

Abstract
High-risk solid tumors continue to pose a tremendous therapeutic challenge due to multidrug resistance. Biological mechanisms driving chemoresistance in high-risk primary and recurrent disease are distinct: in newly diagnosed patients, non-response to therapy is often associated with a higher level of tumor “stemness” paralleled by overexpression of the ABCG2 drug efflux pump, whereas in tumors relapsing after non-curative therapy, poor drug sensitivity is most commonly linked to the dysfunction of the tumor suppressor protein, p53. In this study, we used preclinical models of aggressive neuroblastoma featuring these characteristic mechanisms of primary and acquired drug resistance to experimentally evaluate a macromolecular prodrug of a structurally enhanced camptothecin analog, SN22, resisting ABCG2-mediated export and glucuronidation. Together with extended tumor exposure to therapeutically effective drug levels via reversible conjugation to Pluronic F-108 (PF108), these features translated into rapid tumor regression and long-term survival in models of both ABCG2-overexpressing and p53-mutant high-risk neuroblastomas, in contrast to a marginal effect of the clinically used camptothecin derivative, irinotecan. Our results demonstrate that pharmacophore enhancement, increased tumor uptake, and optimally stable carrier-drug association integrated into the design of the hydrolytically activatable PF108-[SN22]2 have the potential to effectively combat multiple mechanisms governing chemoresistance in newly diagnosed (chemo-naïve) and recurrent forms of aggressive malignancies. As a macromolecular carrier-based delivery system exhibiting remarkable efficacy against two particularly challenging forms of high-risk neuroblastoma, PF108-[SN22]2 can pave the way to a robust and clinically viable therapeutic strategy urgently needed for patients with multidrug-resistant disease presently lacking effective treatment options.
Keywords: ABCG2, drug resistance, neuroblastoma, SN22, topoisomerase I inhibitor
1 |. INTRODUCTION
Cancer remains the leading cause of disease-related death in children in most developed countries despite recent improvements in diagnostic technologies and the implementation of new therapies. Pediatric patients with high-risk disease have few curative salvage treatment options and most eventually die post-relapse, with a less than 30% chance of surviving 5 years after their diagnosis.1 Drug resistance is the key factor limiting the effectiveness of pharmacotherapy in patients with high-risk tumors. The increased rate of recurrences and a lack of significant improvement in survival rates despite aggressive multimodal therapy point to a need for novel and much more efficient therapeutic approaches for this patient group.2 However, primary (or de novo) and acquired forms of chemoresistance in newly diagnosed and recurrent variants of high-risk malignancies rely on different biological mechanisms. A major process governing reduced responsiveness of chemo-naïve cancer cells to a broad range of chemotherapeutics is drug efflux mediated by proteins of the ATP-binding cassette (ABC) transporter family frequently overexpressed in de novo resistant tumors.3 Therefore, while there may be no actual shift in the intracellular drug concentration required for triggering a robust antiproliferative response, reaching and maintaining this concentration threshold may not be practically achievable without overcoming this mechanism of drug evasion. In contrast, acquired resistance seen at relapse after non-curative treatment is most often associated with loss-of-function mutations in tumor suppressor genes,4,5 in turn causing a profound change in the intracellular drug levels required to trigger the tumor cell killing action of cytotoxic agents.6
ABCG2 (“breast cancer resistance protein”) is among the ABC transporters most frequently linked to multidrug resistance.7 The expression of the ABCG2 drug efflux pump is associated with a particularly challenging “cancer stem cell” phenotype characterized by inherently high resistance to chemotherapeutic agents.8 ABCG2 is broadly implicated in drug resistance in pediatric cancers,2 and its increased expression levels at diagnosis predict poor response to chemotherapy in several types of malignancies. Susceptibility to ABCG2-driven transport severely compromises the performance of different classes of chemotherapeutics, including clinically used topoisomerase I inhibitors of the camptothecin family,9,10 rendering them ultimately ineffective against chemoresistant, aggressive disease.11–13 As attempts to alleviate ABCG2-mediated multidrug resistance with drug efflux pump inhibitors have so far failed due to poor specificity and high toxicity of the screened candidates,14,15 structure-activity relationship studies have identified SN22 as a camptothecin analog with a particularly low affinity for this transporter, allowing it to kill with similarly high potencies both ABCG2-overexpressing and non-expressing cancer cells.12,16,17 In the present study, we investigated conjugation to a biocompatible polyalkylene glycol block co-polymer of the Poloxamer (Pluronic) family as an experimental strategy that, in addition to allowing formulation of the water-insoluble SN22 for systemic administration, would also enhance uptake and extend the presence of the bioactive drug in the tumor, making possible to fully exploit the pharmacological advantages of SN22.
Several Poloxamer polymers have been used clinically for a variety of applications as excipients18,19 and, more recently, as biological response modifiers.20,21 Unlike chemically homogeneous poly(ethylene glycol), the ABA triblock Poloxamers combining intermediate lengths of the middle hydrophobic poly(propylene oxide) (PPO) block with comparatively high hydrophilic/lipophilic balance (HLB) values are capable of stably associating with cell membranes,22,23 which provides them with an effective mechanism for tumor penetration and for extending their intratumoral presence.20,24 Although several of these polymers exhibit prolonged residence times in circulation,25 Poloxamers are sized below the threshold of glomerular filtration (i.e., 30–50 kDa26) and are fully eliminated intact, without forming degradation products. Their proven biocompatibility, metabolic and chemical stability, and bioeliminability obviate concerns for significant acute or delayed adverse effects and meet the essential requirements for parenterally administered pharmaceutical materials.25,27 This makes Poloxamers particularly well-suited as scaffolds for creating polymer-linked prodrugs designed to enhance drug delivery to solid tumors. However, while used extensively in their chemically unmodified form,21,24 their utility for making prodrug-based delivery systems to improve cancer pharmacotherapy has remained largely unexplored.28
In this study, we developed a hydrolytically cleavable prodrug of SN22 using carboxylated Pluronic F-108 (Poloxamer 338) as a macromolecular carrier (Figure 1) and investigated its effectiveness in the therapeutic context of neuroblastoma (NB), the most common and deadly extracranial pediatric solid tumor responsible for 15% of all childhood cancer-related deaths.29 The antitumor potency of this prodrug and its dependence on the individual and combined effects of enhanced delivery and optimized pharmacology of the inactivation- and elimination-resisting drug cargo were examined in clinically relevant models of high-risk disease recapitulating the key features of primary and acquired multidrug resistance. Experimentally showing the effectiveness of this delivery strategy against aggressive tumor types protected from chemotherapy by different biological mechanisms is anticipated to facilitate further development and optimization of Poloxamer-linked SN22 and analogously constructed prodrugs as novel therapeutics for refractory malignancies.2,30
FIGURE 1.
