
Figure 3: The Biochemistry of Parkinson’s Disease Initiation and Progression and How to Disrupt It.
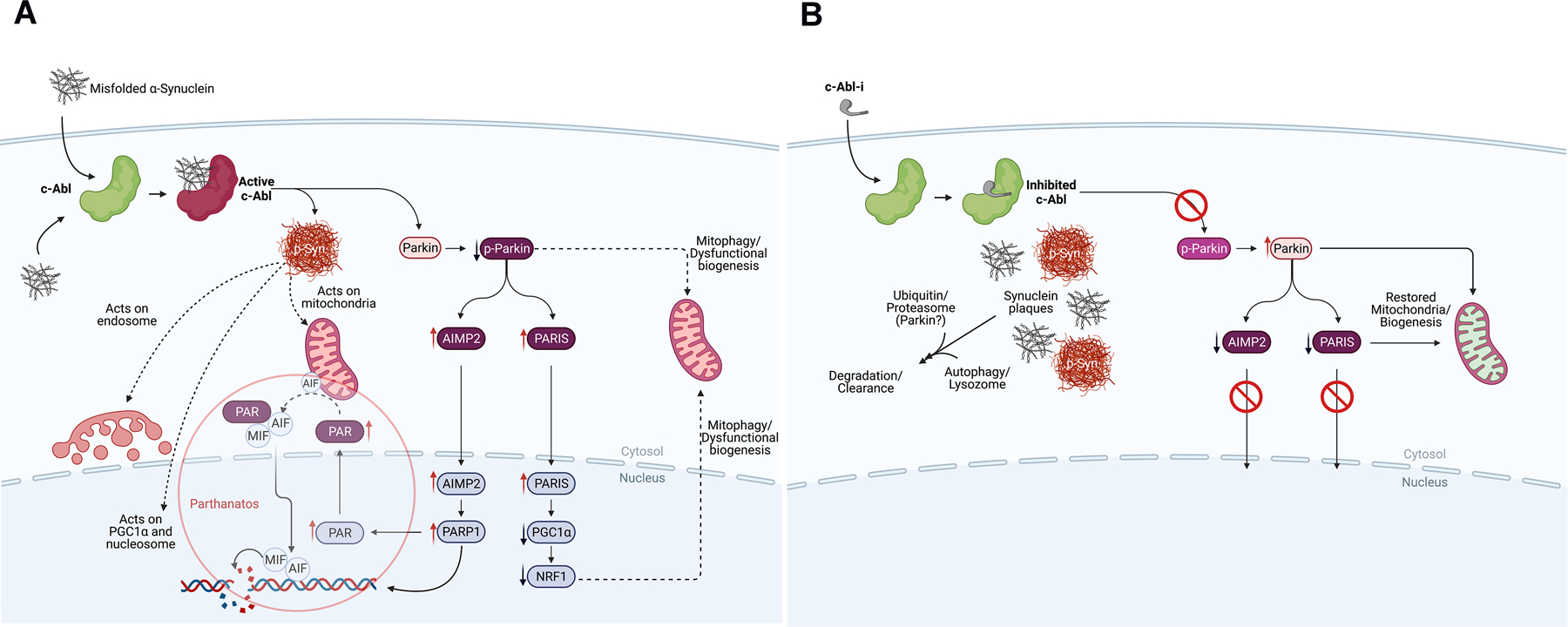
A. The process of neurodegeneration. Misfolded α-synuclein can arise from a variety of factors (see text). Misfolded α-synuclein may form within the neuron or by transfer through cell surface receptors or by crossing membrane bilayers. Within a neuron, misfolded α-synuclein is ‘sensed’ and c-Abl activated, driving the formation of pathologic α-synuclein by chemical modification (p-Syn).28 Chemical modification creates a form of α-synuclein that represents the pathologic species of the disease leading to disruption of mitochondrial integrity, negatively impact the endosome, disrupt nucleosomal structure and modulate transcription of certain genes.32–38, 63–73 C-Abl also inactivates parkin by chemical modification, which affects mitochondrial quality control and suppresses protein clearance mechanisms.28–34, 78–80 Parkin inactivation suppresses the complex interplay between parkin and pink1 at the mitochondrion, which act in concert to maintain mitochondrial integrity, quality and regulate mitochondrial biogenesis.78 Parkin inactivation leads to the accumulation of toxic parkin substrates PARIS (PARkin Interacting Substrate), aminoacyl tRNA synthetase complex-interacting multifunctional protein 2 (AIMP2) and far upstream element-binding protein 1 (FBP1).28–34, 78–80 PARIS and AIMP2 accumulate in adult conditional parkin knockout mice and MPTP-intoxicated mice as well as in patients with PD. Increased levels of PARIS can lead to mitochondrial dysfunction through down-regulation of PGC-1α and loss of DA neurons in a PARIS-dependent manner. Overexpression of AIMP2 leads to an age-dependent, selective degeneration of DA neurons through activation of poly (ADP-ribose) polymerase 1 (PARP1), driving PARP1-mediated parthanatos. This suggests that PARIS and AIMP2 may be important contributors to the loss of DA neurons and possibly other vulnerable neurons following parkin inactivation. Inactivation of parkin also disrupts protein clearance mechanisms through autophagy, lysosomal and proteasomal degradation pathways.
B. The consequences of c-Abl inhibitor treatment on the process of neurodegenerative disease. Inhibition of c-Abl precludes c-Abl activation, blocking the build-up of toxic parkin substrates PARIS and AIMP2 and terminating downstream events. This also re-establishes normal mitochondrial quality control and biogenesis. Model studies demonstrate that modified and unmodified α-synuclein aggregates are shunted to lysosomal or proteasomal degradation pathways for clearance with concomitant recovery of motor function.