

Abstract
Pancreatic ductal adenocarcinomas (PDAC) depend on autophagy for survival; however, the metabolic substrates that autophagy provides to drive PDAC progression are unclear. Ferritin, the cellular iron storage complex, is targeted for lysosomal degradation (ferritinophagy) by the selective autophagy adaptor NCOA4, resulting in release of iron for cellular utilization. Using patient-derived and murine models of PDAC we now demonstrate that ferritinophagy is upregulated in PDAC to sustain iron availability thereby promoting tumor progression. Quantitative proteomics reveals that ferritinophagy fuels iron-sulfur cluster protein synthesis to support mitochondrial homeostasis. Targeting NCOA4 leads to tumor growth delay and prolonged survival but with development of compensatory iron acquisition pathways. Finally, enhanced ferritinophagy accelerates PDAC tumorigenesis, and an elevated ferritinophagy expression signature predicts for poor prognosis in PDAC patients. Together, our data reveal that maintenance of iron homeostasis is a critical function of PDAC autophagy, and we define NCOA4-mediated ferritinophagy as a therapeutic target in PDAC.
Keywords: NCOA4, ferritinophagy, iron, PDAC
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the third most common cause of cancer-related death in the US with dismal five-year survival rates of 10% (1). Thus, PDAC patients are in desperate need of new treatment options. Oncogenic KRAS, the genetic driver of 90% of PDAC, induces a metabolic rewiring characterized in part by dependency on elevated levels of iron and autophagy, a catabolic process that degrades cellular components in the lysosome for recycling (2–4). Human PDACs have elevated basal autophagy and genetic inhibition (e.g. ATG5/7 ablation) or pharmacologic inhibition of autophagy with chloroquine (CQ, a weak inhibitor of lysosomal acidification) decreases PDAC growth in vitro and in vivo in mouse models of PDAC (3,5). However, to date, clinical trial results with hydroxychloroquine monotherapy are disappointing (4). Iron addiction and biomarkers of high iron content portend shorter overall survival for patients with PDAC; however, there are no effective therapies targeting cancer-specific upregulated iron metabolic pathways. NCOA4 (Nuclear Receptor Coactivator 4) is a selective autophagy adaptor that targets ferritin, the cellular iron storage complex, via autophagy to the lysosome for degradation and release of iron to the cytosol for utilization (e.g. ‘ferritinophagy’), thereby providing a molecular link between autophagy and iron metabolism (6,7). NCOA4 was initially discovered as one of the most highly enriched selective autophagy receptors in PDAC autophagosomes; however, whether PDAC are reliant on NCOA4-mediated ferritinophagy for maintenance of iron metabolism, tumor proliferation, or tumor survival is unknown.
While free or ‘labile’ iron (Fe2+) can drive cancer proliferation, excess labile iron promotes ferroptosis (8), an iron-dependent form of cell death. Therefore, cells have evolved mechanisms whereby iron can be sequestered and released from protein complexes in response to changing iron levels. Ferritin, a 24-subunit protein complex (consisting of ferritin heavy and light chains: FTH1/FTL), can store up to 4,500 iron atoms. To access ferritin-bound iron for cellular use, NCOA4 mediates ferritinophagy promoting iron release from ferritin to maintain the cellular labile iron pool (6). Systemic ablation of Ncoa4 in murine models leads to accumulation of tissue ferritin, reduced systemic iron availability and a functional iron deficiency that manifests as a mild hypochromic microcytic anemia (9–11). As a block in ferritinophagy decreases labile iron, ablation of NCOA4 decreases sensitivity to ferroptosis induction in cell culture models of PDAC (12) and in vivo in the context of murine models of ischemia-induced ferroptosis, for instance (13). On the other hand, NCOA4 over-expression in PDAC cell lines promotes sensitivity to ferroptosis, which is a vulnerability in PDAC (12).