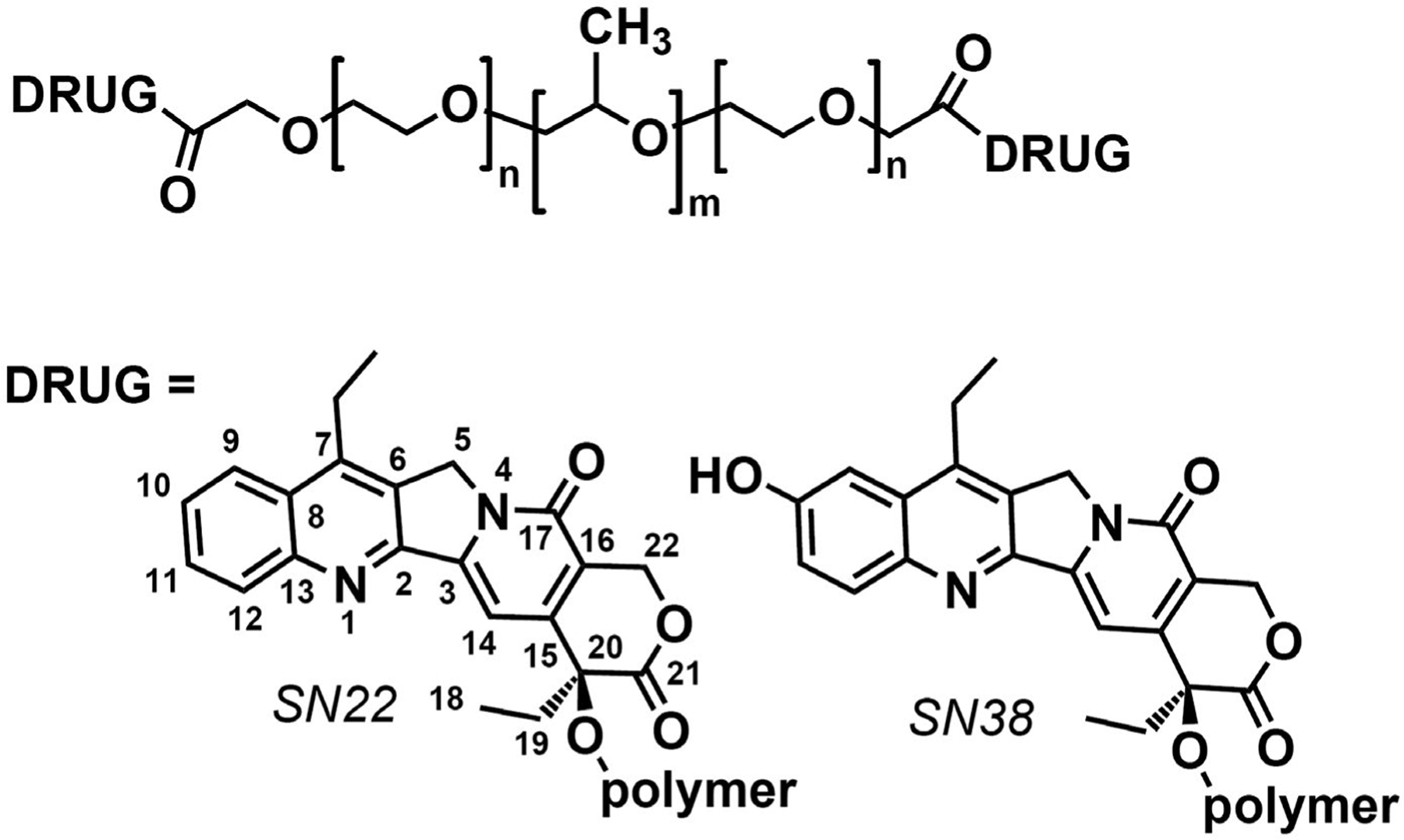
Structures of Poloxamer-linked SN22 and SN38 derivatives investigated in this study
2 |. MATERIALS AND METHODS
2.1 |. Synthesis of Poloxamer-[drug]2 derivatives
Pluronic F108 (Poloxamer 338, Sigma–Aldrich, St. Louis, MO) was first oxidized with Jones reagent in tetrahydrofuran (THF) at 22–25°C to transform terminal CH2OH into α-alkoxy carboxylic groups. Briefly, polymer (4.0 g) was dissolved in THF (20 ml), protected with argon, and Jones reagent (1.5 ml, containing 3.80 mmol of CrO3 and 5.87 mmol of H2SO4) was added dropwise. The mixture was stirred at ambient temperature under argon for 24 h, the reaction was stopped by the addition of bisulfate solution (4.0 ml, containing 6.0 mmol of Na2SO3 and 3.0 mmol of H2SO4), the mixture was acidified with 2M H2SO4 (5.0 ml) and diluted with water (60 ml). After removal of THF and most of the water by distillation, the polymer was precipitated from an aqueous solution with Na2SO4 and purified from Cr3+ by boiling with aqueous EDTA, reprecipitation with Na2SO4, and extraction with chloroform. The soft residue obtained after evaporating chloroform was solidified by adding pentane. The resultant chemically activated polymer contained 0.18 mmol/g of carboxylic groups, as was determined using 1H NMR (400 MHz, CDCl3) by the signal of OCH2CO protons. Further conjugation of the carboxylated Pluronic F-108 with SN22 (AK Scientific, Union City, CA) using 1,3-dicyclohexylcarbodiimide as an activating agent, 4-dimethylaminopyridine tosylate as a catalyst, and methylene chloride as a solvent formed a polymeric derivative containing 0.13 mmol/g (4.8% wt) of the drug. The conjugate was purified by several precipitations from solution in THF with methyl tert-butyl ether until TLC (silica gel, chloroform-acetonitrile 7:3) failed to detect any mobile compounds. Finally, the polymer was dissolved in a 4:1 mixture of benzene-methylene chloride (30 ml) and washed with a 21% aqueous solution of Na2SO4 to remove any residual catalyst. 1H NMR (Supporting Information Figure S1) confirmed covalent binding of SN22 via ester bonds between the carboxylic groups of the carboxylated Pluronic and 20-OH of the SN22 molecule.
To prepare PF108-[SN38]2, the phenolic 10-OH of SN38 (AstaTech, Bristol, PA) was first protected with a 10-tert-butyldiphenylsilyl group by the action of tert-butyl(chloro)diphenylsilane in the presence of imidazole in N-methylpyrrolidone. The protected SN38 (obtained in a 97% yield) was reacted with carboxylated polymer as above yielding after deprotection (with pyridinium fluoride in methylene chloride) the target conjugate containing 0.12 mmol/g (4.8% wt) of SN38 covalently bound via ester bonds by the 20-OH according to 1H NMR.
For animal studies, the prodrugs were dissolved in cold aqueous glucose solution (5% w/v, 4°C), sterilized by passing through a 0.2 μm PTFE membrane (Millipore Sigma, Burlington, MA), and lyophilized for 24 h.
2.2 |. In vitro neuroblastoma cell growth inhibition studies
A TH-MYCN NB cell line was established from a spontaneous tumor in a 129X1/SvJ transgenic mouse carrying the human MYCN transgene in two concatamers as previously described.31 Human neuroblastoma [IMR-32 and BE(2)C] cells purchased from the American Type Culture Collection (Manassas, VA, USA) were stably transduced with firefly luciferase and characterized as described elsewhere.32 For growth inhibition studies, the cells were seeded on day −1 on 96-well plates at ~5% confluence. DMEM (for IMR-32 cells) or 1:1 DMEM/F-12 (for TH-MYCN NB and BE(2)C cells) supplemented with 10% FBS were used as media in all experiments. Cell viability was longitudinally monitored by measuring the activity of firefly luciferase by luminometry using D-luciferin potassium salt (PerkinElmer, Bridgeville, PA, USA) as a substrate (50 μg/ml). On day 0, cells were treated with indicated doses of free drugs dissolved in dimethylsulfoxide (0.4 mg/ml) prior to further sequential dilutions, or with equivalent doses of the Poloxamer-linked prodrugs diluted directly in the culture medium. At indicated times corresponding to exposure duration periods, the medium was carefully removed, cells were washed, and their incubation was continued in a fresh medium. Cell viability was measured at predetermined time points against untreated cells included as a reference.
2.3 |. Neuroblastoma animal model experiments
Animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Children’s Hospital of Philadelphia. Human NB cells (IMR-32 or BE(2)C, purchased from the American Type Culture Collection, Manassas, VA) were stably transduced with firefly luciferase and characterized as described elsewhere.32 For orthotopic inoculation, one million cells suspended in 20 μl of Cultrex Basement Membrane Extract (Trevigen, Gaithersburg, MD) were implanted into the adipose tissue surrounding the adrenal capsule of anesthetized athymic nude (nu/nu) mice.33 Tumor burden was monitored by bioluminescent imaging using a Xenogen IVIS Imaging System (Caliper Life Sciences, Hopkinton, MA). Homozygous TH-MYCN mice genetically engineered to exhibit key molecular and biological characteristics of MYCN-driven aggressive human NB34,35 and developing clearly palpable tumors at 5–6 weeks of age were used as a model of de novo chemoresistant disease. After reaching an estimated tumor size of 1.0 cm3, animals were randomized into groups of 5 mice per treatment arm (therapeutic efficacy studies) or 4 mice per time point (biodistribution analysis). Tissue distribution was determined using a modification of a previously reported fluorimetric assay.36 In brief, the drugs and prodrug constructs were extracted from tissue homogenates and blood or serum samples (100 μl) in two steps in acetonitrile (200 μl) in the presence of aqueous sodium sulfate as a salting-out agent (30% w/v, 0.5 ml). For measuring the total SN38, the extracts were additionally alkalized by adding a methanolic solution of sodium hydroxide (1 mg/ml, 100 μl). The analytes were determined against suitable calibration curves (λex/λem = 377 nm/440 nm, 430 nm/565 nm, and 370 nm/450 nm for SN22, SN38, and irinotecan, respectively). In therapeutic efficacy experiments, the prodrugs or irinotecan were administered intravenously over 4 weeks at indicated frequencies and at doses equivalent to 10 mg/kg of SN22 or SN38. Therapeutic response and tumor progression were monitored longitudinally in orthotopically inoculated animals by measuring the tumor-associated bioluminescent signal. The tumor size of TH-MYCN animals was assessed twice a week by palpation. Animals were sacrificed when tumors exceeded 2 cm in diameter or if signs of distress, discomfort, lethargy, or weight loss were apparent. Animals reaching the endpoint or sacrificed after surviving beyond 180 days were examined by necropsy for tumor presence.
