

Abstract
Although several monoclonal antibodies (mAbs) targeting SARS-CoV-2 have been approved for COVID-19 therapy, their development was generally inefficient, with lead generation often requiring the production and testing of numerous antibody candidates. Here, we report that the integration of target–ligand blocking with a previously described B-cell receptor sequencing approach (LIBRA-seq) enables the rapid and efficient identification of multiple neutralizing mAbs that prevent the binding of SARS-CoV-2 Spike (S) protein to angiotensin-converting enzyme 2 (ACE2). The combination of target–ligand blocking and high-throughput antibody sequencing promises to increase the throughput of programs aimed at discovering novel neutralizing antibodies.
Technologies for developing preventive and therapeutic measures that can counteract potential pandemics are of utmost significance for public health. The COVID-19 pandemic has emphasized the importance of rapid countermeasure development. Through pandemic preparedness initiatives, effective SARS-CoV-2 neutralizing antibodies were discovered and validated within months1–6, as were SARS-CoV-2 vaccine candidates7. However, even with such unprecedented speed of vaccine and therapeutic development, the pandemic has inflicted devastating worldwide effects. Accelerating actions by weeks or months can make an enormous difference in an exponentially evolving pandemic. Therefore, efficient methods for discovery of effective countermeasures against emerging pathogens can play a critical role in pandemic preparedness for future infectious disease outbreaks.
Antibodies are a major modality for therapy and vaccine design strategies for a wide range of diseases; however, the functional antibody discovery process can be inefficient. Typically, at the screening step, B cells are prioritized based on antigen-recognition, but this often requires time-intensive subsequent monoclonal antibody validation steps for discovery of functional, neutralizing antibodies. This limitation was exemplified by SARS-CoV-2 antibody discovery initiatives, as testing of large numbers of antibodies (frequently hundreds to thousands) was generally required to identify a small fraction of neutralizing antibodies, with a wide range of hit rates when using Spike (S) as an antigen bait (about 2 to 23%) or when using RBD and/or S1 (about 2–55%)1–6, 8–12 in various studies.
To overcome this limitation, we developed LIBRA-seq with ligand blocking, a second-generation LIBRA-seq technology that incorporates a functional readout into the antibody discovery process13. LIBRA-seq (linking B cell receptor to antigen specificity through sequencing) uses DNA-barcoded antigens to map antibody sequence to antigen specificity using next-generation sequencing13. For LIBRA-seq with ligand blocking, a ligand and its cognate target antigen(s) are each labeled with a unique oligonucleotide barcode (Extended Data Figure 1A), enabling the transformation of antigen-ligand interactions into sequence-able events. In these experiments, B cells that can block antigen-ligand interactions are expected to have high LIBRA-seq scores for the target antigen(s) and low LIBRA-seq scores for the ligand (Extended Data Figure 1A). Therefore, a single high-throughput LIBRA-seq with ligand blocking experiment provides both antigen recognition and ligand blocking information simultaneously for many B cells.
To evaluate this technology, we sought to discover SARS-CoV-2-specific antibodies from B cells from subjects with past SARS-CoV-2 infection, since antibodies that block the interactions of the SARS-CoV-2 S protein with its host receptor angiotensin-converting enzyme 2 (ACE2) are among the most potently neutralizing identified to date2–6, 8, 9. We performed three LIBRA-seq experiments, with screening libraries that included: experiment 1, ACE2 and SARS-CoV-2 S; experiment 2, a titration series of different aliquots of SARS-CoV-2 S, each labeled with a unique barcode; and experiment 3, ACE2 and a titration series of S (Figure 1A). The incorporation of a titration series of S antigen in the screening library for experiments 2 and 3 aimed to assess the strength of BCR-antigen interactions (Extended Data Figure 1B, C).
Figure 1. Antibody discovery using LIBRA-seq with ligand blocking.
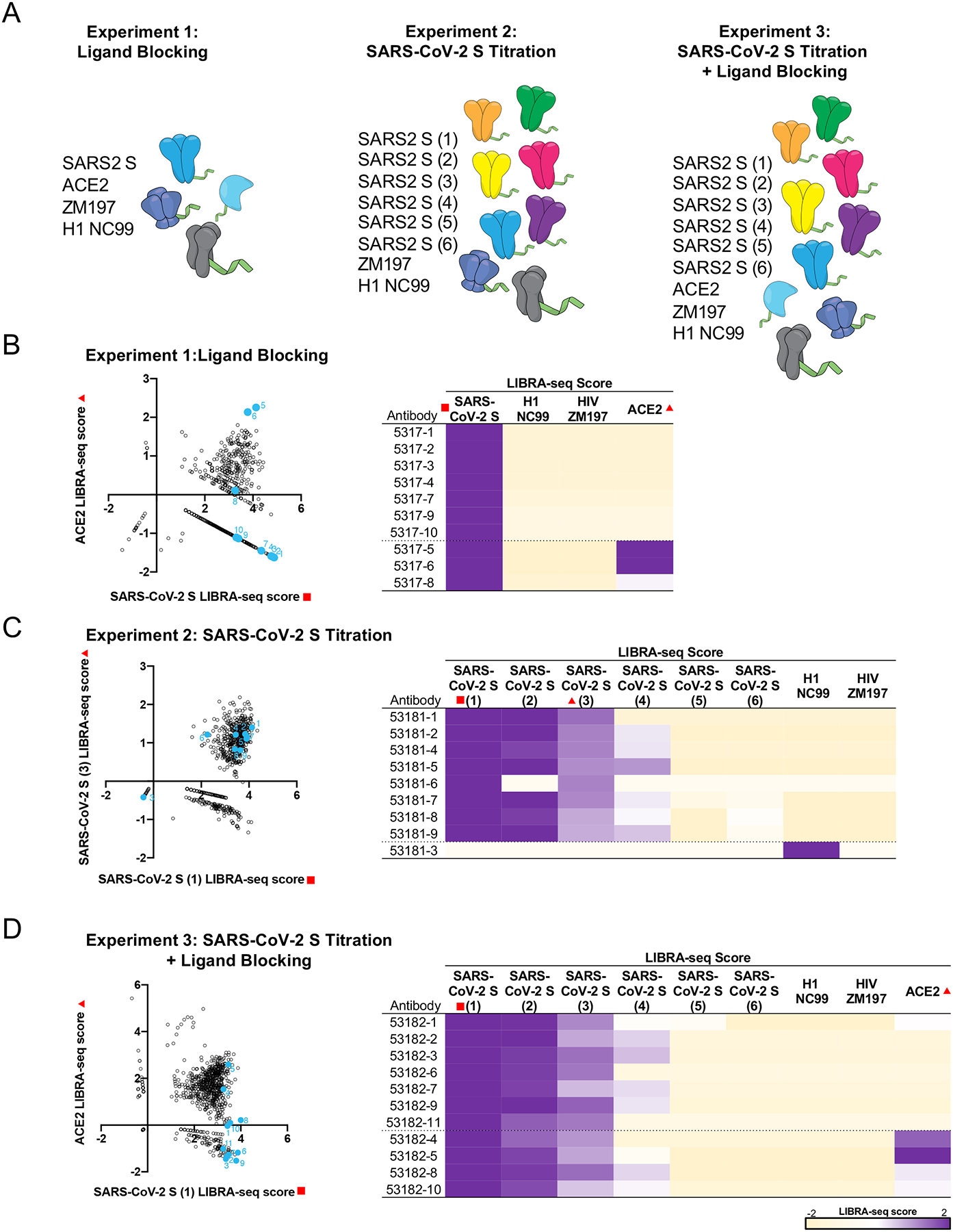
A. Experimental setup of three LIBRA-seq experiments: experiment 1, LIBRA-seq with ligand blocking; experiment 2, LIBRA-seq with a SARS-CoV-2 S titration; and experiment 3, LIBRA-seq with a SARS-CoV-2 S titration and ligand blocking. For experiment 2 and 3, six different aliquots of S protein were added in a titration series (1–6). (B-D) (left) After next-generation sequencing, hundreds of B cells (dots) were recovered that had paired heavy/light chain sequencing information and antigen reactivity information for the three experiments. For experiment 1 (B), 2 (C), and 3 (D), select LIBRA-seq scores for all cells per experiment are shown as open circles (n=828, 829, 957, respectively). Antibodies selected for expression and validation are highlighted and numbered in light blue. (right) LIBRA-seq scores for the selected antibodies for all antigens from each experiment are shown as a heatmap from −2 to 2 (tan to purple); scores outside of this range are shown as the minimum and maximum values. For experiments 1 and 3, antibodies with negative scores for ACE2 are shown above the dotted line while antibodies with positive scores for ACE2 are shown below the dotted line and are controls. For experiment 2, all SARS-CoV-2 reactive antibodies are shown above the dotted line, whereas influenza specific antibody 53181–3 is shown as a control below the dotted line.
The application of LIBRA-seq resulted in 828, 829, and 957 antigen-specific B cells for the three experiments, respectively. We prioritized a set of B cells for monoclonal antibody production and validation based on the following conditions: for experiments 1 and 3 (with ACE2 in the screening library), we selected B cells with high LIBRA-seq scores for S and low scores for ACE2; and for experiment 2, we selected B cells that had positive scores for multiple aliquots of S (Figure 1B–D). B cells with high S and high ACE2 scores were also selected as controls from experiments 1 and 3, along with an influenza-specific B cell from experiment 2 (Figure 1B–D). We further aimed to prioritize antibodies with diverse sequence features, although some of the selected antibodies appeared to be clonally related (Extended Data Figure 2A).
We confirmed the predicted antigen specificity for 26/27 (96%) antibodies and mapped the general antibody epitope regions by testing antibodies for binding to recombinant SARS-CoV-2 subdomain proteins (Figure 2A, Extended Data Figure 2B). The majority of antibodies from experiments 1 and 3 (but none from experiment 2) recognized the RBD (Figure 2A, Extended Data Figure 2B). Further, the antibodies had a wide range of affinities for RBD or NTD, including several antibodies with KD <1 nM, although we did not observe a correlation between LIBRA-seq spike score and affinity (Figure 2B). Next, we tested the ability of the antibodies to block ACE2 binding to spike. For antibodies predicted to block ACE2 by LIBRA-seq, 57% from experiment 1 and 67% from experiment 3 demonstrated ACE2 blocking via ELISA, whereas no antibodies from experiment 2 blocked ACE2 binding (Figure 2C, Extended Data Figure 2C).