Given the importance of iron metabolism and autophagy in PDAC, we investigated the role of NCOA4-mediated ferritinophagy in PDAC. Using patient-derived and murine PDAC models, we demonstrate that NCOA4 as well as ferritinophagy are upregulated in PDAC to support iron metabolism and stability and activity of iron-sulfur (Fe-S) cluster-containing proteins. Ablation of NCOA4-mediated ferritinophagy delays tumor progression and improves survival in an autochthonous genetically engineered mouse model (GEMM) of PDAC. Conversely, enhanced ferritinophagy via Ncoa4 over-expression in the context of a PDAC GEMM accelerates tumor initiation and worsens survival. These findings demonstrate the important link between autophagy and iron metabolism in PDAC and nominate NCOA4 for further investigation as a targeted approach for inhibition of autophagy and iron metabolism in PDAC.
RESULTS
NCOA4 is a pancreatic cancer dependency
NCOA4 levels have been shown to dictate ferritinophagic flux (14). We observed that NCOA4 protein expression is higher in established PDAC cell lines versus pancreatic ductal epithelial cell lines (Fig. 1A, Supplementary Fig. S1A) (15). Likewise, NCOA4 mRNA expression is significantly increased in human PDAC versus normal human pancreas (Fig. 1B) (16). These data suggest higher flux and potential reliance on ferritinophagy in PDAC compared to normal pancreas. Ferritinophagic flux was elevated in PDAC cell lines as measured by accumulation of NCOA4 and FTH1 after blocking autophagic flux with chloroquine (Fig. 1C).
Figure 1. NCOA4 is a dependency in PDAC.
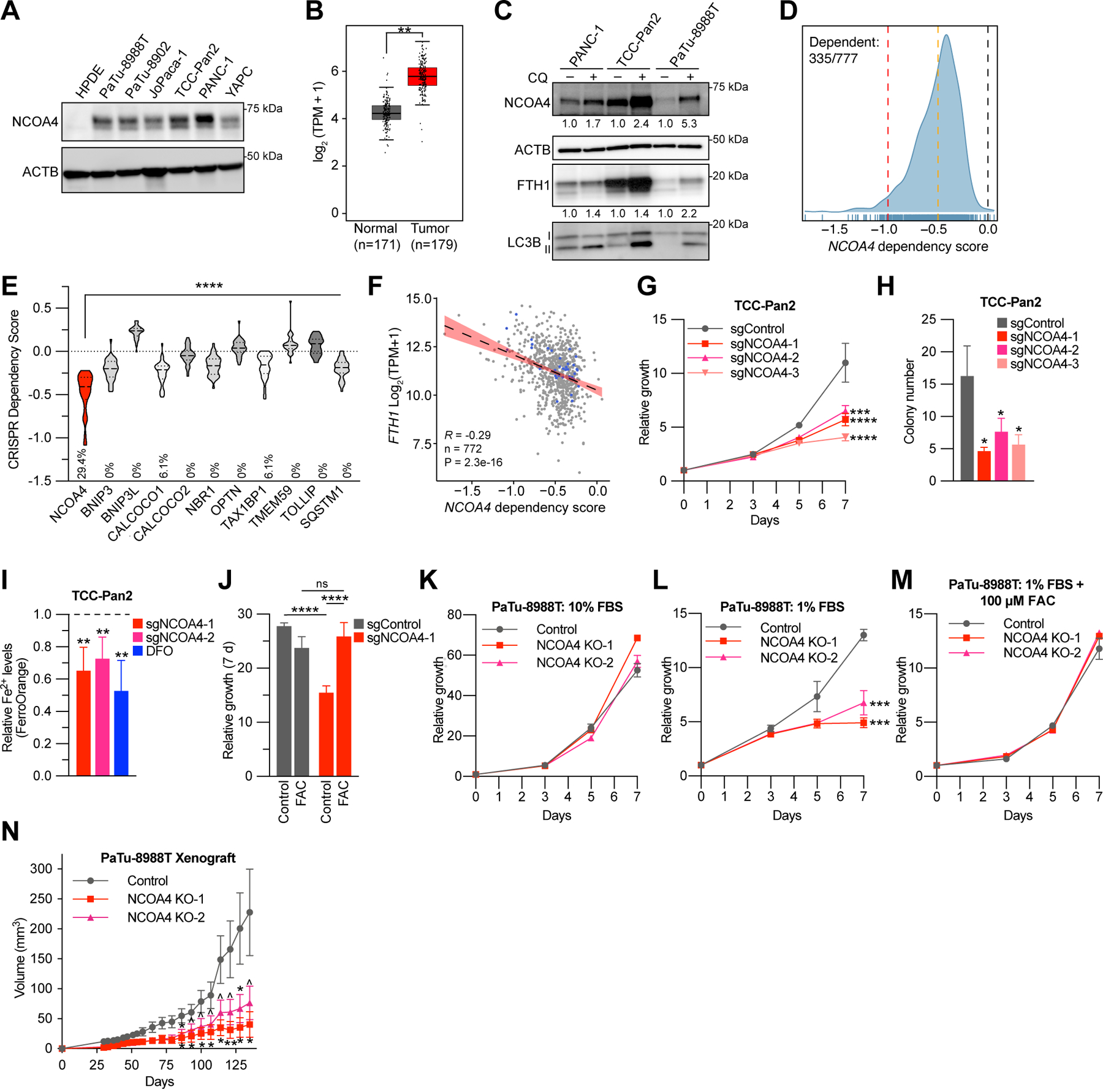
A, Immunoblot showing NCOA4 protein levels in lysates from HPDE (human pancreatic ductal epithelial) and PDAC cell lines, as indicated. B, NCOA4 mRNA expression levels in human normal pancreas tissue (GTEx and TCGA normal, n=171 patients) versus human PDAC tumor (TCGA, n=179 patients). The data are presented as box-and-whisker plots, boxes represent the median and the 25th and 75th percentiles (** p<0.01, t-test, as calculated in GEPIA2 database). C, Immunoblot showing NCOA4, FTH1, and MAP1LC3B (LC3B) protein levels in lysates from PDAC cell lines treated with or without