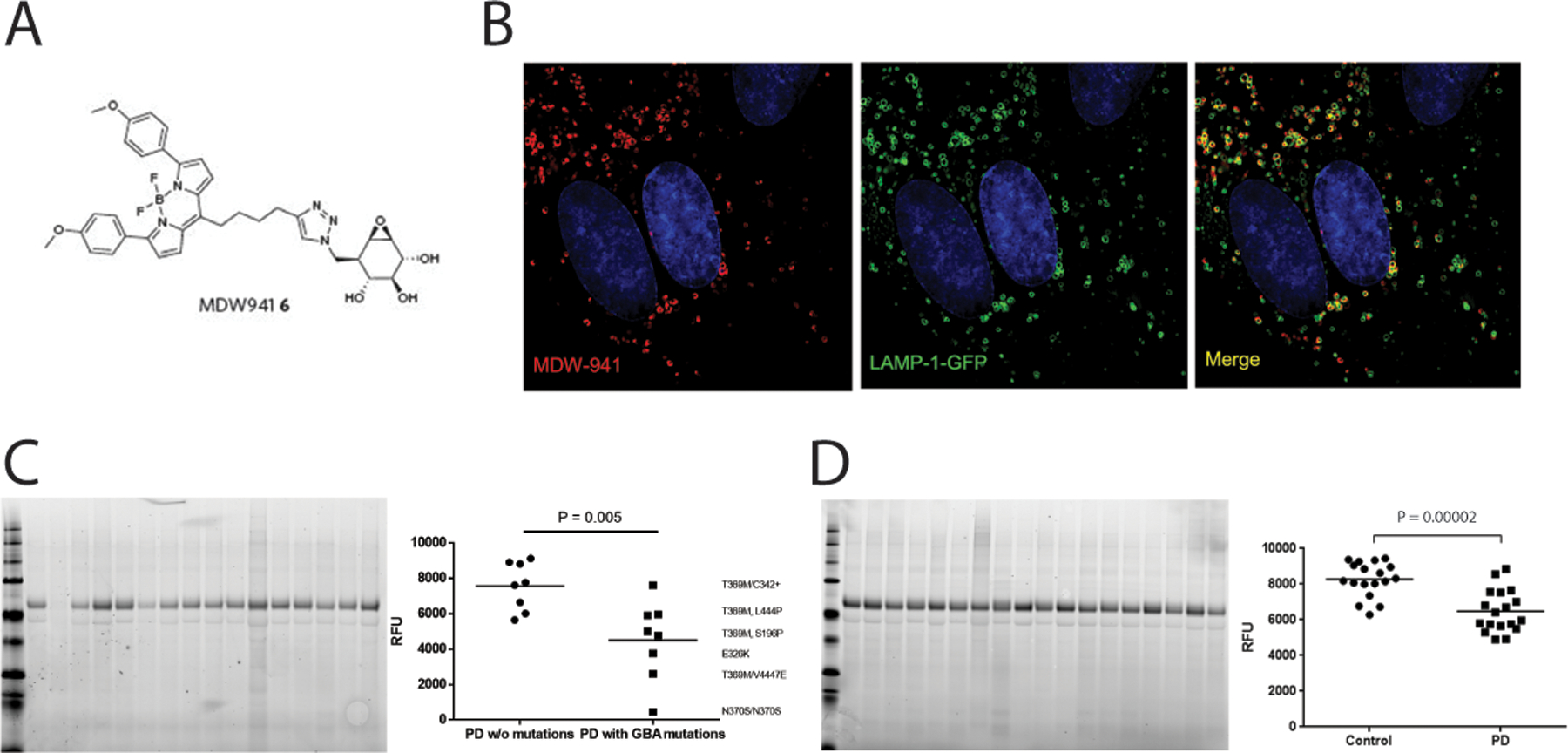
(A) Chemical structure of MDW941. (B) Representative images from super-resolution microscopy imaging of cultured human fibroblasts expressing Lamp-1-GFP stained with the GCase probe MDW-941. (C) Representative SDS PAGE analysis of STG lysates derived from PD patients with and without GBA1 mutations treated with MDW-941. Genotypes for each data point are shown on the right (D) SDS PAGE analysis of STG lysates derived from healthy control or PD patients without GBA1 mutations. Data are presented as the mean fluorescence signal from MDW-941-modified GCase with individual data points representing unique samples. Data were analyzed using two-way ANOVA followed by a Bonferroni post hoc test.