Figure 2. Validation and characterization of selected antibodies.
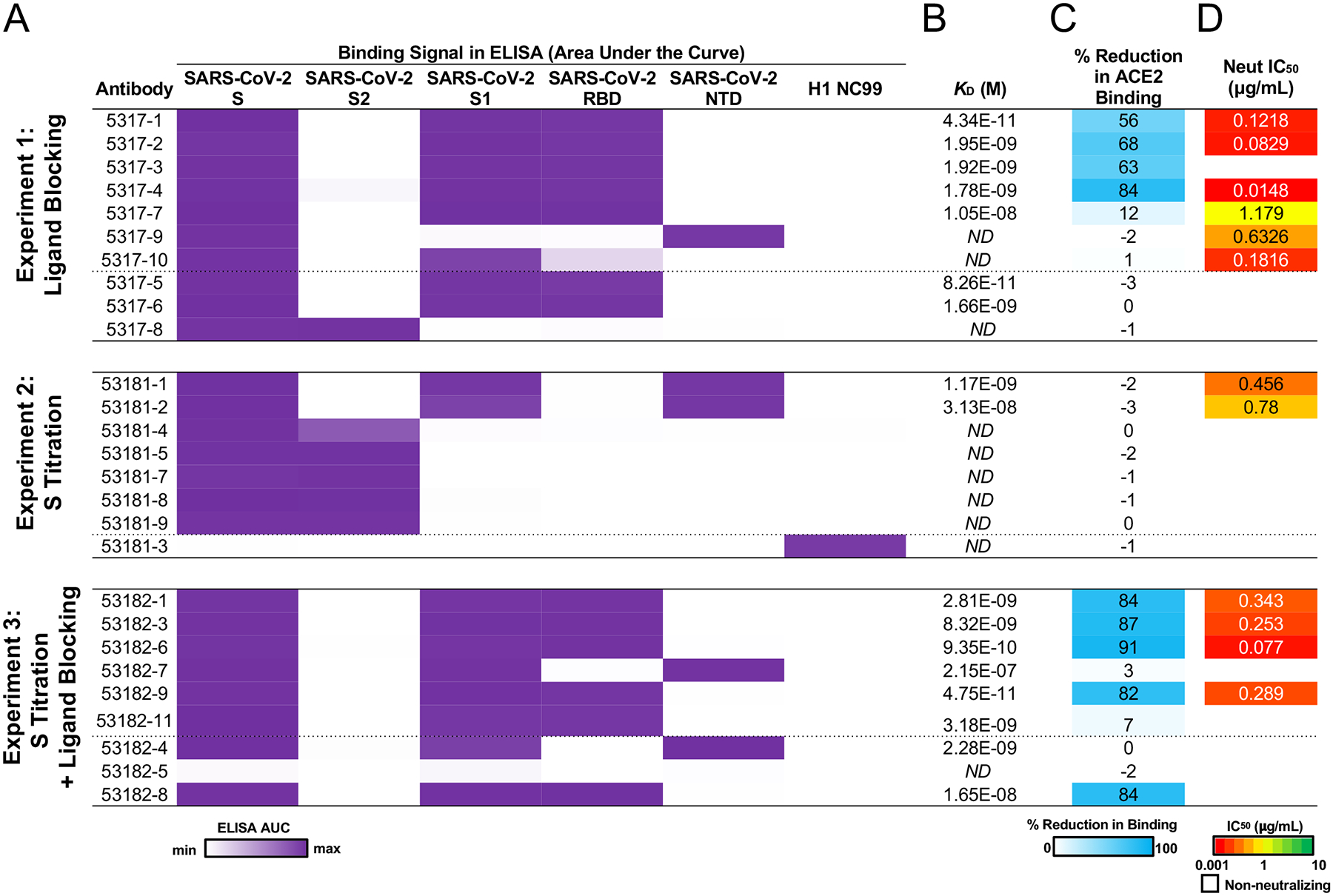
A. ELISA area under the curve (AUC) values for binding to SARS-CoV-2 recombinant antigen proteins and a negative control influenza hemagglutinin protein are shown for antibodies (rows) in each experiment, calculated from data in Extended Data Figure 2B. B. KD (M) of antibodies for SARS-CoV-2 RBD or NTD (based on epitope shown in A) was determined by biolayer interferometry. ND, not done. C. Percent reduction in ACE2 binding by ELISA is shown as a heatmap from 0 to 100% (white to blue) reduction in binding compared to SARS-CoV-2 binding only. D. VSV SARS-CoV-2 neutralization IC50 values are shown as a heatmap from high potency (red) to low potency (green). Non-neutralizing antibodies are shown as white.
Next, we tested the antibodies in a VSV SARS-CoV-2 chimeric virus neutralization assay (Figure 2D, Extended Data Figure 2D). For antibodies predicted to block ACE2 by LIBRA-seq, 86% from experiment 1 and 67% from experiment 3 were neutralizing, while only two clonally related antibodies (29%) from experiment 2 were neutralizing (Figure 3A–B). For the antibodies from experiments 1 and 3, the ACE2 LIBRA-seq scores were correlated with the percent reduction in ACE2 binding (Figure 3C, Spearman r = −0.54, p = 0.017). Furthermore, several antibodies also showed potent neutralization against authentic SARS-CoV-2 virus in a plaque reduction assay, and in some cases against multiple SARS-CoV-2 variants (Figure 4). Together, these results highlight the importance of including ligand blocking in LIBRA-seq for selectively identifying potent neutralizing antibodies.
Figure 3. Assessment of LIBRA-seq with ligand blocking.
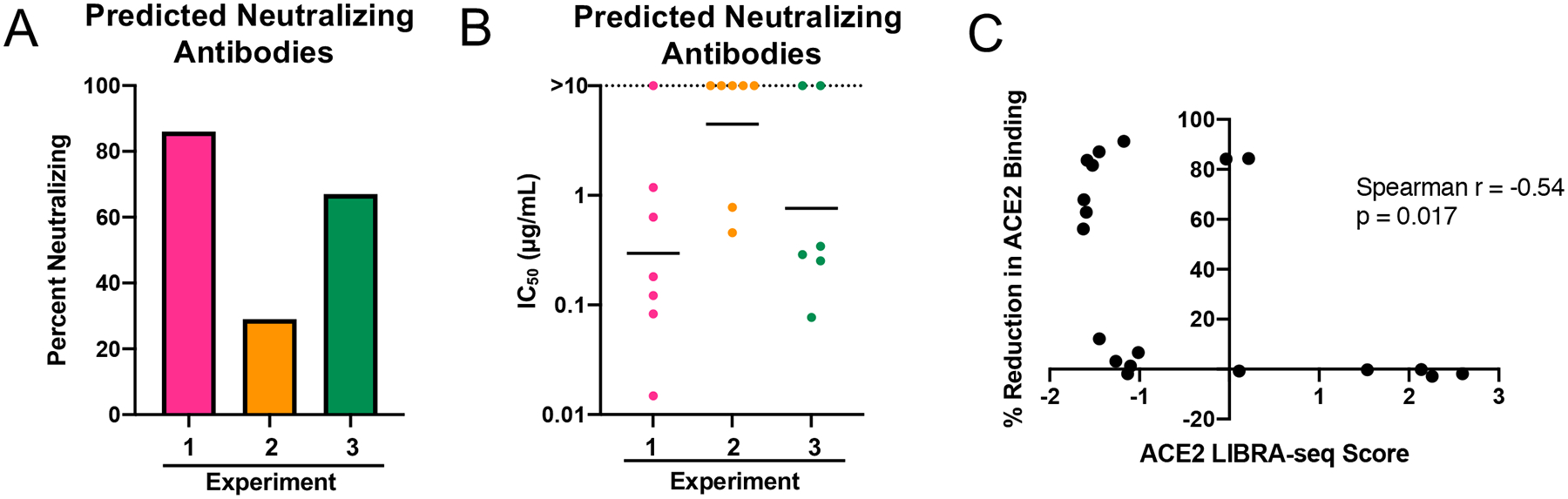
A. “Predicted Neutralizing Antibodies” were defined as the subset of selected antibodies with negative ACE2 LIBRA-seq scores from experiments 1 (n=7 antibodies) and 3 (n=6 antibodies), and all antibodies with high LIBRA-seq scores (>1) for SARS-CoV-2 S from experiment 2 (n=7 antibodies). The percent of neutralizing antibodies from the set of predicted neutralizers is shown for each experiment. B. The IC50 values (μg/mL) for SARS-CoV-2 neutralization by RTCA with VSV-SARS-CoV-2 (IC50 value for each antibody shown as single dot) are plotted for the set of predicted neutralizers. Horizontal line shown is geometric mean for each experiment. Non-neutralizing antibodies are shown as >10 μg/mL. C. Spearman correlation of ACE2 LIBRA-seq score (x-axis) and % Reduction in ACE2 Binding to SARS-CoV-2 (y-axis) for antibodies from experiments 1 and 3. Spearman r = −0.54, p = 0.017 (two-tailed, 95% confidence interval).
Figure 4. Antibody neutralization of SARS-CoV-2 variants.