2.4 |. Statistical analysis
Experimental data are expressed as mean ± standard deviation. Regression analysis and the Kruskal-Wallis non-parametric ANOVA with Dunn’s post hoc test were used to compare tumor progression rates and biodistribution/elimination patterns, respectively. Animal survival in experimental groups was compared using the Kaplan-Meier method with the Holm-Sidak post hoc test. Differences were termed significant at p < .05.
3 |. RESULTS
Single-step oxidation of Pluronic polymers with Jones reagent using a modification of a process previously applied to functionalize poly(ethylene glycol)37 transforms the polymer’s terminal primary alcohols into α-alkoxy carboxylic groups, which then can be used for reversible covalent binding of a hydroxyl-containing drug via hydrolyzable ester bonds (Supporting Information Figure S1). Oxidation of Pluronic F-108 resulted in 0.18 mmol/g of carboxylic groups, as was determined from the signal of OCH2CO protons at 4.11 ppm, which was absent in the unmodified polymer. Subsequent drug attachment to the carboxylated polymer could be carried out under mild reaction conditions using the nearly neutral N,N-dimethylaminopyridine tosylate as a catalyst that allows direct carbodiimide-based coupling of alcohols and carboxylic acids at room temperature with minimal generation of byproducts (corresponding acylureas).38 Precipitations with methyl tert-butyl ether, in which the polymer is insoluble, effectively removed any traces of the activating agent and catalyst. After confirming its purity and structure, the prodrug construct containing 4.8% (w/w) of SN22 covalently bound to the polymer via hydrolytically cleavable ester bonds formed by its 20-OH was evaluated for its antitumor effects in vitro and in vivo.
To reproduce the distinct challenges posed by different mechanisms of chemoresistance in association with a high-risk tumor phenotype, we selected three representative models of NB driven by MYCN, a protooncogene whose amplification is an established marker of high-risk disease.39 We used two human-derived NB cell lines, IMR-32 and BE(2)C, originating from metastatic sites and sharing MYCN amplification but differing in the disease phase. The chemo-naïve IMR-32 line was derived at diagnosis prior to any treatment,40 whereas the parent SK-N-BE(2) cells exhibiting an acquired loss-of-function mutation in the tumor suppressor protein p53 and high-level multidrug resistance6 were obtained following several courses of radiotherapy and intensive multiagent chemotherapy.41 We also included TH-MYCN NB cells derived from tumors spontaneously developing in mice genetically engineered to overexpress human MYCN in a tissue-specific manner and recapitulating the key histological features and gene expression profile of MYCN-amplified, de novo resistant aggressive human NB.34,35
A comparison between the two human cell lines confirmed a marked reduction in drug sensitivity associated with the acquired p53 loss-of-function mutation: while the growth of chemo-naïve IMR-32 was inhibited uniformly and with high potency by both free SN22 and PF108-[SN22]2 within the entire studied dose range even at the shortest exposure duration of 30 min (Figure 2A), suppressing the proliferation of BE(2)C cells by more than 90% required 24 h of exposure to either the free compound or the prodrug (Figure 2B,C), consistent with strongly reduced drug sensitivity of recurrent NB tumors exhibiting the loss of p53 function.6 Furthermore, in contrast to free SN22 able to partially but significantly inhibit BE(2)C cell growth at higher concentrations when the exposure was limited to 30 min, the antiproliferative effect of PF108-[SN22]2 was negligible following a 30-min exposure regardless of its concentration (Figure 2B), suggesting that the prodrug remains largely intact in the serum-containing medium at shorter times. This lag is expected to play a beneficial role by limiting premature prodrug activation, reducing the clearance rate, and allowing a larger fraction of the drug to reach the tumor in the form of the macromolecular precursor. Contrastingly, however, when the exposure was extended to 24 h, PF108-[SN22]2 durably suppressed the growth of BE(2)C cells in a concentration-dependent manner, with over 80% inhibition seen at concentrations corresponding to 80–100 nM of SN22 (Figure 2C and Supporting Information Figure S2A).
FIGURE 2.
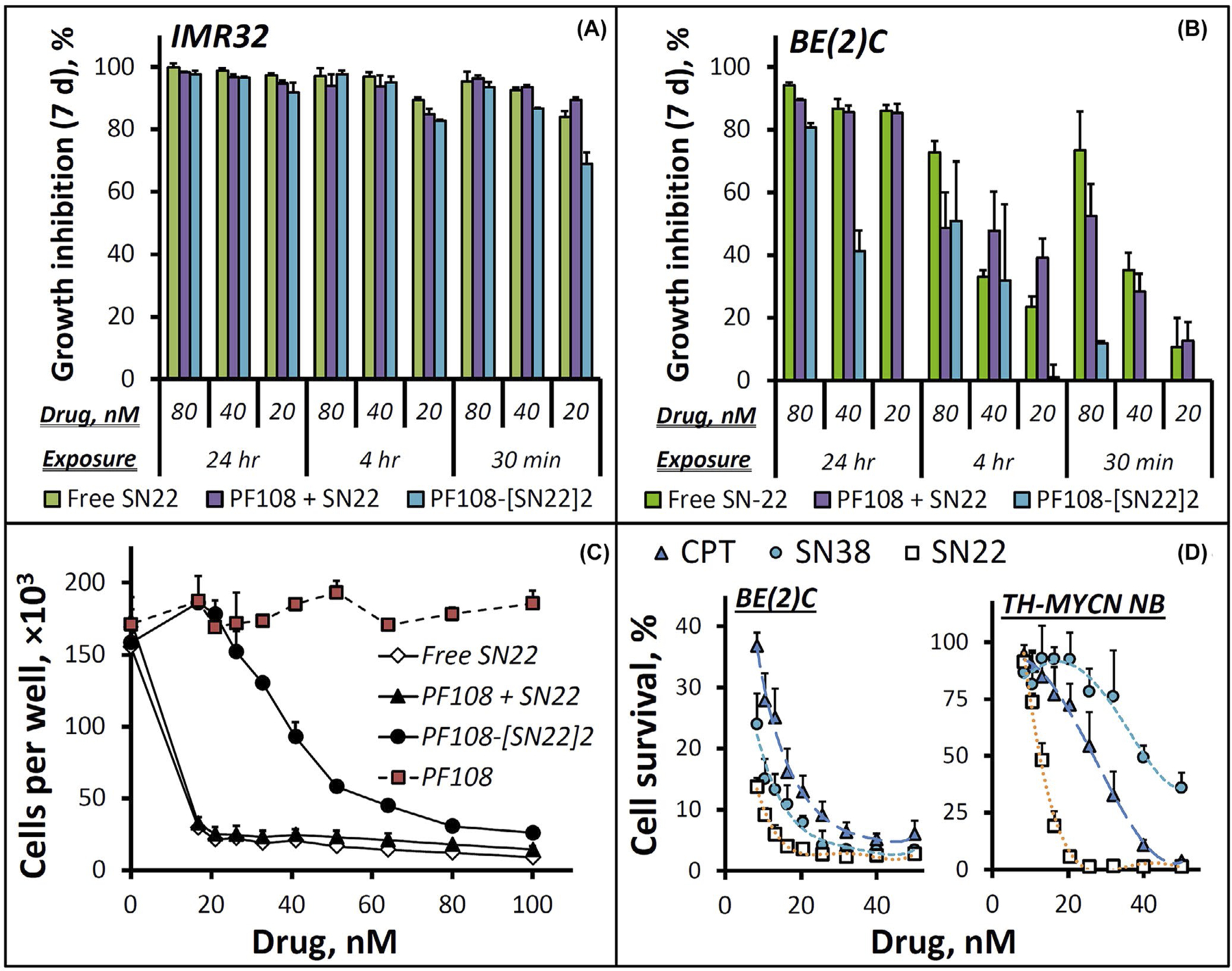
In vitro growth inhibition effects of camptothecins and a Poloxamer-linked prodrug of SN22 (PF108-[SN22]2) on cultured NB cells. Human NB cells, IMR-32 (A) and BE(2)C (B), exhibit MYCN amplification and characteristic features of different disease states: newly diagnosed versus recurrent after intensive chemoradiotherapy. The antiproliferative effects of PF108-[SN22]2 were determined in comparison to free SN22 with or without unmodified polymer (PF108) 7 days post-treatment as a function of drug concentration (20–80 nM) and exposure duration (30 min, 4 h, and 24 h). A detailed profile of BE(2)C cell growth inhibition by PF108-[SN22]2 versus controls as a function of drug concentration is shown in (C) (BE(2)C cell growth monitored over 7 days is included in Figure S2 in Supporting Information). The distinct antiproliferative effects of three camptothecin derivatives (camptothecin [CPT], SN38, SN22) on BE(2)C cells and tumor cells derived from mice genetically engineered to develop NB exhibiting key features of MYCN-driven de novo resistant human disease (TH-MYCN NB cells) are shown in comparison in (D). Cell survival is expressed as a percent of cells per treatment condition determined at 7 days in reference to untreated cells. Data are presented as mean ± SD
To elucidate the specific contribution of the respective resistance mechanisms, we comparatively tested the survival of BE(2)C versus TH-MYCN NB cells challenged with camptothecin, SN22, or SN38. SN38 is a hydroxylated analog of SN22 and the biologically active component of the clinically used camptothecin derivative, irinotecan.42,43 The 10-hydroxy substituent distinguishing SN38 from SN22 makes the former highly susceptible to ABCG2-mediated efflux strongly affecting the performance of irinotecan in the settings of high-risk disease.11,12 While SN38 and SN22, both being more effective than camptothecin, showed similar activities (IC90: 16 and 10 nM, respectively) at inhibiting the growth of BE(2)C cells, a major change in response pattern to these agents was observed when tested in TH-MYCN NB cells expressing ABCG2 at four times higher levels (Supporting Information Figure S3). Camptothecin inhibited TH-MYCN NB cell growth by 90% at 40 nM, and its 7-ethyl derivative, SN22, had IC90 further reduced to 18 nM. In contrast, SN38 exhibited the lowest antiproliferative effect, failing to reach 90% of TH-MYCN NB cell growth inhibition in the studied concentration range (8–50 nM), consistent with a key role of ABCG2 transporter protecting TH-MYCN NB cells against SN38 but not SN22.44
The contribution of the delivery enhancement due to the use of the Poloxamer-linked prodrug construct was first examined in a model of MYCN-amplified, newly diagnosed disease. In a control group, irinotecan given twice a week at 15 mg/kg was initially effective at shrinking orthotopically induced chemo-naïve NB tumors (Figure 3A). However, the tumors were not eradicated and started to regrow immediately following a 4-week treatment period, with all animals reaching the endpoint within 70–90 days (Supporting Information Figure S4A,B). In contrast, PF108-[SN22]2 given once a week at an equivalent dose (i.e., 10 mg of SN22 per kg) fully cleared the tumors in 4 out of 5 mice and extended the survival of all animals beyond 180 days. Furthermore, the prodrug was also highly effective when administered to animals approaching the endpoint with large-sized tumors, causing rapid disappearance of the tumor-associated signal and preventing subsequent tumor recurrence (Figure 3B).