Advantages/Disadvantages
The use of inhibody based probes has an advantage over in vitro activity assays and western blotting as it allows for relative quantification of active GCase protein levels in live cells or tissue lysates (Figure2 c,d). This enables the use of less biased high content imaging and flow cytometry-based approaches to quantify GCase levels. This could be especially useful for evaluation of target engagement of molecular chaperone-based approaches. However, it is unclear what effect lysosomal pH could have on the fluorescent intensity of the BODIPY fluorophore, as this would have implications for quantification. Although the probes were shown to be predominantly active at lower pH, they retain modest inhibitory activity at neutral pH40. Therefore, it is unclear to what extent they will react with ER-retained GCase, although preliminary data show strong lysosomal localization of the probe in treated cells (Figure 2b). Lastly, while these probes do label GCase in live cells, they have similar limitations as the in vitro GCase activity measurements as they will only measure the total amount of GCase in the lysosome and not account for endogenous lysosomal conditions that could affect GCase activity.
Measurement of in situ GCase activity using PFB-FDGlu
As mentioned above, a major disadvantage of in vitro assays to measure GCase activity is that they do not account for changes in the lysosomal microenviroment that could impact GCase activity. To overcome this limitation the cell permeable GCase substrate 5-(Pentafluorobenzoylamino) Fluorescein Di-beta-D-Glucopyranoside (PFB-FDGlu) can be used. PFB-FDGlu is a fluorescent quenched probe that yields green fluorescence upon hydrolysis by GCase. The probe is taken up in the cell by pinocytosis and trafficked through the endosomal system to the lysosome where it can be cleaved by lysosomal GCase42. To correct for background fluorescence and potential off target hydrolysis of PFB-FDGlu by cytosolic GCase, cells can be incubated with GCase selective inhibitors CBE or isofagomine (Figure 3a).
FIG. 3. Dose dependent reduction in live-cell GCase activity in the presence of isofagomine or the GCase chaperone Ambroxol.
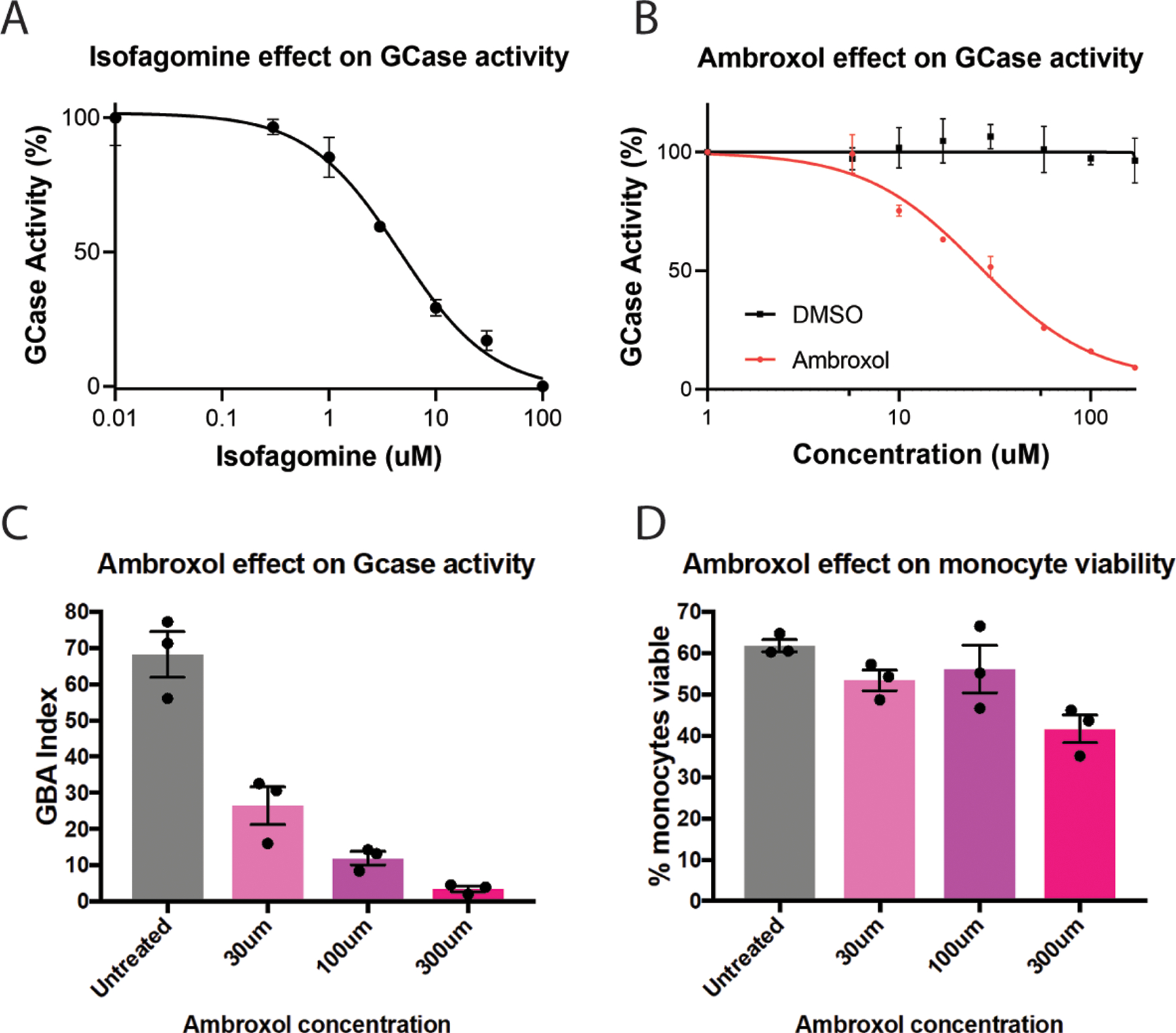