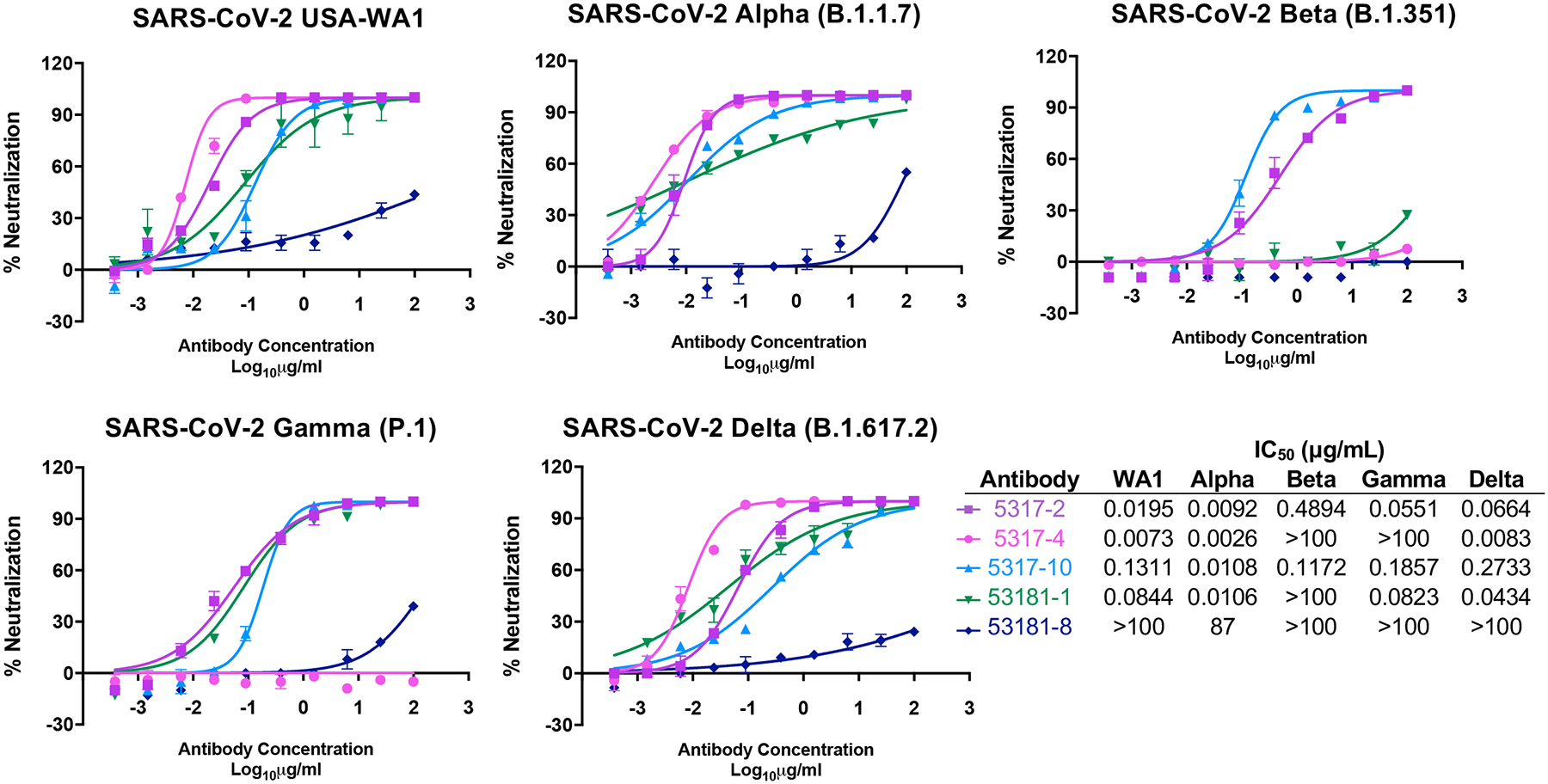
Authentic SARS-CoV-2 neutralization for a panel of antibodies is shown against USA-WA1 and variants (Alpha, Beta, Gamma, and Delta). Data represent the % neutralization as mean ± SD. The IC50 values calculated in GraphPad prism software by 4-parameter best-fit analysis are shown to the right of the panel.
To investigate antibody recognition of SARS-CoV-2 S, we determined a 9 Å-resolution Cryo-EM structure of the antigen-binding fragments of antibodies 5317–4 and 5317–10 bound to the SARS-CoV-2 S extracellular domain (Figure 5A). We chose 5317–4 based on its potent neutralization (IC50 value of 7.3 ng/mL against authentic SARS-CoV-2, Figure 4) and ACE2 competition. The 3D reconstruction revealed that 5317–4 binds to RBD in the “up” and “down” conformations, and its epitope partially overlaps the ACE2 binding footprint (Figure 5A–B). When bound to the RBD in the down conformation, 5317–4 competes with ACE2 binding to the adjacent up RBD (Figure 5B). We investigated 5317–10 because of its inconclusive epitope, as it bound to S1 but not individual RBD or NTD constructs (Figure 2A). The map revealed that 5317–10 binds a quaternary epitope that bridges an RBD in the down position and the NTD of an adjacent protomer (Figure 5A). This mode of recognition may prevent the RBD from transitioning into an ACE2-accessible up position, thereby preventing binding by ACE2.
Figure 5. Structural characterization of antibodies 5317–4 and 5317–10.
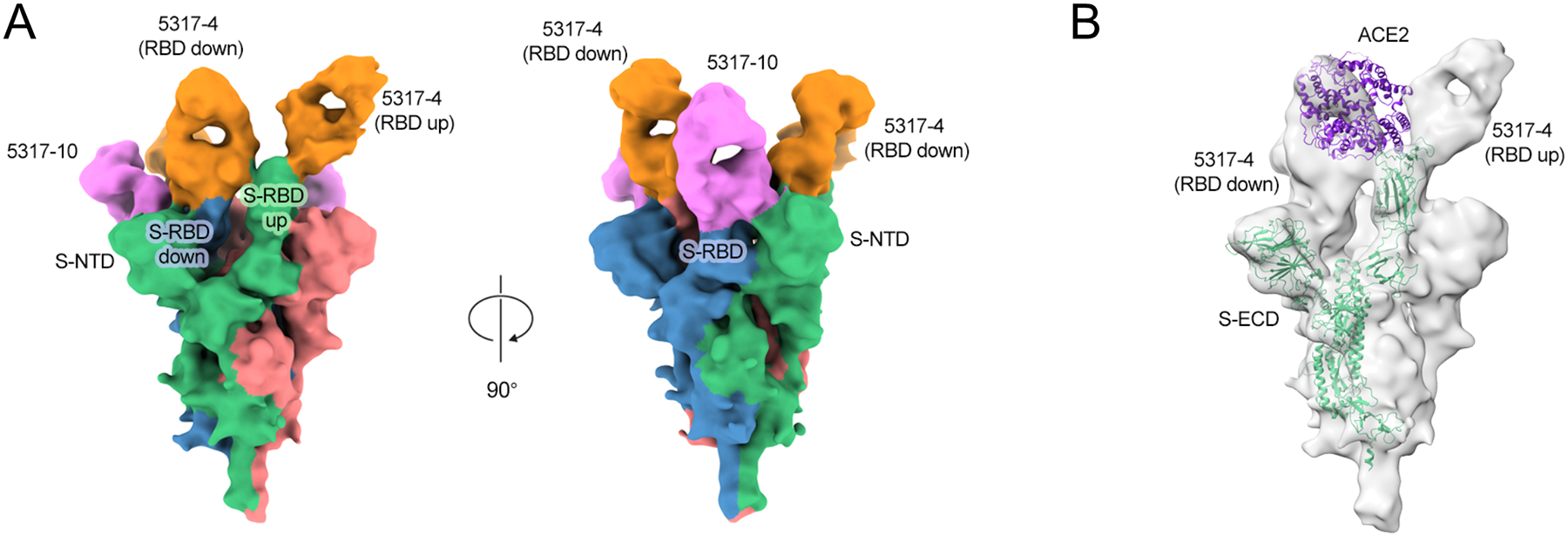
A. 9 Å-resolution cryo-EM structure of Fab-spike complex for 5317–4 Fab (orange) and 5317–10 Fab (pink). Spike protomers are shown in green, blue, and red. B. Fab-spike complex structure modeled with ACE2 (purple).
To further demonstrate the utility of LIBRA-seq with ligand blocking, we sought to identify antibodies that show cross-reactivity between SARS-CoV-2 and SARS-CoV, and that are capable of blocking spike-ACE2 interactions. To that end, we applied LIBRA-seq to B cells from a subject with past SARS-CoV-2 infection, using an antigen library that included SARS-CoV-2 S, SARS-CoV S, and ACE2 (Figure 6A). This resulted in 120 IgG+ B cells with high LIBRA-seq scores for both SARS-CoV-2 S and SARS-CoV S (Figure 6B). Only 8% of these cells were associated with low LIBRA-seq scores for ACE2 (Extended Data Figure 3A, highlighting the advantage of including ligand blocking to screen for such rare cells (although we also note that information about B cells that show cross-reactivity but are not ACE2 blocking is also retained, enabling characterization of B cells with alternative phenotypes as well). Based on LIBRA-seq antigen and ligand blocking scores, we produced and validated a set of antibodies, including 8 with high scores for both S antigens and low scores for ACE2 (Figure 6C–D). Of these, 100% bound SARS-CoV-2 S, 88% showed the predicted SARS-CoV-2/SARS-CoV cross-reactivity, and 63% demonstrated strong ACE2 blocking ability via ELISA (Figure 6E–H, Extended Data Figure 3B–C), confirming that LIBRA-seq with ligand blocking efficiently identified ACE2-blocking antibodies with cross-reactivity between multiple coronaviruses.
Figure 6. Discovery of cross-reactive ACE2-blocking coronavirus antibodies using LIBRA-seq with ligand blocking.