FIGURE 3.
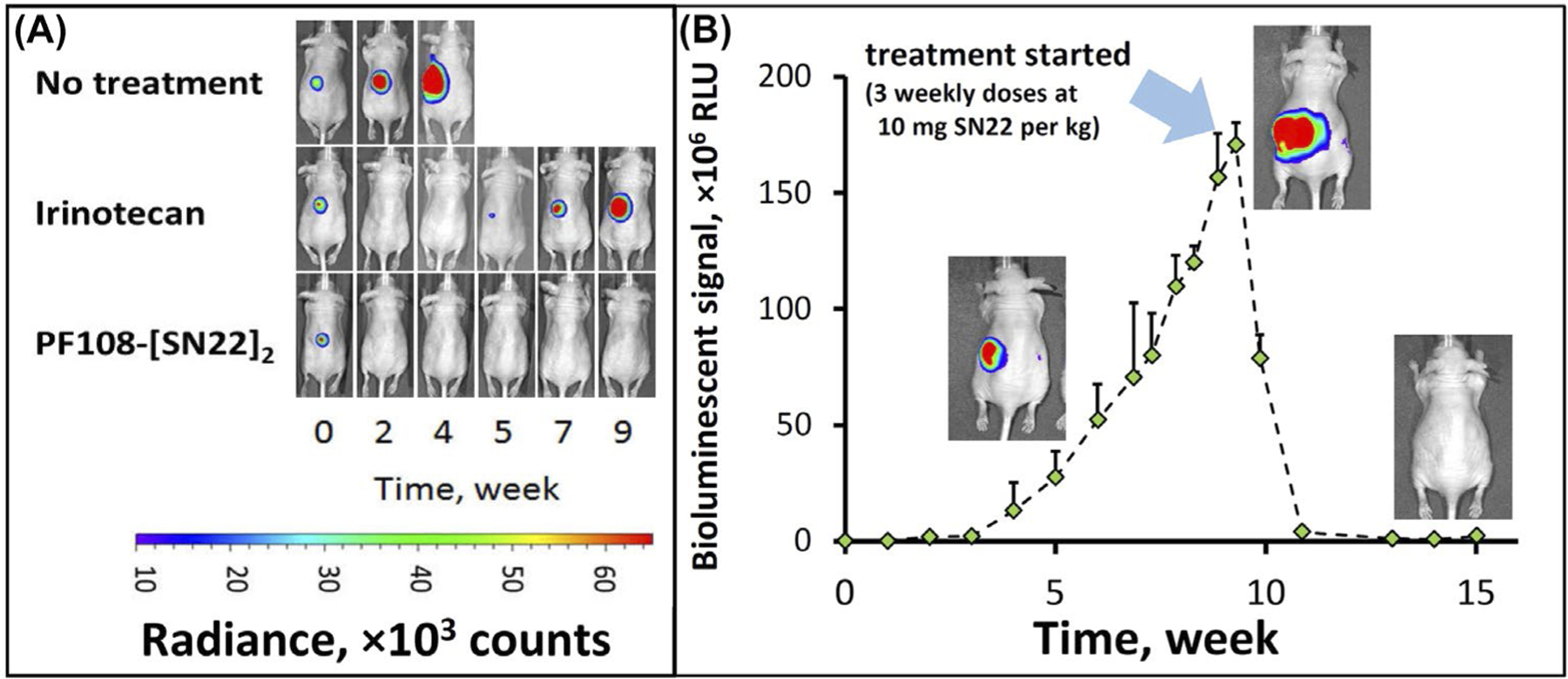
Therapeutic efficacy of PF108-[SN22]2 in an orthotopic mouse model of newly diagnosed, MYCN-driven NB. Athymic nude mice (nu/nu) were inoculated with 106 luciferase-expressing IMR-32 cells in the perirenal fat pad. Treatment with irinotecan or PF108-[SN22]2 at doses corresponding to 10 mg of SN38 or SN22 per kg (2× and 1× week, respectively, over 4 weeks) was initiated 3 weeks after inoculation (A). Tumor-associated signal was monitored by quantitative bioluminescence. Although animals in the irinotecan group initially responded by tumor regression, the response did not extend beyond the treatment period. In contrast, PF108-[SN22]4 caused complete tumor disappearance in 4 and a durable partial response in 1 out of 5 animals (the detailed tumor monitoring analysis and animal survival are shown in Figure S4 in Supporting Information). In an additional experiment, PF108-[SN22]2 administered 1× week over 3 weeks to animals approaching the endpoint with 10-fold larger NB tumors achieved durable responses and extended event-free survival (B). Data are shown as mean ± SD
Unlike IMR-32 cells derived prior to treatment, BE(2)C originating from an SK-N-BE(2) cell line derived at relapse show marked drug resistance, which is likely driven primarily by an acquired loss-of-function p53 mutation that strongly affects both the scale and duration of the pharmacological effect.45 The sensitivity of SK-N-BE(2) cells toward SN38 was indeed shown to be altered by more than an order of magnitude in comparison to the SK-N-BE(1) cell line with the wild-type p53 derived from the same patient at diagnosis [IC90 of 25 vs. 2 ng/ml46], in agreement with the strong difference in responsiveness between BE(2)C and IMR-32 cells in our study (Figure 2A,B). Based on the in vitro antiproliferative potency results, BE(2)C cells require exposure to 80–100 nM (i.e., ~32–40 ng/ml) of SN22 formulated as the prodrug for at least 24 h in order to durably inhibit their growth (Figure 2C and Supporting Information Figure S2A). In vivo, systemic delivery of SN22 as the PF108-linked prodrug exceeded this threshold by a wide margin, with drug concentrations in the tumor found to be ≥40 times higher 4–72 h after administration (Figure 4A). The enhanced drug uptake and sustained tumor exposure were paralleled by the extended residence of the polymer-linked SN22 in the blood, with 290±100 ng/g remaining in the circulation after 3 days. In line with its remarkable longevity in the tumor tissue, PF108-[SN22]2 administered in 4 weekly doses caused rapid regression of BE(2)C-derived tumors and suppressed their regrowth beyond the treatment period (Figure 4B,C). The potency and lasting therapeutic effect of PF108-[SN22]2 contrast starkly with those of SN38 administered as irinotecan: the drug administered twice as frequently had only a marginal and transient effect on the growth of BE(2)C orthotopic xenografts, in accordance with its complete clearance from the blood compartment within 24 h and with its levels in the tumor tissue falling below 25 ng/g at this time point (Figure 4A). However, when SN38 was administered as a PF108-linked construct (i.e., PF108-[SN38]2), its effect on BE(2)C tumors, though smaller in scale than that of PF108-[SN22]2, increased substantially as evidenced by tumor shrinkage, sustained control throughout the treatment period, and extended animal survival (Figure 4B–D). Although the comparison here was not drawn either with the maximum tolerated dose or the standard-of-care dosing schedule of irinotecan, this result shows that greater intratumoral levels and sustained presence of the bioactive agent achievable with a polymer-linked prodrug may lead to a meaningful enhancement in efficacy and allow suppressing tumor growth at a much lower total drug dose, potentially minimizing systemic exposure and toxicity associated with the treatment. Specifically, the contribution of the enhanced delivery and improved pharmacokinetics enabled by this prodrug design is evident from the markedly superior performance of PF108-[SN38]2 in the absence of untoward effects versus twice more frequently dosed irinotecan.