(A,B) Dose response curve showing inhibition of lysosomal GCase activity by isofagomine (A) or by Ambroxol (B) in cultured HeLa cells. (C) GCase activity measured in CD14 positive peripheral blood-derived monocytes treated with increasing concentration of Ambroxol. (D) Evaluation of the effect of Ambroxol on monocyte viability. The data is presented as a GBA activity index, which is the ratio of PFB-FDglu signal without CBE, divided by the PFB-FDglu signal with CBE. Data were analyzed by one-way ANOVA and Dunnet’s multiple comparison test. *= p < 0.05 compared to the untreated group. Graphs show mean +/− SEM with the dots representing individual data points.
The PFB-FDGlu approach has been used to measure in situ GCase activity in a number of cell types including patient-derived fibroblasts43 and liver cells44,45. More recently, PFB-FDGlu was used to examine GCase activity in iPSC-derived dopaminergic neurons using a microplate format46. In this study, it was found that mutations in LRRK2 affect GCase activity, despite not influencing the absolute level of GCase protein. In another recent study, where PFB-FDGlu was used to measure in situ GCase activity in PBMCs from PD patients, the authors found that when correcting for protein content, monocytes from PD patients display reduced GCase activity47. Interestingly, while the raw GCase activity in these cells displayed a trend towards reduced activity, analysis of the protein content revealed in an increase in GCase levels47. This deviation further highlights the disconnect between in situ GCase activity and GCase protein levels and underscores the importance of considering in situ activity when evaluating GCase activity.
Advantages/Disadvantages
The advantage of this approach is that it allows for measurement of in situ GCase activity, which is most relevant to lysosomal function. It also accounts for changes in the lysosomal microenvironment such as changes in pH, ion content, lipid content, accumulation of misfolded protein, and other factors that have been shown to affect the function of lysosomal enzymes. Evaluation of in situ GCase activity will allow for the expansion of studies on GCase regulation in the lysosome, which could lead to the identification of new therapeutic targets to enhance GCase activity independently of the protein. This potential is highlighted by the identification that LRRK2 kinase inhibitors were found to increase GCase activity in neurons46.