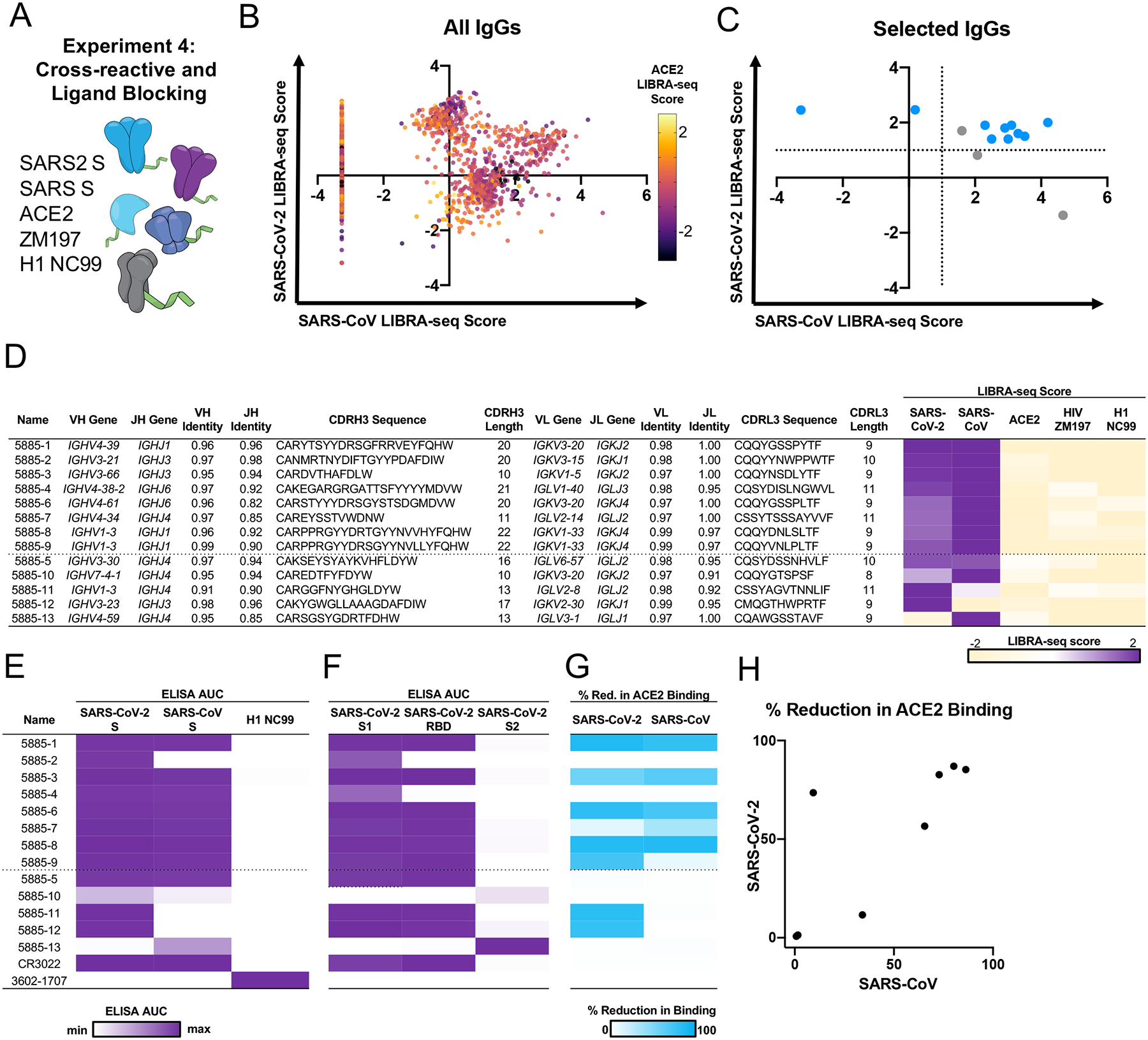
A. Schematic of LIBRA-seq with ligand blocking applied to cross-reactive antibody discovery. B. For identification of cross-reactive coronavirus antibodies with ligand blocking capability, all IgGs recovered from the LIBRA-seq experiment (n=2569) are shown, with LIBRA-seq scores for SARS-CoV (x-axis) and SARS-CoV-2 (y-axis). Each dot represents a cell, and the color of the dots shows the ACE2 LIBRA-seq score, with color heatmap shown on the right. C. Cells selected for expression and validation are shown in blue (ACE2 score <−1) or grey (ACE2 score ≥−1). Of these selected cells, 8 had high LIBRA-seq scores (>1) for SARS-CoV-2 and SARS-CoV and low scores (<−1) for ACE2. Additional candidates with a variety of scores for SARS-CoV-2, SARS-CoV and ACE2 were also selected for expression and validation as controls. D. The 8 IgGs with high LIBRA-seq scores for SARS-CoV-2 and SARS-CoV and low scores for ACE2 are shown above the dotted line. Control antibodies with other LIBRA-seq score patterns are shown below the dotted line. For each antibody, CDR sequences and lengths are shown at the amino acid level and V-gene and J-gene identity are shown at the nucleotide level. LIBRA-seq scores for antigens included in the screening library (SARS-CoV-2 spike, SARS-CoV spike, ACE2, HIV ZM197 Env, influenza hemagglutinin H1 NC99) are shown as a heatmap low (tan) – white – high (purple). Scores outside of this range are shown as the minimum and maximum values. E. ELISA area under the curve (AUC) values from binding to coronavirus spike proteins, influenza hemagglutinin H1 NC99 (negative control), and F. recombinant antigen domains, are shown as a heatmap from minimum (white) to maximum (purple) binding. G. Percent reduction in ACE2 binding by ELISA is shown for SARS-CoV-2 and SARS-CoV spikes, and displayed as a heatmap from 0% (white) to 100% (blue). H. For the 8 IgGs with high LIBRA-seq scores for SARS-CoV-2 and SARS-CoV and low scores for ACE2, the percent reduction in ACE2 binding due to antibody blocking by ELISA is shown for SARS-CoV (x-axis) and SARS-CoV-2 (y-axis).
Together, the results from the four LIBRA-seq experiments reported here showcase the advantages of including ligand blocking as part of the sequencing readout. As with most screening tools, there are limitations to the LIBRA-seq with ligand blocking approach, including the prerequisite for a defined antigen-ligand interaction, as well as the potential for identifying false positives. Nevertheless, through a single high-throughput sequencing experiment, LIBRA-seq with ligand blocking identified potent SARS-CoV-2 antibodies, requiring the subsequent production and validation of less than a dozen antibodies per experiment. The observed hit rates for the discovery of potently neutralizing antibodies are an improvement over what has been reported in the literature, which also typically required the screening of hundreds to thousands of antibody candidates isolated for their reactivity to antigen alone (recombinant S, S1, or RBD)1–6, 8–12. Further, unlike RBD-only discovery efforts, LIBRA-seq with ligand blocking applied to spike antigens has the potential for more comprehensive coverage of antibody epitopes, as evidenced by the discovery of the RBD-NTD antibody in Figure 5A. Overall, the application of LIBRA-seq with ligand blocking can provide critical advantages for rapid development of therapeutic and preventive countermeasures and presents a general platform with applications to virtually any area where targeting the disruption of antigen-ligand interaction is a prime therapeutic goal.
METHODS
Data Availability Statement
All unique reagents generated in this study are available from the corresponding author with a completed Material Transfer Agreement (a template can be found in the manuscript files). Sequences for antibodies identified and characterized in this study have been deposited to GenBank (MZ517191–MZ517250, OM001674 – OM001699). Raw sequencing data has been deposited to Sequence Read Archive (PRJNA744567, SAMN24369247). Further information and requests for resources and reagents should be directed to the corresponding author.
Code Availability
Custom scripts used to analyze data in this manuscript are available at https://github.com/Iglab-repo/LIBRA-seq-with-ligand-blocking.git. An EULA can be found in the manuscript files.
Donor Information
PBMC samples were purchased from Cellero. The PBMCs were from subjects with past SARS-CoV-2 infection at least 14 days post symptom cessation. For experiment 1, three samples were pooled from donors 523, 527, and 528. For experiments 2 and 3, samples from donor 523 were used for LIBRA-seq. Donor 523 had a plaque reduction neutralization test titer of 1:2,560. For experiment 4 (cross-reactive antibody discovery with ligand blocking), a sample from donor 528 was used for LIBRA-seq.
Antigen Purification
A variety of recombinant soluble protein antigens were used in the LIBRA-seq experiment and other experimental assays.
Plasmids encoding residues 1–1208 of the SARS-CoV-2 spike with a mutated S1/S2 cleavage site, proline substitutions at positions 817, 892, 899, 942, 986 and 987, and a C-terminal T4-fibritin trimerization motif, an 8x HisTag, and a TwinStrepTag (SARS-CoV-2 spike HP); residues 1–1190 of the SARS-CoV spike with proline substitutions at positions 968 and 969, and a C-terminal T4-fibritin trimerization motif, an 8x HisTag, and a TwinStrepTag (SARS-CoV S-2P); and 1−615 of human ACE2 with a C-terminal HRV3C protease cleavage site, a TwinStrepTag and an 8XhisTag (ACE2) were transiently transfected in Expi293F cells using polyethylenimine. Transfected supernatants were harvested 5 days after expression and purified over a StrepTrap column (Cytiva Life Sciences). Both recombinant SARS-CoV-2 S HP and ACE2 were further purified to homogeneity using a Superose6 Increase column (Cytiva Life Sciences).
For the HIV-1 gp140 SOSIP variant from strain ZM197 (clade C) and hemagglutinin from strain A/New Caledonia/20/99 (H1N1) (GenBank ACF41878), recombinant, soluble antigens contained an AviTag and were expressed in Expi293F cells using polyethylenimine transfection reagent and cultured. FreeStyle F17 expression medium supplemented with pluronic acid and glutamine was used. The cells were cultured at 37°C with 8% CO2 saturation and shaking. After 5–7 days, cultures were centrifuged and supernatant was filtered and run over an affinity column of agarose-bound Galanthus nivalis lectin. The column was washed with PBS and antigens were eluted with 30 mL of 1M methyl-a-D-mannopyranoside. Protein elutions were buffer exchanged into PBS, concentrated, and run on a Superdex 200 Increase 10/300 GL Sizing column on the AKTA FPLC system. Fractions corresponding to correctly folded protein were collected, analyzed by SDS-PAGE and antigenicity was characterized by ELISA using known monoclonal antibodies specific to each antigen. AviTagged antigens were biotinylated using BirA biotin ligase (Avidity LLC).
SARS-CoV-2 S1, SARS-CoV-2 S2, SARS-CoV-2 RBD and SARS-CoV-2 NTD proteins were purchased from the commercial vendor, Sino Biological.