FIGURE 4.
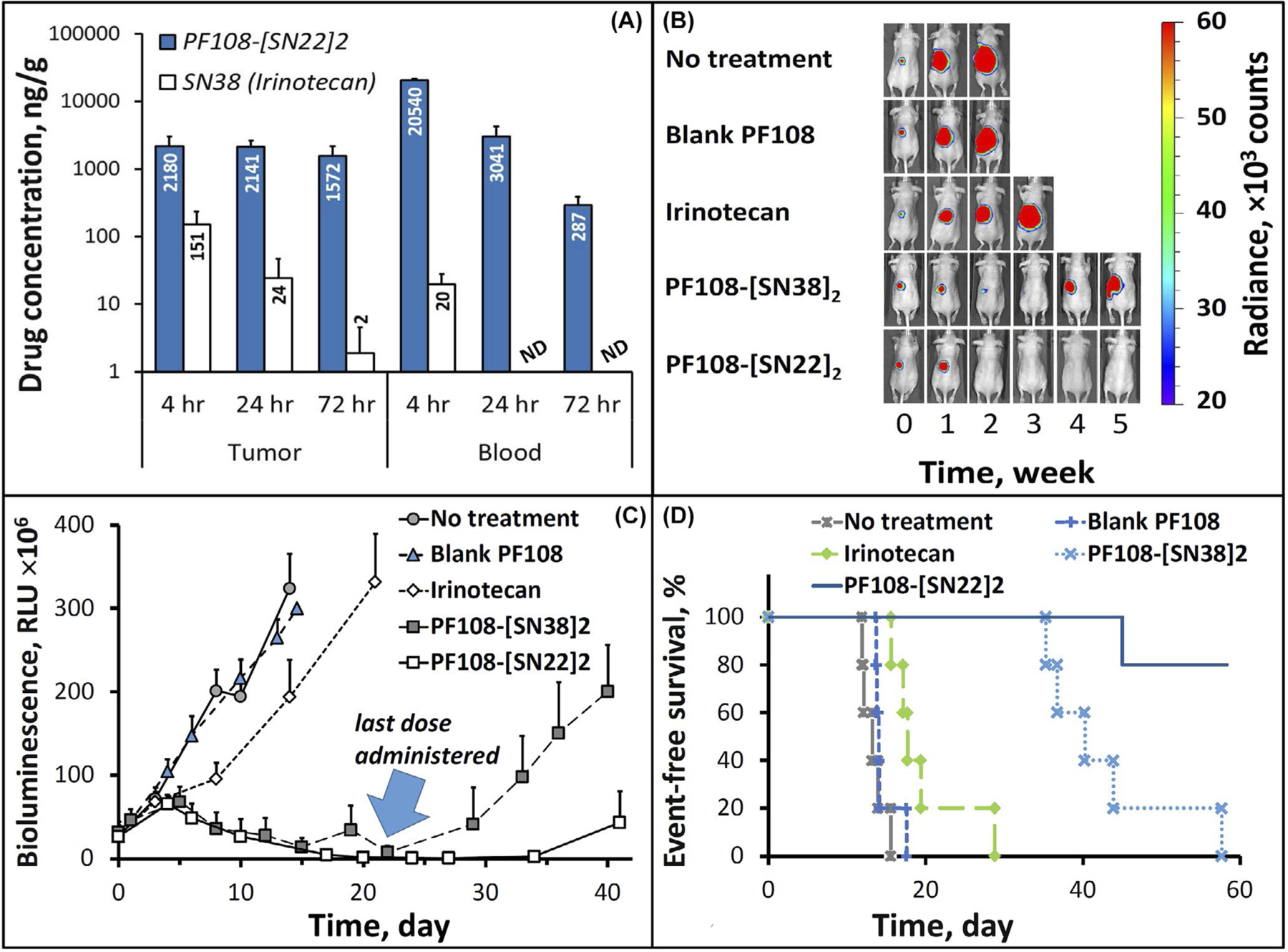
Tumor and blood residence profiles and therapeutic efficacy of PF108-[SN22]2 in an orthotopic model of relapsed, MYCN-amplified NB. Athymic nude mice (nu/nu) were inoculated in the perirenal fat pad with 106 BE(2)C cells stably expressing luciferase. After reaching the tumor size of ~1 cm3, PF108-[SN22]2 was administered intravenously at a dose equivalent to 10 mg/kg of SN22 (a single dose for drug analysis (A) or 1× week for 4 weeks in an efficacy study (B–D). Control groups included animals administered with saline, chemically unmodified Pluronic F-108, analogously constructed PF108-[SN38]2, or irinotecan (1× and 2× week, respectively, over 4 weeks at 10 mg/kg of SN38). Tumor-associated signal was monitored by quantitative bioluminescence (representative images taken at 0–5 weeks are included in B). Quantitative data shown in (C) are expressed as mean ± SD. The survival curves for respective animal groups are shown in (D)
The biodistribution analysis in TH-MYCN mice with established NB tumors confirmed a profound difference in drug delivery efficiency in favor of the PF108-linked constructs versus SN38 administered as irinotecan: after 24 h, the tissue weight-normalized tumor levels of the drugs delivered as polymeric prodrugs were about two orders of magnitude higher than that of irinotecan-derived SN38. Whereas the polymer-linked and free forms of SN22 or SN38 could not be quantified separately using the bioanalytical assay employed in this study, the two prodrug constructs exhibited similar biodistribution profiles, with comparable levels of the total (PF108-linked and free) drugs seen in the organs of the reticuloendothelial system (Figure 5A), a relatively small difference in the tumor concentrations slightly favoring PF108-[SN22]2 versus PF108-[SN38]2 (2.4 ± 0.4 and 1.7 ± 0.8 μg/g, respectively, p = 0.71), and a similar minor difference in serum levels (3.9 ± 0.8 vs. 3.6±0.5 μg/ml, Figure 5B). However, notwithstanding the relatively small difference in their tumor uptake, the antitumor effects of the two prodrug constructs in this animal model were consistent with the much greater in vitro growth-inhibitory potency of SN22 versus SN38 seen in cultured TH-MYCN NB cells. PF108-[SN22]2 was most effective at reducing the number of cycling cells 24 h after administration, reaching a more than sixfold difference versus irinotecan (Supporting Information Figure S5D, p = .005). Tumors harvested from animals treated with PF108-[SN22]2 revealed a different staining pattern with mostly single Ki67-positive cells in contrast to multiple clusters of actively proliferating cells following administration of PF108-[SN38]2 (Supporting Information Figure S5A vs. B). In vivo efficacy studies showed that both prodrug-mediated delivery enhancement and the use of the pharmacologically optimized SN22 contributed to significantly improved outcomes. In control groups, all untreated TH-MYCN animals reached the endpoint within four weeks, while the last animal administered with irinotecan (2× week over 4 weeks) was sacrificed after eight weeks. In comparison, PF108-[SN38]2 or PF108-[SN22]2 administered over a 4-week period at 2× week and 1× week, respectively, both markedly extended survival, with a majority of the animals alive after 10 weeks and with one tumor-free survivor in each group at 180 days. Increasing the dosing frequency of PF108-[SN22]2 to 2× week resulted in an additional improvement, with 100% and 60% animal survival at 19 and 26 weeks, respectively, and with two animals showing no evidence of the tumor presence at 180 days.
FIGURE 5.