However, a weakness of the PFB-FDGlu approach is that the substrate requires uptake by pinocytosis which leads to several concerns that must be considered when evaluating relative enzyme activity. As with any enzymatic assay, the rate of hydrolysis of PFB-FDGlu is dependent on substrate concentration42. Genetic or chemical perturbations that affect the pinocytosis pathway could lead to reduced loading of substate which may falsely produce differences in GCase activity readout. This also applies in the evaluation of different cell types as the rate of pinocytosis could vary greatly between different cells leading to artifacts of apparent’ differences in GCase activity but may simply reflect the differences in pinocytosis rates.
Use in therapeutic development
In situ GCase activity is the most accurate measurement of GCase activity occurring in the lysosome. For this reason, use of in situ measurements are well suited to evaluate the effects of all therapeutic strategies targeting GCase in cell culture models. This is especially important in the identification of GCase chaperones, as molecular chaperones can often inhibit enzyme activity at elevated concentrations48. This inhibitory effect is observed for Ambroxol at micromolar concentrations in cell culture models (Figure 3b–d). For preclinical animal models, the PFB-FDGlu assay is more limited. The ability to measure GCase activity in PBMCs would allow measurement of target engagement for both GCase chaperones and activators in blood. However, it is currently not possible to perform in situ measurement in the CNS limiting the use of this technique for gene therapy approaches that are CNS administered. Therefore, for early clinical trials, measurement of GCase activity in patient PBMCs could allow measurement of target engagement for GCase chaperones and activators in blood, although this may not be feasible in multisite studies for logistical reasons. Perhaps the best role of in situ GCase activity measurements for clinical development is in patient selection. Pre-screening PD patients to identify individuals that have significantly reduced GCase activity in the presence or absence of GBA1 mutations could increase the likelihood of seeing a significant effect of therapeutic intervention. The assumption is that patients with low PBMC GCase activity will also have low activity in the CNS. This has not yet been established but may warrant further investigation given the potential benefits of this approach.
Measurement of GCase activity in dry blood spots
Dried blood spot assays are currently being used for the identification of a range of lysosomal storage disorders including Gaucher Disease49,50. This technique uses blood blotted onto filter paper to enable simple storage and banking of samples for future analysis. Recent iterations of this technique use mass spectrometer-based detection instead of fluorescent detection, which allows for measurement of multiple lysosomal enzymes concurrently.
The dry blood spot analysis has been applied to assess GCase activity in PD patients with and without GBA1 mutations18. In this study, the researchers included a natural substrate C12-glucosylceramide for measurement of enzymatic activity. Specifically, they used punches from stored dried blood spots and upon initial extraction in a neutral buffer, samples were then incubated in an acidic assay buffer containing C12-glucosylceramide. The samples were analyzed by mass spectrometry to measure the hydrolysis of C12-glucosylceramide. More recently, this dry blood spot assay was used to assess GCase activity in a three-year longitudinal study of 1559 samples from the Parkinson’s Progression Markers Initiative (PPMI) cohort51. In concurrence with previous studies, this study reported a significant reduction in GCase activity in samples from patients with PD relative to healthy control.
Advantage/Disadvantage
The use of dried blood spots to measure enzyme activity is advantageous due to ease of sampling, shipping, and stability of the samples. The use of mass spectrometer-based approaches is also advantageous as it allows for concurrent measurement of multiple lysosomal enzymes. Additionally, this method examines hydrolysis of a natural substrate mimic, C12 glucosylceramide, which avoids concerns associated with artificial substrates, as discussed above31. A disadvantage of this approach is that there is that more advanced instrumentation is required in contrast to the quick, fluorescence-based detection methods. Additionally, this approach cannot easily account for sampling differences in cell types that may change dramatically from day to day or may exist in a disease population. While the recent study was able to correct for white blood cell count, future studies could focus on further refinement to specifically account for different cell populations.