DNA-barcoding of Antigens
We used oligos that possess 15 bp antigen barcode, a sequence capable of annealing to the template switch oligo that is part of the 10X bead-delivered oligos and contain truncated TruSeq small RNA read 1 sequences in the following structure: 5′-CCTTGGCACCCGAGAATTCCANNNNNNNNNNNNNCCCATATAAGA*A*A-3′, where Ns represent the antigen barcode1. For each antigen, a unique DNA barcode was directly conjugated to the antigen itself. For Experiment 1, the barcodes included SARS-CoV-2 S (GACAAGTGATCTGCA), H1 NC99 (TCATTTCCTCCGATT), ZM197 (TACGCCTATAACTTG), and ACE2 (CTTCACTCTGTCAGG). For Experiment 2, the barcodes included SARS-CoV-2 S aliquot 1 (GACAAGTGATCTGCA), SARS-CoV-2 S aliquot 2 (TGTGTATTCCCTTGT), SARS-CoV-2 S aliquot 3 (GCAGCGTATAAGTCA), SARS-CoV-2 S aliquot 4 (GCTCCTTTACACGTA), SARS-CoV-2 S aliquot 5 (AGACTAATAGCTGAC), SARS-CoV-2 S aliquot 6 (GGTAGCCCTAGAGTA), H1 NC99 (TCATTTCCTCCGATT), and ZM197 (TACGCCTATAACTTG). For Experiment 3, the same barcodes were included as Experiment 2 and also included ACE2 (CTTCACTCTGTCAGG). For Experiment 4, the barcodes included SARS-CoV-2 S (GCAGCGTATAAGTCA), SARS-CoV S (GCTCCTTTACACGTA), ACE2 (TACGCCTATAACTTG), ZM197 (TCATTTCCTCCGATT), and H1 NC99 (CTTCACTCTGTCAGG). In particular, 5′ amino-oligonucleotides were conjugated directly to each antigen using the SoluLINK Protein-Oligonucleotide Conjugation Kit (TriLink cat no. S-9011) according to manufacturer’s instructions. Briefly, the oligo and protein were desalted, and then the amino-oligo was modified with the 4FB crosslinker, and the biotinylated antigen protein was modified with S-HyNic. Then, the 4FB-oligo and the HyNic-antigen were mixed. This process causes a stable bond to form between the protein and the oligonucleotide. The concentration of the antigen-oligo conjugates was determined by a BCA assay, and the HyNic molar substitution ratio of the antigen-oligo conjugates was analyzed using the NanoDrop according to the SoluLINK protocol guidelines. AKTA FPLC was used to remove excess oligonucleotide from the protein-oligo conjugates, which were also verified using SDS-PAGE with a silver stain. Antigen-oligo conjugates were also used in flow cytometric titration experiments to determine optimal amounts for antigen-specific B cell sorting.
Antigen-specific B Cell Sorting
Cells were stained and mixed with DNA-barcoded antigens and other antibodies, and then sorted using fluorescence activated cell sorting (FACS). First, cells were counted, and viability was assessed using trypan blue. Then, cells were washed three times with DPBS supplemented with 0.1% bovine serum albumin (BSA). Cells were resuspended in DPBS-BSA and stained with cell markers including viability dye (Ghost Red 780), CD14-APC-Cy7, CD3-FITC, CD19-BV711, and IgG-PE-Cy5. Additionally, antigen-oligo conjugates were added to the stain. For experiment 1, oligo-labeled SARS-CoV-2 S and three-fold molar excess of oligo-labeled ACE2 was added. For experiment 2, six aliquots of S protein that were each labeled with a unique DNA oligonucleotide were added in a titration series from 5 μg to 0.0016 μg (in 5-fold dilutions). For experiment 3, the same titration series of S was added along with three fold molar excess of ACE2. For experiment 4, SARS-CoV-2 S, SARS-CoV S and three-fold molar excess of oligo-labeled ACE2 was added. The antigen screening library for each of the four experiments also included an influenza virus hemagglutinin and an HIV-1 envelope variant protein as controls.
After staining in the dark for 30 minutes at room temperature, cells were washed three times with DPBS-BSA at 300 × g for five minutes. Cells were then incubated for 15 minutes at room temperature with Streptavidin-PE to label cells with bound antigen. Cells were washed three times with DPBS-BSA, resuspended in DPBS, and sorted by FACS. Antigen positive cells were bulk sorted and delivered to the Vanderbilt Technologies for Advanced Genomics (VANTAGE) sequencing core at an appropriate target concentration for 10X Genomics library preparation and subsequent sequencing. Flow cytometry data were analyzed using FlowJo.
Sample Preparation, Library Preparation, and Sequencing
Single-cell suspensions were loaded onto the Chromium Controller microfluidics device (10X Genomics) and processed using the B-cell Single Cell V(D)J solution according to manufacturer’s suggestions for a target capture of 10,000–20,000 B cells, with minor modifications to intercept, amplify and purify the antigen barcode libraries1. The 10X Genomics single cell VDJ human B cell assay and target enrichment protocol were completed. cDNA was amplified and additive primers were added to increase the yield of antigen derived transcript products. After cDNA amplification, the antigen derived transcript products were size separated from the mRNA-derived cDNA products using SPRI selection and further purification (per manufacturers protocol). The supernatant fraction contained the antigen-oligo derived cDNA whereas the beads fraction contained the full-length mRNA-derived cDNAs. After purification, the antigen-derived transcripts sequencing library was prepared using a PCR reaction and purified using SPRI purification. The antigen and VDJ libraries were then analyzed, quantified, and sequenced using the Illumina NovaSeq platform.
Sequence processing and bioinformatic analysis
We used our previously described pipeline to use paired-end FASTQ files of oligo libraries as input, process and annotate reads for cell barcode, UMI, and antigen barcode, and generate a cell barcode - antigen barcode UMI count matrix. BCR contigs were processed using Cell Ranger (10X Genomics) using GRCh38 as reference. Antigen barcode libraries were also processed using Cell Ranger (10X Genomics). The overlapping cell barcodes between the two libraries were used as the basis of the subsequent analysis. We removed cell barcodes that had only non-functional heavy chain sequences as well as cells with multiple functional heavy chain sequences and/or multiple functional light chain sequences, reasoning that these may be multiplets. Additionally, we aligned the BCR contigs (filtered_contigs.fasta file output by Cell Ranger, 10X Genomics) to IMGT reference genes using HighV-Quest2. The output of HighV-Quest was parsed using ChangeO3 and merged with an antigen barcode UMI count matrix. Finally, for experiments 1–3, we determined the LIBRA-seq score for each antigen in the library by calculating the centered-log ratios (CLR) of each antigen UMI count for each cell. A psedo-count of 1 was added to each UMI count and then the CLR was taken for each antigen for each cell. For experiment 4, the LIBRA-seq scores were calculated as previously described1. Briefly, the CLR of each antigen UMI count for each cell was calculated and a Z-score transformation was also performed.
Antibody Expression and Purification
For each antibody, variable genes were inserted into custom plasmids encoding the constant region for the IgG1 heavy chain as well as respective lambda and kappa light chains (pTwist CMV BetaGlobin WPRE Neo vector, Twist Bioscience). Antibodies were expressed in Expi293F mammalian cells (Thermo Fisher Scientific) by co-transfecting heavy chain and light chain expressing plasmids using polyethylenimine transfection reagent and cultured for 5 to 7 days. Cells were maintained in FreeStyle F17 expression medium supplemented at final concentrations of 0.1% Pluronic Acid F-68 and 20% 4 mM L-Glutamine. These cells were cultured at 37°C with 8% CO2 saturation and shaking. After transfection and 5–7 days of culture, cell cultures were centrifuged and supernatant was 0.45 μm filtered with Nalgene Rapid Flow Disposable Filter Units with PES membrane. Filtered supernatant was run over a column containing Protein A agarose resin equilibrated with PBS. The column was washed with PBS, and then antibodies were eluted with 100 mM Glycine HCl at 2.7 pH directly into a 1:10 volume of 1M Tris-HCl pH 8.0. Eluted antibodies were buffer exchanged into PBS 3 times using Amicon Ultra-centrifugal filter units and concentrated. Antibody plasmids were sequenced. If antibody sequences did not match expected heavy or light chain, antibody was excluded from downstream analysis.
High-throughput Antibody Expression
For high-throughput production of recombinant antibodies, approaches were used that are designated as microscale. For antibody expression, microscale transfection was performed (~1 mL per antibody) of CHO cell cultures using the Gibco ExpiCHO Expression System and a protocol for deep 96-well blocks (Thermo Fisher Scientific). In brief, synthesized antibody-encoding DNA (~2 μg per transfection) was added to OptiPro serum free medium (OptiPro SFM), incubated with ExpiFectamine CHO Reagent and added to 800 μL of ExpiCHO cell cultures into 96-deep-well blocks using a ViaFlo 384 liquid handler (Integra Biosciences). The plates were incubated on an orbital shaker at 1,000 r.p.m. with an orbital diameter of 3 mm at 37°C in 8% CO2. The next day after transfection, ExpiFectamine CHO Enhancer and ExpiCHO Feed reagents (Thermo Fisher Scientific) were added to the cells, followed by 4 d incubation for a total of 5 d at 37°C in 8% CO2. Culture supernatants were collected after centrifuging the blocks at 450 × g for 5 min and were stored at 4°C until use. For high-throughput microscale antibody purification, fritted deep-well plates were used containing 25 μL of settled protein G resin (GE Healthcare Life Sciences) per well. Clarified culture supernatants were incubated with protein G resin for antibody capturing, washed with PBS using a 96-well plate manifold base (Qiagen) connected to the vacuum and eluted into 96-well PCR plates using 86 μL of 0.1 M glycine-HCL buffer pH 2.7. After neutralization with 14 μL of 1 M Tris-HCl pH 8.0, purified antibodies were buffer-exchanged into PBS using Zeba Spin Desalting Plates (Thermo Fisher Scientific) and stored at 4°C until use.