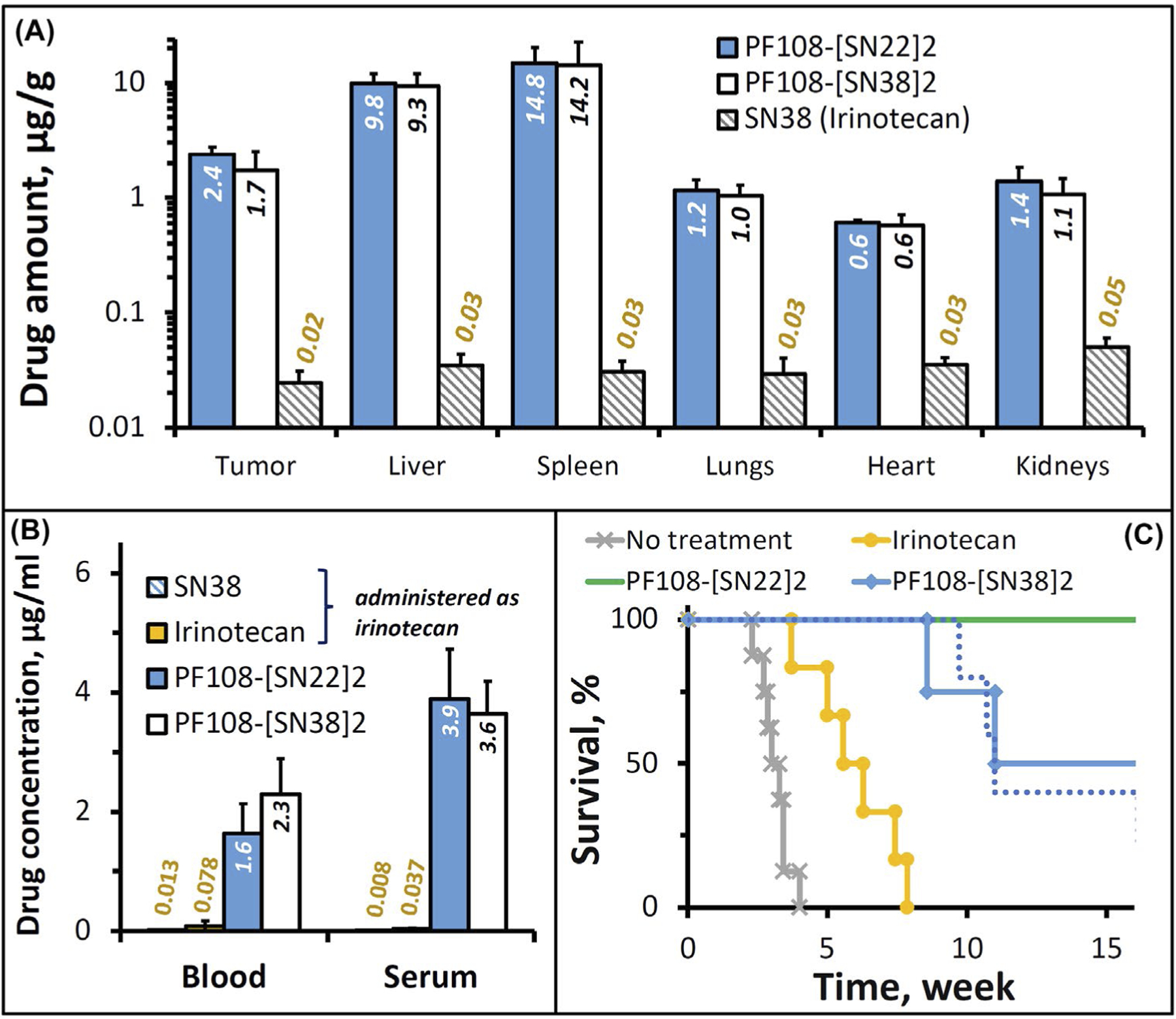
Biodistribution and therapeutic efficacy of PF108-[SN22]2 in a genetically engineered TH-MYCN mouse model of de novo resistant, MYCN-driven NB characterized by overexpression of the ABCG2 transporter. Homozygous 5-week-old TH-MYCN animals with verified large NB tumors were administered with PF108-[SN22]2, PF108-[SN38]2 or irinotecan intravenously at a dose equivalent to 10 mg/kg of SN22 or SN38. For biodistribution analysis, solid tissue samples (A), blood, and serum (B) were collected 24 h after administering a single dose of the prodrugs or irinotecan (shown as mean ± SD). Alternatively, the compounds were given 2× week for 4 weeks in a therapeutic efficacy experiment (C). The survival of animals administered 1× week for 4 weeks with PF108-[SN22]2 is shown for comparison as a dotted line
4 |. DISCUSSION
The results of our study support the rationale for integrating pharmacophore structure optimization with enhanced delivery mediated by a Poloxamer-linked prodrug as part of an experimental approach for treating aggressive, MYCN-amplified NB. Several Poloxamer polymers exhibit high hydrophilicity and molecular sizes greater than 8 kDa allowing for significantly extended blood residence. Within the small group of the hydrophilic Poloxamers with HLB > 24, Pluronic F-108 is the largest-sized polymer (13–18 kDa), which makes it potentially more effective at slowing down renal excretion of its cargo and preventing its extravasation across normal endothelium, both contributing toward stronger on-target antineoplastic activity and lower systemic toxicity.26,47 At the same time, the molecular size of this polymer is within the range where sequestration and accumulation of the polymeric carrier in splenic and renal interstitial macrophages and choroid plexus epithelial cells is expected to be avoided or markedly reduced compared to larger-sized polymers,48 such as 40-kDa poly(ethylene glycol) previously employed to solubilize and deliver SN38.49,50 In the present study, we used a one-step oxidation strategy to chemically activate the polymer and convert it into a carboxylated form for subsequent drug attachment via a hydrolytically cleavable α-alkoxy ester bond. While the α-alkoxy ester-based carrier-drug linkage employed in PF108-[SN22]2 is versatile and can be applied to different molecular cargoes, SN22 as a camptothecin analog offers an important advantage of the highly selective action mechanism, where DNA damage-associated apoptosis is essentially restricted to cycling cells.51,52 Combined with the favorably modulated biodistribution of the macromolecular prodrug construct, this selectivity toward dividing cells is expected to further reduce the untoward effects on normal tissues with low proliferative activity, in particular the organs of the mononuclear phagocytic system, where a significant fraction of the polymer-linked SN22 was detected 24 h after administration.
Among the three models chosen for this study, IMR-32 orthotopic xenografts recapitulate the drug responsiveness pattern of MYCN-amplified, chemo-naïve NB. The clinically used irinotecan included as a control was able to shrink these tumors, even though its therapeutic effect at the dosing regimen used in the present study did not extend beyond the treatment period. While often achieving significant early responses in newly diagnosed high-risk patients, conventional therapies apply strong selection pressure promoting the acquisition of drug resistance by the initially chemosensitive tumors. As a result, the non-curatively treated high-risk disease is likely to recur in a multidrug-resistant form, with a major shift in chemosensitivity severely compromising the efficacy of rescue therapy.53 The goal at this stage is, therefore, to maximize tumor regression and, whenever possible, to eliminate residual disease and to prevent its recurrence.54 In accordance with this approach, PF108-[SN22]2 achieved complete elimination of most IMR-32 tumors, in contrast to the transient response to irinotecan observed in this model. Although the profound and lasting tumor growth inhibitory effect of a polymer-linked camptothecin analog on MYCN-amplified chemosensitive tumors is not unexpected and has previously been reported for SN38 reversibly linked to poly(ethylene glycol),55 it is noteworthy that in our study PF108-[SN22]2 administered in three weekly doses also caused rapid shrinkage and suppressed regrowth of large orthotopic IMR-32 tumors as evidenced by a steep decline in the tumor-associated signal below detectable levels in this group.