Use in therapeutic development
The dry blood spot assay allows for very simple sample collection and storage. This makes the assay well-suited to perform longitudinal assessments of GCase activity. As discussed, measurements of GCase activity in dried blood spots are likely to reflect the amount of GCase protein in the sample. As a result, this assay could serve as an excellent strategy to evaluate GCase accumulation resulting from molecular chaperone exposure. This could be applied to preclinical animal studies, as well as clinical trials in humans. The ability to collect samples from multiple sites over multiple time points and perform the analysis at a single location is a clear advantage. It may even be possible to adapt this method to measure GCase activity in CSF of individuals treated with chaperones or gene therapy. However, the sample dilution required in this assay would result in dilution of the active compound, therefore, this assay is unlikely to capture effects of GCase activators on enzyme activity.
Conclusion:
There is an increasing recognition that the lysosomal enzyme GCase plays a critical role in the onset of familial and also sporadic Parkinson’s Disease and Dementia with Lewy Bodies. As a result, there is considerable interest in development of therapies that target GCase to slow or stop progression of these diseases. To enable measurement of GCase activity in disease modeling systems and patient populations, a growing number of techniques have been established. This review provides a framework for how these techniques can be used in preclinical and clinical development of GCase targeted therapies.
Supplementary Material
Acknowledgements:
This work was supported by the Michael J. Fox Foundation’s LRRK2 and GBA Consortium Call Series. We are grateful to Paolo Loos who provided Figure 2a,b and Peter Buckett for Figure 2c,d.
Financial Disclosures:
D. Y. and M.N. are employees of Vanqua Bio. T.Y. has nothing to declare. N.D. was supported by the Michael J. Fox Foundation and the Shake It Up Australia foundation. W.D.H. is a Biogen employee and shareholder. D.K is a venture partner with Orbimed Advisors, scientific advisor for Intellia Therapeutics, AcureX, The Silverstein Foundation, Prevail Therapeutics, and the Founder of Vanqua Bio and Lysosomal Therapeutics. D.K. was supported by NIH grants R01 NS076054, R37 NS096241, R01 NS096240-01, U01 NS 094148 and grants from the Michael J. Foundation for Parkinson’s Research
Footnotes
Relevant Conflicts of interest/financial disclosures: D.Y. and M.N are employees of Vanqua Bio, W.D.H. is a Biogen employee and shareholder, D.K is a venture partner with Orbimed Advisors, scientific advisor for Intellia Therapeutics, AcureX, The Silverstein Foundation, Prevail Therapeutics, and the Founder of Vanqua Bio and Lysosomal Therapeutics.
References
- 1.Ho MW & O’Brien JS Gaucher’s disease: deficiency of ‘acid’ -glucosidase and reconstitution of enzyme activity in vitro. Proc Natl Acad Sci U S A 68, 2810–2813, doi: 10.1073/pnas.68.11.2810 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yildiz Y et al. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J Clin Invest 116, 2985–2994, doi: 10.1172/JCI29224 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayashi Y et al. Klotho-related protein is a novel cytosolic neutral beta-glycosylceramidase. J Biol Chem 282, 30889–30900, doi: 10.1074/jbc.M700832200 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Erickson AH, Ginns EI & Barranger JA Biosynthesis of the lysosomal enzyme glucocerebrosidase. J Biol Chem 260, 14319–14324 (1985). [PubMed] [Google Scholar]
- 5.Do J, McKinney C, Sharma P & Sidransky E Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener 14, 36, doi: 10.1186/s13024-019-0336-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boven LA et al. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am J Clin Pathol 122, 359–369, doi: 10.1309/BG5V-A8JR-DQH1-M7HN (2004). [DOI] [PubMed] [Google Scholar]
- 7.Ferraz MJ et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett 590, 716–725, doi: 10.1002/1873-3468.12104 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Revel-Vilk S, Fuller M & Zimran A Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int J Mol Sci 21, doi: 10.3390/ijms21197159 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaye EM, Ullman MD, Wilson ER & Barranger JA Type 2 and type 3 Gaucher disease: a morphological and biochemical study. Ann Neurol 20, 223–230, doi: 10.1002/ana.410200208 (1986). [DOI] [PubMed] [Google Scholar]