ELISA
To assess antibody binding, soluble protein was plated at 2 μg/mL overnight at 4°C. The next day, plates were washed three times with PBS supplemented with 0.05% Tween-20 (PBS-T) and coated with 5% milk powder in PBS-T. Plates were incubated for one hour at room temperature and then washed three times with PBS-T. Primary antibodies were diluted in 1% milk in PBS-T, starting at 10 μg/mL with a serial 1:5 dilution and then added to the plate. The plates were incubated at room temperature for one hour and then washed three times in PBS-T. The secondary antibody, goat anti-human IgG conjugated to peroxidase, was added at 1:10,000 dilution in 1% milk in PBS-T to the plates, which were incubated for one hour at room temperature. Plates were washed three times with PBS-T and then developed by adding TMB substrate to each well. The plates were incubated at room temperature for ten minutes, and then 1N sulfuric acid was added to stop the reaction. Plates were read at 450 nm.
Data are represented as mean ± SEM for one ELISA experiment. ELISAs were repeated 2 or more times. If ELISA replicates were inconsistent over more than three experiments, antibody was excluded from in vitro characterization analysis. The area under the curve (AUC) was calculated using Prism software version 8.0.0 (GraphPad).
ACE2 Binding Inhibition Assay
96-well plates were coated with 2 μg/mL purified recombinant SARS-CoV-2 at 4°C overnight. The next day, plates were washed three times with PBS supplemented with 0.05% Tween-20 (PBS-T) and coated with 5% milk powder in PBS-T. Plates were incubated for one hour at room temperature and then washed three times with PBS-T. Purified anti were diluted in blocking buffer at 10 μg/mL in triplicate, added to the wells, and incubated at room temperature. Without washing, recombinant human ACE2 protein with a mouse Fc tag was added to wells for a final 0.4 μg/mL concentration of ACE2 and incubated for 40 minutes at room temperature. Plates were washed three times with PBS-T, and bound ACE2 was detected using HRP-conjugated anti-mouse Fc antibody and TMB substrate. The plates were incubated at room temperature for ten minutes, and then 1N sulfuric acid was added to stop the reaction. Plates were read at 450 nm. ACE2 binding without antibody served as a control. Experiment was done in biological replicate and technical triplicates.
BioLayer Interferometry (BLI)
Purified antibodies were immobilized to AHC sensortips (FortéBio) to a response level of approximately 1.4 nm in a buffer composed of 10 mM HEPES pH 7.5, 150 mM NaCl, 3 mM EDTA, 0.05% Tween 20 and 0.1% (w/v) BSA. Immobilized antibodies were then dipped into wells containing two-fold dilutions of either SARS-CoV-2 RBD-SD1 (residues 306–577) or SARS-CoV-2 NTD, ranging in concentration from 10–0.156 nM, to measure association kinetics. Dissociation kinetics were measured by dipping sensortips into wells containing only buffer. Data were reference subtracted and kinetics were calculated in Octet Data Analysis software v10.0 using a 1:1 binding model.
RTCA Method for Initial Screening of Antibody Neutralizing Activity
To screen for neutralizing activity in the panel of recombinantly expressed antibodies, we used a high-throughput and quantitative RTCA assay and xCelligence RTCA HT Analyzer (ACEA Biosciences) that assesses kinetic changes in cell physiology, including virus-induced cytopathic effect (CPE). Twenty μL of cell culture medium (DMEM supplemented with 2% FBS) was added to each well of a 384-well E-plate using a ViaFlo384 liquid handler (Integra Biosciences) to obtain background reading. Six thousand (6,000) Vero-furin cells in 20 μL of cell culture medium were seeded per well, and the plate was placed on the analyzer. Sensograms were visualized using RTCA HT software version 1.0.1 (ACEA Biosciences). For a screening neutralization assay, equal amounts of virus were mixed with micro-scale purified antibodies in a total volume of 40 μL using DMEM supplemented with 2% FBS as a diluent and incubated for 1 h at 37°C in 5% CO2. At ~17–20 h after seeding the cells, the virus–antibody mixtures were added to the cells in 384-well E-plates. Wells containing virus only (in the absence of antibody) and wells containing only Vero cells in medium were included as controls. Plates were measured every 8–12 h for 48–72 h to assess virus neutralization. Micro-scale antibodies were assessed in four 5-fold dilutions (starting from a 1:20 sample dilution), and their concentrations were not normalized. Neutralization was calculated as the percent of maximal cell index in control wells without virus minus cell index in control (virus-only) wells that exhibited maximal CPE at 40–48 h after applying virus–antibody mixture to the cells. An antibody was classified as fully neutralizing if it completely inhibited SARS-CoV-2-induced CPE at the highest tested concentration, while an antibody was classified as partially neutralizing if it delayed but did not fully prevent CPE at the highest tested concentration4, 5.
Real-time Cell Analysis (RTCA) Neutralization Assay
To determine neutralizing activity of IgG, we used real-time cell analysis (RTCA) assay on an xCELLigence RTCA MP Analyzer (ACEA Biosciences Inc.) that measures virus-induced cytopathic effect (CPE)6. Briefly, 50 μL of cell culture medium (DMEM supplemented with 2% FBS) was added to each well of a 96-well E-plate using a ViaFlo384 liquid handler (Integra Biosciences) to obtain background reading. A suspension of 18,000 Vero-E6 cells in 50 μL of cell culture medium was seeded in each well, and the plate was placed on the analyzer. Measurements were taken automatically every 15 min, and the sensograms were visualized using RTCA software version 2.1.0 (ACEA Biosciences Inc). VSV-SARS-CoV-2 (0.01 MOI, ~120 PFU per well) was mixed 1:1 with a dilution of antibody in a total volume of 100 μL using DMEM supplemented with 2% FBS as a diluent and incubated for 1 h at 37°C in 5% CO2. At 16 h after seeding the cells, the virus-antibody mixtures were added in replicates to the cells in 96-well E-plates. Triplicate wells containing virus only (maximal CPE in the absence of antibody) and wells containing only Vero cells in medium (no-CPE wells) were included as controls. Plates were measured continuously (every 15 min) for 48 h to assess virus neutralization. Normalized cellular index (CI) values at the endpoint (48 h after incubation with the virus) were determined using the RTCA software version 2.1.0 (ACEA Biosciences Inc.). Results are expressed as percent neutralization in a presence of respective antibody relative to control wells with no CPE minus CI values from control wells with maximum CPE. RTCA IC50 values were determined by nonlinear regression analysis using Prism software.
Plaque Reduction Neutralization Test (PRNT)
The virus neutralization with live authentic SARS-CoV-2 virus (USA-WA1) was performed in the BSL-3 facility of the Galveston National Laboratory using Vero E6 cells (ATCC CRL-1586) following the standard procedure. Vero E6 cells were cultured in 96-well plates (104 cells/well). Next day, 4-fold serial dilutions of antibodies were made using MEM-2% FBS, as to get an initial concentration of 100 μg/mL. Equal volume of diluted antibodies (60 μL) were mixed gently with original SARS-CoV-2 (USA-WA1) (60 μL containing 200 pfu) and incubated for 1 h at 37°C/5% CO2 atmosphere. The virus-serum mixture (100 μL) was added to cell monolayer in duplicates and incubated for 1 at h 37°C/5% CO2 atmosphere. Later, the virus-serum mixture was discarded gently, and cell monolayer was overlaid with 0.6% methylcellulose and incubated for 2 days. The overlay was removed, and the plates were fixed in 4% paraformaldehyde twice following BSL-3 protocol. The plates were stained with 1% crystal violet and virus-induced plaques were counted. The percent neutralization and/or NT50 of antibody was calculated by dividing the plaques counted at each dilution with plaques of virus-only control. For antibodies, the inhibitory concentration at 50% (IC50) values were calculated in Prism software (GraphPad) by plotting the midway point between the upper and lower plateaus of the neutralization curve among dilutions. The Alpha variant virus incorporates the following substitutions: Del 69–70, Del 144, E484K, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H. The Beta variant incorporates the following substitutions: Del 24, Del 242–243, D80A, D215G, K417N, E484K, N501Y, D614G, H665Y, T1027I. The Gamma variant incorporates the following substitutions: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I. The Delta variant incorporates the following substitutions: T19R, G142D, Del 156–157, R158G, L452R, T478K, D614G, P681R, Del 689–691, D950N; the deletion at positions 689–691 has not been observed in nature, and was identified upon one passage of the virus.
Fab Preparation
To generate Fabs, IgGs were incubated with Lys-C at 1:4,000 (weight:weight) overnight at 37 °C. EDTA free protease inhibitor (Roche) was dissolved to 25X and then added to the sample at a final 1X concentration. The sample was passed over a Protein A column. The flow-through was collected run on a Superdex 200 Increase 10/300 GL Sizing column on the AKTA FPLC system. Fabs were visualized on SDS-PAGE.
Biolayer Interferometry
Purified mAbs were immobilized to AHC sensortips (FortéBio) to a response level of approximately 1.4 nm in a buffer composed of 10 mM HEPES pH 7.5, 150 mM NaCl, 3 mM EDTA, 0.05% Tween 20 and 0.1% (w/v) BSA. Immobilized mAbs were then dipped into wells containing two-fold dilutions of either SARS-CoV-2 RBD-SD1 or SARS-CoV-2 NTD, ranging in concentration from 10–0.15625 nM, to measure association. Dissociation was measured by dipping sensortips into wells containing only running buffer. Data were reference subtracted and kinetics were calculated in Octet Data Analysis software v10.0 using a 1:1 binding model.
Electron Microscopy Sample Preparation and Data Collection
Purified SARS-CoV-2 S HexaPro ectodomain7 and Fabs 5317–4 and 5317–10 were combined at a final complex concentration of 0.4 mg/mL. Fab 5317–10 was added to spike and incubated on ice for 30 minutes before the addition of Fab 5317–4 immediately prior to grid deposition and freezing. The complex was deposited on Au-300 1.2/1.3 grids that had been plasma cleaned for 4 minutes in a Solarus 950 plasma cleaner (Gatan) with a 4:1 ratio of O2/H2. Excess liquid was blotted for 3 seconds with a force of −4 using a Vitrobot Mark IV (Thermo Fisher) and plunge frozen into liquid ethane. 2,655 micrographs were collected from a single grid with the stage at a 30° tilt using a Titan Krios (Thermo Fisher) equipped with a K3 detector (Gatan). Movies were collected using SerialEM8 at 29,000X magnification with a corresponding calibrated pixel size of 0.81 Å/ pixel.