Recurrent high-risk tumors with an acquired loss of p53 function exhibit a substantial reduction in chemosensitivity, requiring sustained drug presence in the tumor at levels exceeding by orders of magnitude those achievable clinically.56 Thus, irinotecan capable of initially reducing the size of IMR-32 tumors showed only a marginal effect against BE(2)C orthotopic xenografts used in our study to model relapsed MYCN-amplified NB with an acquired p53 mutation. However, in contrast to the rapid loss of SN38 delivered as irinotecan with only trace levels detectable in the blood and tumor tissue after 24 h, a single dose of PF108-[SN22]2 achieved stable drug presence over 72 h at levels at least 40 times higher than its concentration shown to stably suppress the growth of cultured BE(2)C cells. The increased exposure, in turn, translated into the rapid disappearance of the chemoresistant tumors and led to lasting suppression of their regrowth after four weekly doses of the prodrug. Whereas the notably more pronounced antitumor activity and improved survival may primarily be attributed to the enhancement in drug delivery, it is noteworthy that PF108-[SN22]2 was also found to be more effective in this model than the control construct, PF108-[SN38]2, likely due to a combination of the greater antiproliferative potency of SN22 against BE(2)C cells and its lack of susceptibility to glucuronidation that contributes to intrinsic resistance by accelerating clearance and by compromising biological activity of SN38.57 These results emphasize the important advantages of the Poloxamer-linked SN22 prodrug, including protection of the labile pharmacophore from chemical and biological inactivation, reduced clearance rates, and a major increase in the drug fraction accumulated and stably retained in the tumor. As shown in the models of both pre-therapy and relapsed MYCN-amplified NB, these concomitant improvements built into the design of PF108-[SN22]2 have the potential to markedly prolong survival and strengthen therapeutic efficacy against newly diagnosed high-risk disease and recurrent tumors harboring tumor suppressor protein mutations. The dismal prognosis and poor survival rates of children with relapsed/refractory high-risk NB showing minimal or no response to standard chemotherapeutics58 stress the clinical significance of these improvements and the urgency of their practical implementation in the settings of aggressive disease with limited treatment options.
The therapeutically relevant benefits of the optimized pharmacophore construction in the Poloxamer-SN22 prodrug were most evident in the TH-MYCN genetically engineered mouse model of de novo resistant high-risk disease. Whereas several mechanisms may contribute to the distinct chemoresistance pattern in this model, it was shown to recapitulate the increased ABCG2 expression driving intrinsically low levels of drug sensitivity and linked to non-response to induction chemotherapy.59 Restoring chemosensitivity in poorly differentiated tumors exhibiting high “stemness” and enriched in ABCG2-overexpressing cancer progenitor cells requires chemical modifications in the drug structure directed toward reducing the binding affinity and susceptibility to efflux mediated by this transporter.15 The phenolic 10-hydroxy substituent of the hydrophobic SN38 introduced to allow its conversion to a water-soluble precursor, irinotecan, also makes SN38 vulnerable to this resistance mechanism.9,10 SN22 devoid of the 10-hydroxy function but otherwise analogous to SN38 exhibited a markedly stronger cytotoxic effect on cultured TH-MYCN NB-derived tumor cells, paralleled in vivo by the substantially increased antiproliferative activity detected in NB tumors harvested from PF108-[SN22]2-treated TH-MYCN animals. Although using the more effective Poloxamer prodrug-based delivery in the absence of pharmacophore adjustments was already sufficient for markedly improving therapeutic results and achieving better survival rates in PF108-[SN38]2- versus irinotecan-treated animals, the combination of enhanced delivery with the pharmacophore structure optimization abolishing ABCG2-mediated drug export and restoring tumor cell sensitivity resulted in the most substantial survival extension and the largest number of animals confirmed to be tumor-free after the completion of the study (180 days). Thus, mitigated ABCG2-mediated resistance in combination with an extended residence of the bioactive drug in the tumor is expected to provide the greatest therapeutic gain when applied against chemoresistant, aggressive tumors with complex high-risk features, including MYCN oncogene amplification and enrichment in ABCG2-overexpressing cancer progenitor cells.
Despite aggressive multimodal therapy, less than half of high-risk NB patients survive. NB tumors overexpressing ABCG2 or featuring loss-of-function p53 mutations (the hallmarks of primary or acquired drug resistance, respectively) and showing no durable response to conventional cancer therapies continue to pose a tremendous therapeutic challenge. Our results demonstrate that by exerting potent antiproliferative effects on chemoresistant tumor cells and by favorably modulating the biodistribution and intratumoral residence of its cargo, the Poloxamer-linked prodrug of SN22 can lead to rapid tumor regression and lasting therapeutic responses in models of newly diagnosed and relapsed, MYCN-amplified NB. The experimentally shown ability of PF108-[SN22]2 to achieve long-term survival and to overcome mechanisms governing drug resistance at different phases of refractory disease is an important step toward addressing the urgent need for more robust therapeutic strategies effective against high-risk tumors.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the U.S. National Cancer Institute grant R01-CA251883, the U.S. Department of Defense grant W81XWH2110536, Solving Kids Cancer Foundation, Alex’s Lemonade Stand Foundation, the CURE Childhood Cancer Foundation, Peel Therapeutics (M.C., I.S.A., G.M.B.), and the Audrey E. Evans Endowed Chair (G.M.B.). We are grateful to Dr. Joshua Schiffman for his advice on preparing this manuscript.
Funding information
HHS | NIH | National Cancer Institute (NCI), Grant/Award Number: R01-CA251883; Solving Kids’ Cancer (SKC); Alex’s Lemonade Stand Foundation for Childhood Cancer (ALSF); CURE Childhood Cancer (CURE); Peel Therapeutics; Audrey E. Evans Endowed Chair; U.S. Department of Defense (DOD), Grant/Award Number: W81XWH2110536
DISCLOSURES
M. Chorny, G.M. Brodeur, and I.S. Alferiev are inventors of a patent 2020061007A1 licensed to Peel Therapeutics. They also report a grant from Peel Therapeutics for preclinical research studies. No potential conflicts of interest were disclosed by the other authors.
Abbreviations:
- HLB
hydrophylic/lipophilic balance
- NB
neuroblastoma
- PF108
Pluronic F-108
- PF108-[SN22]2
SN22 conjugate with Pluronic F-108
- PF108-[SN38]2
SN38 conjugate with Pluronic F-108
- THF
tetrahydrofuran
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the main text and Supporting Information of this article.
REFERENCES
- 1.Wong M, Mayoh C, Lau LMS, et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med. 2020;26:1742–1753. [DOI] [PubMed] [Google Scholar]
- 2.Shah N Dodging the bullet: therapeutic resistance mechanisms in pediatric cancers. Cancer Drug Resist. 2019;2:428–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu CP, Hsieh CH, Wu YS. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol Pharm. 2011;8:1996–2011. [DOI] [PubMed] [Google Scholar]
- 4.Keshelava N, Zuo JJ, Chen P, et al. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001;61:6185–6193. [PubMed] [Google Scholar]
- 5.Tweddle DA, Malcolm AJ, Bown N, Pearson AD, Lunec J. Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res. 2001;61:8–13. [PubMed] [Google Scholar]
- 6.Keshelava N, Seeger RC, Groshen S, Reynolds CP. Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Res. 1998;58:5396–5405. [PubMed] [Google Scholar]
- 7.Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85:1219–1226. [DOI] [PubMed] [Google Scholar]
- 8.Hirschmann-Jax C, Foster AE, Wulf GG, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228–14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas A, Pommier Y. Targeting topoisomerase I in the era of precision medicine. Clin Cancer Res. 2019;25:6581–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zage PE. Novel therapies for relapsed and refractory neuroblastoma. Children (Basel). 2018;5:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bates SE, Medina-Perez WY, Kohlhagen G, et al. ABCG2 mediates differential resistance to SN-38 (7-ethyl-10-hydroxyca mptothecin) and homocamptothecins. J Pharmacol Exp Ther. 2004;310:836–842. [DOI] [PubMed] [Google Scholar]
- 12.Gandhi YA, Morris ME. Structure-activity relationships and quantitative structure-activity relationships for breast cancer resistance protein (ABCG2). AAPS J. 2009;11:541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schellens JH, Maliepaard M, Scheper RJ, et al. Transport of topoisomerase I inhibitors by the breast cancer resistance protein. Potential clinical implications. Ann N Y Acad Sci. 2000;922:188–194. [DOI] [PubMed] [Google Scholar]