- 10.Farfel-Becker T et al. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum Mol Genet 23, 843–854, doi: 10.1093/hmg/ddt468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verity MA & Montasir M Infantile Gaucher’s disease: neuropathology, acid hydrolase activities and negative staining observations. Neuropadiatrie 8, 89–100, doi: 10.1055/s-0028-1091508 (1977). [DOI] [PubMed] [Google Scholar]
- 12.Grafe M, Thomas C, Schneider J, Katz B & Wiley C Infantile Gaucher’s disease: a case with neuronal storage. Ann Neurol 23, 300–303, doi: 10.1002/ana.410230315 (1988). [DOI] [PubMed] [Google Scholar]
- 13.Clark LN et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 69, 1270–1277, doi: 10.1212/01.wnl.0000276989.17578.02 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sidransky E et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361, 1651–1661, doi: 10.1056/NEJMoa0901281 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nalls MA et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 70, 727–735, doi: 10.1001/jamaneurol.2013.1925 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiger JT et al. Next-generation sequencing reveals substantial genetic contribution to dementia with Lewy bodies. Neurobiol Dis 94, 55–62, doi: 10.1016/j.nbd.2016.06.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazzulli JR et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52, doi: 10.1016/j.cell.2011.06.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alcalay RN et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 138, 2648–2658, doi: 10.1093/brain/awv179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parnetti L et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov Disord 32, 1423–1431, doi: 10.1002/mds.27136 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Gegg ME et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 72, 455–463, doi: 10.1002/ana.23614 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rocha EM et al. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol 2, 433–438, doi: 10.1002/acn3.177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huebecker M et al. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol Neurodegener 14, 40, doi: 10.1186/s13024-019-0339-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burbulla LF et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261, doi: 10.1126/science.aam9080 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas R, Moloney EB, Macbain ZK, Hallett PJ & Isacson O Fibroblasts from idiopathic Parkinson’s disease exhibit deficiency of lysosomal glucocerebrosidase activity associated with reduced levels of the trafficking receptor LIMP2. Mol Brain 14, 16, doi: 10.1186/s13041-020-00712-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glew RH, Daniels LB, Clark LS & Hoyer SW Enzymic differentiation of neurologic and nonneurologic forms of Gaucher’s disease. J Neuropathol Exp Neurol 41, 630–641, doi: 10.1097/00005072-198211000-00006 (1982). [DOI] [PubMed] [Google Scholar]
- 26.Owada M, Sakiyama T & Kitagawa T Neuropathic Gaucher’s disease with normal 4-methylumbelliferyl-beta-glucosidase activity in the liver. Pediatr Res 11, 641–646, doi: 10.1203/00006450-197705000-00004 (1977). [DOI] [PubMed] [Google Scholar]
- 27.Hays WS, Wheeler DE, Eghtesad B, Glew RH & Johnston DE Expression of cytosolic beta-glucosidase in guinea pig liver cells. Hepatology 28, 156–163, doi: 10.1002/hep.510280121 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Lee JC et al. Synthesis and evaluation of eight- and four-membered iminosugar analogues as inhibitors of testicular ceramide-specific glucosyltransferase, testicular beta-glucosidase 2, and other glycosidases. J Org Chem 77, 3082–3098, doi: 10.1021/jo202054g (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang L et al. A rare form of Gaucher disease resulting from saposin C deficiency. Blood Cells Mol Dis 68, 60–65, doi: 10.1016/j.bcmd.2017.04.001 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Urban DJ et al. Optimization and validation of two miniaturized glucocerebrosidase enzyme assays for high throughput screening. Comb Chem High Throughput Screen 11, 817–824, doi: 10.2174/138620708786734244 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf P et al. Tandem mass spectrometry assay of beta-glucocerebrosidase activity in dried blood spots eliminates false positives detected in fluorescence assay. Mol Genet Metab 123, 135–139, doi: 10.1016/j.ymgme.2017.10.