Cryogenic Electron Microscopy (Cryo-EM)
Motion correction, CTF estimation, particle picking, and 2D classification were performed using cryoSPARC v3.2.09. The final iteration of 2D class averaging distributed 17,710 particles into 50 classes using an uncertainty factor of 3. From that, 13,232 particles were selected and an ab inito reconstruction was performed with four classes followed by heterogeneous refinement of those four classes. 6,803 particles from the highest-quality class were used for homogenous refinement of the best volume without imposed symmetry. The resulting volume was used for an additional round of homogenous refinement. To filter out additional junk particles, an ab initio reconstruction was performed with three classes followed by heterogeneous refinement of those three classes. 5,171 particles from the highest-quality class were used for homogenous refinement of the best volume without imposed symmetry, resulting in a final 9 Å map.
QUANTIFICATION AND STATISTICAL ANALYSIS
ELISA error bars (standard error of the mean) were calculated using GraphPad Prism version 8.0.0. Spearman r correlation was performed using GraphPad Prism 8.0.0. ANOVA analysis was performed for neutralization potency comparisons using GraphPad Prism version 8.0.0.
Extended Data
Extended Data Fig. 1. Schematic representation of LIBRA-seq experiments.
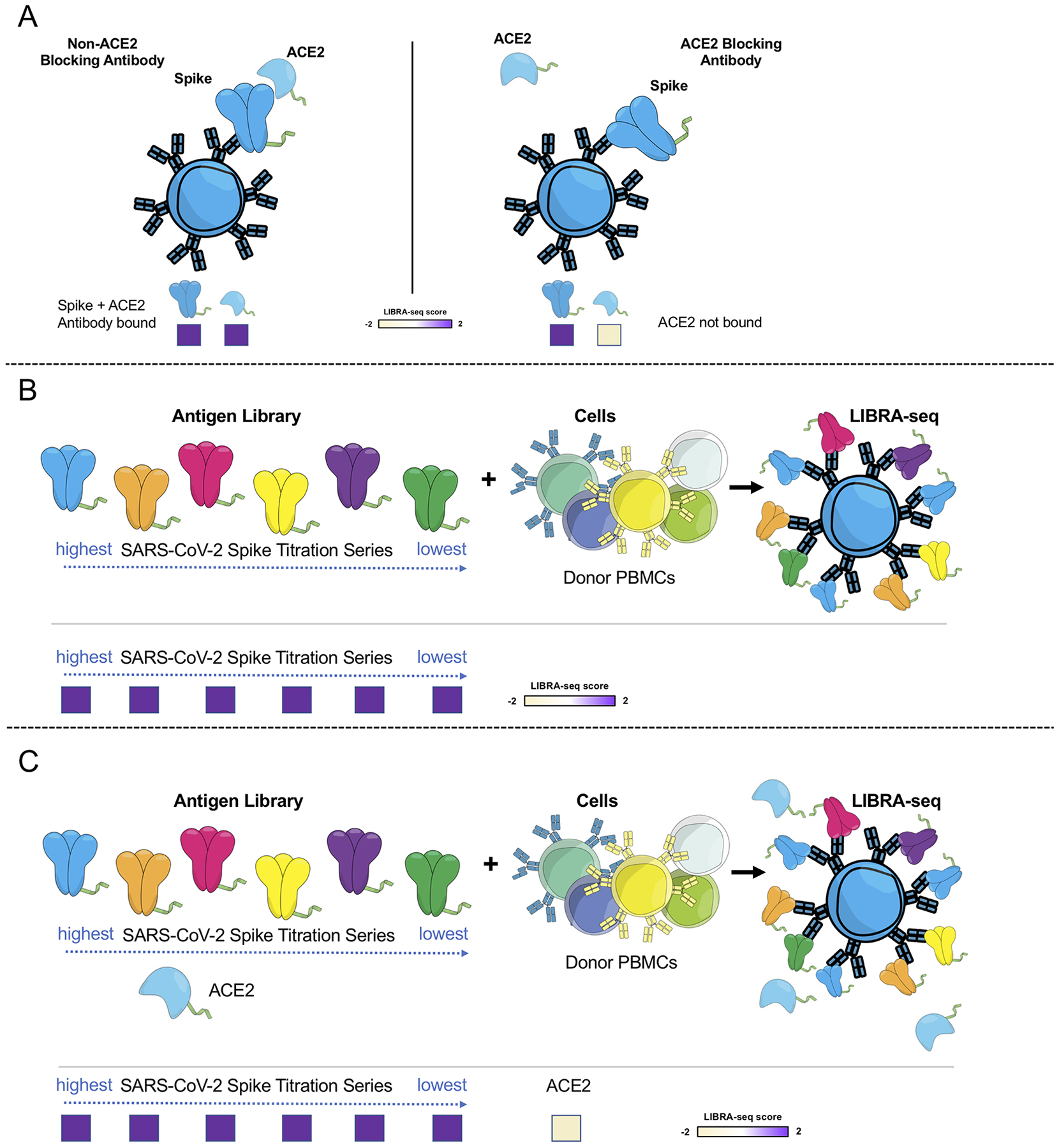
A. An antigen screening library of oligonucleotide-labeled antigens was generated. This library consisted of SARS-CoV-2 spike antigens and negative controls. Additionally, oligo-labeled ACE2 (the SARS-CoV-2 spike host cell receptor) was included. The antigen screening library was mixed with donor PBMCs. This approach allowed for assessment of B cell ligand blocking functionality from the sequencing experiment. B. An antigen screening library containing an antigen titration was generated, with a goal of identifying high affinity antibodies from LIBRA-seq. In this experiment, six different amounts of oligo-labeled SARS-CoV-2 S protein, each labeled with a different barcode, were included in a screening library. C. Schematic of LIBRA-seq with S titrations and ACE2 included for ligand blocking.
Extended Data Fig. 2. Characterization of LIBRA-seq-identified antibodies.
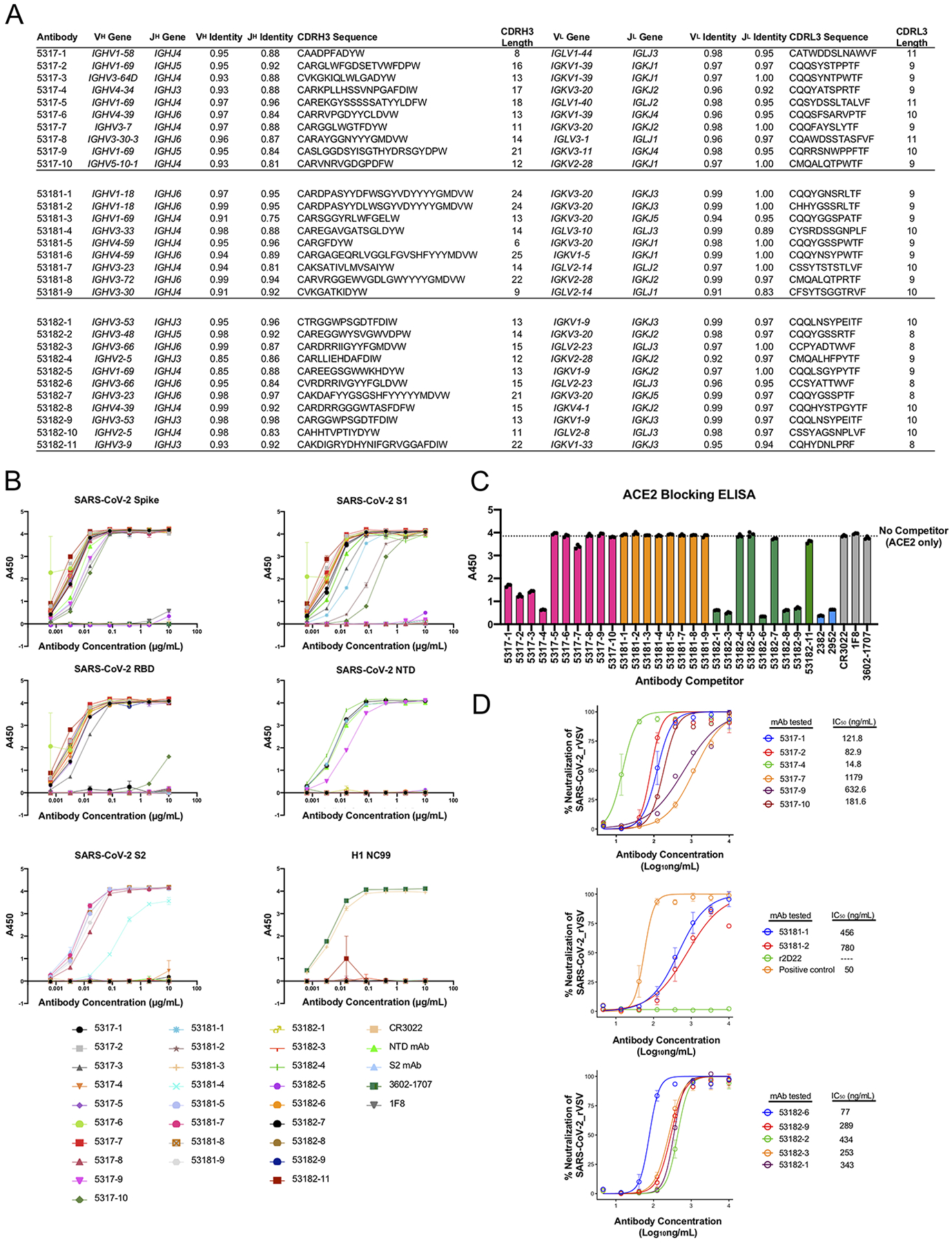
A. Genetic characteristics for monoclonal antibodies prioritized for expression and validation. VH, JH, VL, JL inferred gene segment identity is shown at the nucleotide level. CDRH3 and CDRL3 amino acid sequence and length are also shown. B. ELISA binding of antibodies to SARS-CoV-2 spike, SARS-CoV-2 S1, SARS-CoV-2 RBD, SARS-CoV-2 NTD, SARS-CoV-2 S2 and influenza hemagglutinin H1 NC99. Data are represented as mean ± SEM of technical duplicates and represent one of at least two independent experiments (n=2). C. ACE2 blocking ELISA. Antibodies were added to spike, and recombinant ACE2 was added and detected. Antibodies that block ACE2 binding show a reduction in absorbance compared to ACE2 binding without competitor (dotted line). ELISAs were performed at one antibody concentration, and data are represented as mean ± SEM of technical triplicates and represent one of at least two independent experiments (n=2). D. Antibodies were tested in a VSV SARS-CoV-2 real time cell analysis (RTCA) neutralization assay. Neutralization curves and IC50 values are shown. Data are represented as mean ± S.D. of technical triplicates, and represent one of two independent experiments (n=2).