- 14.Toyoda Y, Takada T, Suzuki H. Inhibitors of human ABCG2: from technical background to recent updates with clinical implications. Front Pharmacol. 2019;10:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westover D, Li F. New trends for overcoming ABCG2/BCRP-mediated resistance to cancer therapies. J Exp Clin Cancer Res. 2015;34:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshikawa M, Ikegami Y, Hayasaka S, et al. Novel camptothecin analogues that circumvent ABCG2-associated drug resistance in human tumor cells. Int J Cancer. 2004;110:921–927. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawa H, Saito H, Ikegami Y, Aida-Hyugaji S, Sawada S, Ishikawa T. Molecular modeling of new camptothecin analogues to circumvent ABCG2-mediated drug resistance in cancer. Cancer Lett. 2006;234:81–89. [DOI] [PubMed] [Google Scholar]
- 18.Nema S, Brendel RJ. Excipients and their role in approved injectable products: current usage and future directions. PDA J Pharm Sci Technol. 2011;65:287–332. [DOI] [PubMed] [Google Scholar]
- 19.Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21:201–230. [DOI] [PubMed] [Google Scholar]
- 20.Alvarez-Lorenzo C, Sosnik A, Concheiro A. PEO-PPO block copolymers for passive micellar targeting and overcoming multidrug resistance in cancer therapy. Curr Drug Targets. 2011;12:1112–1130. [DOI] [PubMed] [Google Scholar]
- 21.Pitto-Barry A, Barry NPE. Pluronic® block-copolymers in medicine: from chemical and biological versatility to rationalisation and clinical advances. Polym Chem. 2014;5:3291–3297. [Google Scholar]
- 22.Moloughney JG, Weisleder N. Poloxamer 188 (p188) as a membrane resealing reagent in biomedical applications. Recent Pat Biotechnol. 2012;6:200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maskarinec SA, Lee KYC. Comparative study of Poloxamer insertion into lipid monolayers. Langmuir. 2003;19:1809–1815. [Google Scholar]
- 24.Alakhova DY, Kabanov AV. Pluronics and MDR reversal: an update. Mol Pharm. 2014;11:2566–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh-Joy SD, McLain VC. Safety assessment of poloxamers 101, 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403, and 407, poloxamer 105 benzoate, and poloxamer 182 dibenzoate as used in cosmetics. Int J Toxicol. 2008;27(suppl 2):93–128. [DOI] [PubMed] [Google Scholar]
- 26.Fox ME, Szoka FC, Frechet JM. Soluble polymer carriers for the treatment of cancer: the importance of molecular architecture. Acc Chem Res. 2009;42:1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis SS. Parenteral polymers. Drug Discov Today. 2002;7:1159–1161. [DOI] [PubMed] [Google Scholar]
- 28.Singla P, Garg S, McClements J, Jamieson O, Peeters M, Mahajan RK. Advances in the therapeutic delivery and applications of functionalized Pluronics: a critical review. Adv Colloid Interface Sci. 2022;299:102563. [DOI] [PubMed] [Google Scholar]
- 29.Swift CC, Eklund MJ, Kraveka JM, Alazraki AL. Updates in diagnosis, management, and treatment of neuroblastoma. Radiographics. 2018;38:566–580. [DOI] [PubMed] [Google Scholar]
- 30.Laetsch TW, DuBois SG, Bender JG, Macy ME, Moreno L. Opportunities and challenges in drug development for pediatric cancers. Cancer Discov. 2021;11:545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L, Esfandiari A, Reaves W, et al. Characterisation of the p53 pathway in cell lines established from TH-MYCN transgenic mouse tumours. Int J Oncol. 2018;52:967–977. [DOI] [PubMed] [Google Scholar]
- 32.Patterson DM, Shohet JM, Kim ES. Preclinical models of pediatric solid tumors (neuroblastoma) and their use in drug discovery. Curr Protoc Pharmacol. Chapter 14. 2011;52:14.17.11–14.17.18. [DOI] [PubMed] [Google Scholar]
- 33.Byrne FL, McCarroll JA, Kavallaris M. Analyses of tumor burden in vivo and metastasis ex vivo using luciferase-expressing cancer cells in an orthotopic mouse model of neuroblastoma. Methods Mol Biol. 2016;1372:61–77. [DOI] [PubMed] [Google Scholar]
- 34.Chesler L, Weiss WA. Genetically engineered murine models—contribution to our understanding of the genetics, molecular pathology and therapeutic targeting of neuroblastoma. Semin Cancer Biol. 2011;21:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teitz T, Stanke JJ, Federico S, et al. Preclinical models for neuroblastoma: establishing a baseline for treatment. PLoS One. 2011;6:e19133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serrano LA, Yang Y, Salvati E, Stellacci F, Krol S, Guldin S. pH-Mediated molecular differentiation for fluorimetric quantification of chemotherapeutic drugs in human plasma. Chem Commun (Camb). 2018;54:1485–1488. [DOI] [PubMed] [Google Scholar]
- 37.Lele BS, Kulkarni MG. Single step room temperature oxidation of poly(ethylene glycol) to poly(oxyethylene)-dicarboxylic acid. J Appl Polym Sci. 1998;70:883–890. [Google Scholar]
- 38.Moore JS, Stupp SI. Room temperature polyesterification. Macromolecules. 1990;23:65–70. [Google Scholar]
- 39.Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med. 2013;3:a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tumilowicz JJ, Nichols WW, Cholon JJ, Greene AE. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res. 1970;30:2110–2118. [PubMed] [Google Scholar]
- 41.Biedler JL, Spengler BA. A novel chromosome abnormality in human neuroblastoma and antifolate-resistant Chinese hamster cell lives in culture. J Natl Cancer Inst. 1976;57:683–695. [DOI] [PubMed] [Google Scholar]
- 42.Kunimoto T, Nitta K, Tanaka T, et al. Antitumor activity of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxy-camptothecin, a novel water-soluble derivative of camptothecin, against murine tumors. Cancer Res. 1987;47:5944–5947. [PubMed] [Google Scholar]
- 43.Sawada S, Yokokura T, Miyasaka T. Synthesis and antitumor activity of A-ring or E-lactone modified water-soluble prodrugs of 20(S)-camptothecin, including development of irinotecan hydrochloride trihydrate (CPT-11). Curr Pharm Design. 1995;1:113–132. [Google Scholar]
- 44.Nguyen F, Guan P, Guerrero DT, et al. Structural optimization and enhanced prodrug-mediated delivery overcomes camptothecin resistance in high-risk solid tumors. Cancer Res. 2020;80:4258–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keshelava N, Zuo JJ, Waidyaratne NS, Triche TJ, Reynolds CP. p53 mutations and loss of p53 function confer multidrug resistance in neuroblastoma. Med Pediatr Oncol. 2000;35:563–568. [DOI] [PubMed] [Google Scholar]
- 46.Keshelava N, Groshen S, Reynolds CP. Cross-resistance of topoisomerase I and II inhibitors in neuroblastoma cell lines. Cancer Chemother Pharmacol. 2000;45:1–8. [DOI] [PubMed] [Google Scholar]
- 47.Ekladious I, Colson YL, Grinstaff MW. Polymer-drug conjugate therapeutics: advances, insights and prospects. Nat Rev Drug Discov. 2019;18:273–294. [DOI] [PubMed] [Google Scholar]
- 48.Ivens IA, Achanzar W, Baumann A, et al. PEGylated biopharmaceuticals: current experience and considerations for non-clinical development. Toxicol Pathol. 2015;43:959–983. [DOI] [PubMed] [Google Scholar]
- 49.Sapra P, Zhao H, Mehlig M, et al. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin Cancer Res. 2008;14:1888–1896. [DOI] [PubMed] [Google Scholar]
- 50.Zhao H, Rubio B, Sapra P, et al. Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjug Chem. 2008;19:849–859. [DOI] [PubMed] [Google Scholar]
- 51.Pommier Y Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. [DOI] [PubMed] [Google Scholar]
- 52.Yamauchi T, Yoshida A, Ueda T. Camptothecin induces DNA strand breaks and is cytotoxic in stimulated normal lymphocytes. Oncol Rep. 2011;25:347–352. [DOI] [PubMed] [Google Scholar]
- 53.Kuosmanen T, Cairns J, Noble R, Beerenwinkel N, Mononen T, Mustonen V. Drug-induced resistance evolution necessitates less aggressive treatment. PLoS Comput Biol. 2021;17:e1009418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith V, Foster J. High-risk neuroblastoma treatment review. Children. 2018;5(9):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pastorino F, Loi M, Sapra P, et al. Tumor regression and curability of preclinical neuroblastoma models by PEGylated SN38 (EZN-2208), a novel topoisomerase I inhibitor. Clin Cancer Res. 2010;16:4809–4821. [DOI] [PubMed] [Google Scholar]
- 56.Polunin Y, Alferiev IS, Brodeur GM, Voronov A, Chorny M. Environment-sensitive polymeric micelles encapsulating SN-38 potently suppress growth of neuroblastoma cells exhibiting intrinsic and acquired drug resistance. ACS Pharmacol Transl Sci. 2021;4:240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brangi M, Litman T, Ciotti M, et al. Camptothecin resistance: role of the ATP-binding cassette (ABC), mitoxantrone-resistance half-transporter (MXR), and potential for glucuronidation in MXR-expressing cells. Cancer Res. 1999;59:5938–5946. [PubMed] [Google Scholar]
- 58.London WB, Bagatell R, Weigel BJ, et al. Historical time to disease progression and progression-free survival in patients with recurrent/refractory neuroblastoma treated in the modern era on Children’s Oncology Group early-phase trials. Cancer. 2017;123:4914–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cialfi S, McDowell HP, Altavista P, et al. ABC drug transporter gene expression in neuroblastoma. J Clin Oncol. 2010;28:9524. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the main text and Supporting Information of this article.