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motabar O et al. A high throughput glucocerebrosidase assay using the natural substrate glucosylceramide. Anal Bioanal Chem 402, 731–739, doi: 10.1007/s00216-011-5496-z (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oftedal L, Maple-Grodem J, Forland MGG, Alves G & Lange J Validation and assessment of preanalytical factors of a fluorometric in vitro assay for glucocerebrosidase activity in human cerebrospinal fluid. Sci Rep 10, 22098, doi: 10.1038/s41598-020-79104-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mullin S et al. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol 77, 427–434, doi: 10.1001/jamaneurol.2019.4611 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prevail Therapeutics. Prevail Therapeutics Second Quarter Clinical and Financial Update [Press Release]. 2020. https://www.prevailtherapeutics.com/wp-content/uploads/2021/01/Prevail-Therapeutics-Reports-Second-Quarter-2020-Financial-Results-and-Business-Highlights.pdf
- 36.Qi W et al. Validation of anti-glucocerebrosidase antibodies for western blot analysis on protein lysates of murine and human cells. Biochem J 476, 261–274, doi: 10.1042/BCJ20180708 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Bergmann JE & Grabowski GA Posttranslational processing of human lysosomal acid beta-glucosidase: a continuum of defects in Gaucher disease type 1 and type 2 fibroblasts. Am J Hum Genet 44, 741–750 (1989). [PMC free article] [PubMed] [Google Scholar]
- 38.Ron I & Horowitz M ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet 14, 2387–2398, doi: 10.1093/hmg/ddi240 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Braunstein H et al. UPR activation and CHOP mediated induction of GBA1 transcription in Gaucher disease. Blood Cells Mol Dis 68, 21–29, doi: 10.1016/j.bcmd.2016.10.025 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Witte MD et al. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat Chem Biol 6, 907–913, doi: 10.1038/nchembio.466 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Herrera Moro Chao D et al. Visualization of Active Glucocerebrosidase in Rodent Brain with High Spatial Resolution following In Situ Labeling with Fluorescent Activity Based Probes. PLoS One 10, e0138107, doi: 10.1371/journal.pone.0138107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lorincz M, Herzenberg LA, Diwu Z, Barranger JA & Kerr WG Detection and isolation of gene-corrected cells in Gaucher disease via a fluorescence-activated cell sorter assay for lysosomal glucocerebrosidase activity. Blood 89, 3412–3420 (1997). [PubMed] [Google Scholar]
- 43.Zheng J et al. Conversion of Quinazoline Modulators from Inhibitors to Activators of beta-Glucocerebrosidase. J Med Chem 62, 1218–1230, doi: 10.1021/acs.jmedchem.8b01294 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Es HH, Veldwijk M, Havenga M & Valerio D A flow cytometric assay for lysosomal glucocerebrosidase. Anal Biochem 247, 268–271, doi: 10.1006/abio.1997.2090 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Chan KW et al. Measurement of lysosomal glucocerebrosidase activity in mouse liver using a fluorescence-activated cell sorter assay. Anal Biochem 334, 227–233, doi: 10.1016/j.ab.2004.08.031 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Ysselstein D et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat Commun 10, 5570, doi: 10.1038/s41467-019-13413-w (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Atashrazm F et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci Rep 8, 15446, doi: 10.1038/s41598-018-33921-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng J et al. beta-Glucocerebrosidase Modulators Promote Dimerization of beta-Glucocerebrosidase and Reveal an Allosteric Binding Site. J Am Chem Soc 140, 5914–5924, doi: 10.1021/jacs.7b13003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang XK et al. Multiplex enzyme assay screening of dried blood spots for lysosomal storage disorders by using tandem mass spectrometry. Clin Chem 54, 1725–1728, doi: 10.1373/clinchem.2008.104711 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Reuser AJ et al. The use of dried blood spot samples in the diagnosis of lysosomal storage disorders--current status and perspectives. Mol Genet Metab 104, 144–148, doi: 10.1016/j.ymgme.2011.07.014 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Alcalay RN et al. Longitudinal Measurements of Glucocerebrosidase activity in Parkinson’s patients. Ann Clin Transl Neurol 7, 1816–1830, doi: 10.1002/acn3.51164 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.