Extended Data Fig. 3. Characterization of selected cross-reactive antibodies.
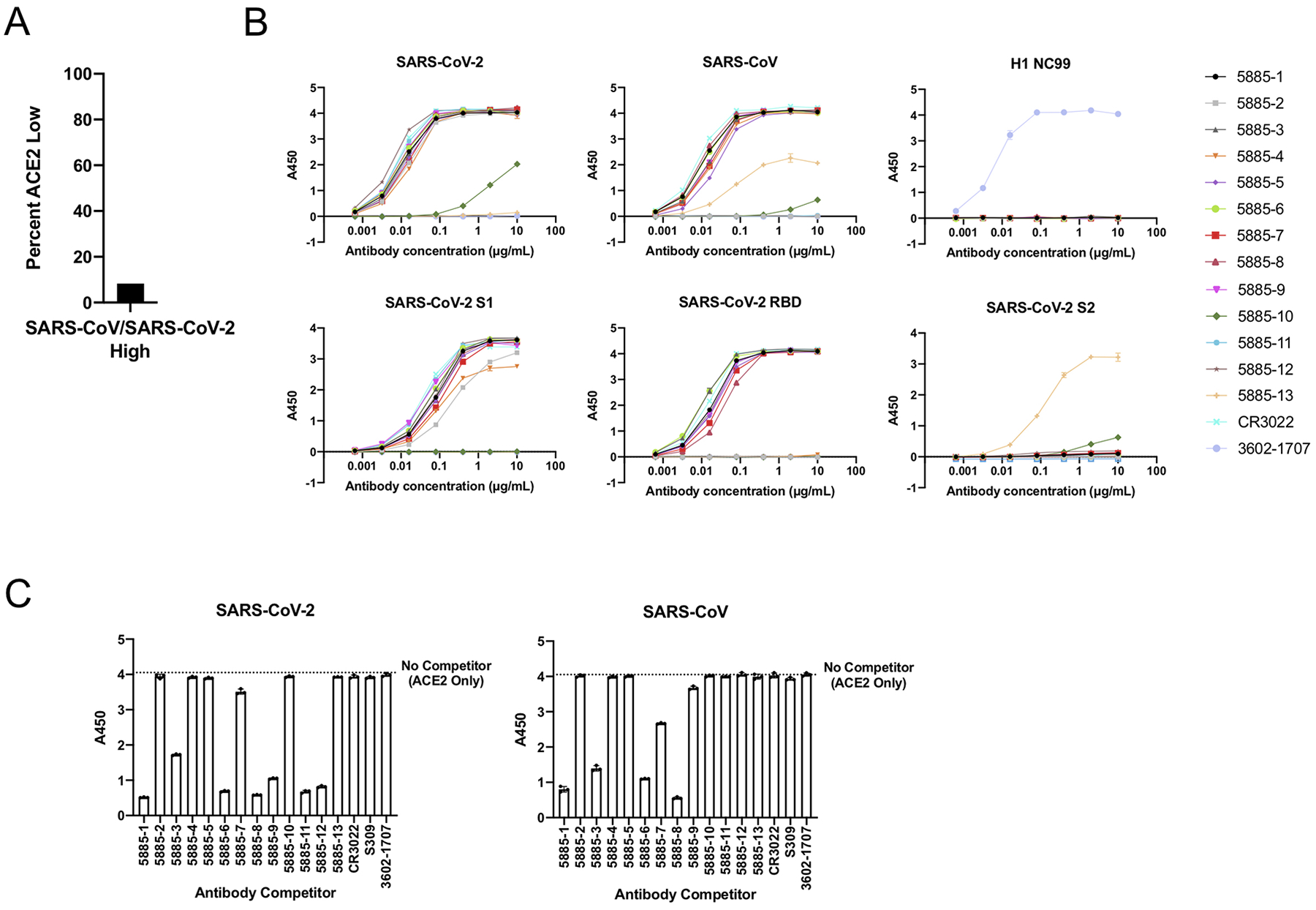
A. For the IgGs that showed high LIBRA-seq scores (>1) for both SARS-CoV-2 and SARS-CoV, the percent of cells with low ACE2 scores (<−1) is shown. B. ELISA binding of antibodies to SARS-CoV-2 spike, SARS-CoV spike, influenza hemagglutinin H1 NC99, SARS-CoV-2 S1, SARS-CoV-2 RBD, and SARS-CoV-2 S2. Data are represented as mean ± SEM of technical duplicates and represent one of at least two independent experiments (n=2). C. ACE2 blocking ELISA. ACE2 binding without competitor is shown as a dotted line. ELISAs were performed at one antibody concentration, and data are represented as mean ± SEM of technical triplicates and represent one of at least two independent experiments (n=2).
Supplementary Material
Acknowledgements
We thank Angela Jones, Latha Raju, and Jamie Roberson of Vanderbilt Technologies for Advanced Genomics for their expertise regarding NGS and library preparation; David Flaherty and Brittany Matlock of the Vanderbilt Flow Cytometry Shared Resource for help with flow panel optimization; and members of the Georgiev laboratory for comments on the manuscript. The Vanderbilt VANTAGE Core provided technical assistance for this work. VANTAGE is supported in part by CTSA grant 5UL1 RR024975-03, the Vanderbilt Ingram Cancer Center (P30 CA68485), the Vanderbilt Vision Center (P30 EY08126), and NIH/NCRR (G20 RR030956). This work was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University (Nashville, TN). Flow cytometry experiments were performed in the VUMC Flow Cytometry Shared Resource. The VUMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
For work described in this manuscript, I.S.G., A.R.S., K.J.K., S.C.W., K.A.P., N.R., and L.M.W. were supported in part by NIH NIAID award R01AI131722-S1, the Hays Foundation COVID-19 Research Fund, Fast Grants, and CTSA award No. UL1 TR002243 from the National Center for Advancing Translational Sciences. J.S.M, N.V.J., and D.W. were supported in part by a National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases (NIAID) grant awarded to J.S.M. (R01-AI127521), Welch Foundation grant F-0003-19620604, and NIH NIAID award R01AI131722-S1. J.E.C., N.S., R.N., R.E.S., and R.H.C. were supported in part by Defense Advanced Research Projects Agency (DARPA) grant HR0011-18-2-0001, U.S. N.I.H. contract 75N93019C00074, N.I.H. grant R01 AI157155, the Dolly Parton COVID-19 Research Fund at Vanderbilt, and a grant from Fast Grants, Mercatus Center, George Mason University. J.E.C. is a recipient of the 2019 Future Insight Prize from Merck KGaA, which supported this work with a grant.
Declaration of Interest
A.R.S. and I.S.G. are co-founders of AbSeek Bio. I.S.G., A.R.S, and K.J.K. are listed as inventors on antibodies described herein. I.S.G., A.R.S, and I.S. are listed as inventors on patent applications for the LIBRA-seq technology. J.E.C. has served as a consultant for Luna Biologics, is a member of the Scientific Advisory Board Meissa Vaccines and is Founder of IDBiologics. The Crowe laboratory has received funding support in sponsored research agreements from AstraZeneca, IDBiologics, and Takeda. The Georgiev laboratory at Vanderbilt University Medical Center has received unrelated funding from Takeda Pharmaceuticals. The remaining authors declare no competing interests.
References
- 1.Jiang S, Hillyer C & Du L Neutralizing Antibodies against SARS-CoV-2 and Other Human Coronaviruses. Trends Immunol 41, 355–359 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zost SJ et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 584, 443–449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chi X et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 369, 650–655 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brouwer PJM et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 369, 643–650 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogers TF et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369, 956–963 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen J et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 369, 1010–1014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krammer F SARS-CoV-2 vaccines in development. Nature 586, 516–527 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Wec AZ et al. Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science 369, 731–736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen MS Monoclonal Antibodies to Disrupt Progression of Early Covid-19 Infection. N Engl J Med 384, 289–291 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ju B et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 584, 115–119 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Robbiani DF et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 584, 437–442 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi R et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 584, 120–124 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Setliff I et al. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell 179, 1636–1646 e1615 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES (METHODS)
- 1.Setliff I et al. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell 179, 1636–1646 e1615 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alamyar E, Duroux P, Lefranc MP & Giudicelli V IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods in molecular biology (Clifton, N.J.) 882, 569–604 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Gupta NT et al. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zost SJ et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 584, 443–449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilchuk P et al. Integrated pipeline for the accelerated discovery of antiviral antibody therapeutics. Nat Biomed Eng 4, 1030–1043 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suryadevara N et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 184, 2316–2331 e2315 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh CL et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369, 1501–1505 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All unique reagents generated in this study are available from the corresponding author with a completed Material Transfer Agreement (a template can be found in the manuscript files). Sequences for antibodies identified and characterized in this study have been deposited to GenBank (MZ517191–MZ517250, OM001674 – OM001699). Raw sequencing data has been deposited to Sequence Read Archive (PRJNA744567, SAMN24369247). Further information and requests for resources and reagents should be directed to the corresponding author.