chloroquine (CQ) (10 μM for 16 h). NCOA4 and FTH1 relative quantification (normalized within each cell line) denoted below each lane. D, Smoothed histogram showing the distribution of NCOA4 dependency scores from the CRISPR-spCas9 Cancer Dependency Map dataset (21Q1) across cancer cell lines. The number of dependent cell lines (CRISPR score less than −0.5, orange line) over the total number of probed cell lines is shown in the top left corner. Red line at −1: CRISPR score for a set of control highly essential genes. Black line at 0: CRISPR score of negative control sgRNAs. E, Violin plot of CRISPR dependency scores of selective autophagy receptor genes in PDAC cell lines represented in the Cancer Dependency Map (n=34), Dashed line: median value, Dotted line: quartile values. Percentage of cell lines scored as dependent (CRISPR score less than −0.5) indicated at bottom of graph (p<0.0001 for all comparisons to NCOA4 using ANOVA with Dunnett’s multiple comparison test). F, Scatter plot showing linear regression with 95% confidence interval (black line) and Pearson’s correlation coefficient between FTH1 mRNA expression (y-axis) and NCOA4 CRISPR dependency scores (x-axis) across 772 cancer cell lines, blue dots represent data for PDAC cell lines. G, Relative proliferation of pools of TCC-Pan2 cells lentivirally transduced with control Cas9-sgRNA targeting the non-essential Rosa26 locus (sgControl) or three independent Cas9-sgRNAs targeting NCOA4 (sgNCOA4). Data are plotted as relative cell proliferation in arbitrary units. Values normalized to Day 0. Error bars represent s.d. of 6 technical replicates (representative of 3 independent experiments). NCOA4 depletion/’knockdown’ level (>90%) is displayed in Supplementary Fig. S1E. H, Clonogenic growth of TCC-Pan2 NCOA4 ‘knockdown’ cells expressing lentiviral sgRNAs as in G. Error bars ± s.d. triplicate wells of a representative experiment of 3 independent experiments. I, Quantification of intracellular Fe2+ by FerroOrange staining and flow cytometry in TCC-Pan2 NCOA4 KD cells. Mean fluorescence intensity was normalized to cells transduced with sgControl. DFO (100 μM) was used as a positive control. Dotted line indicates sgControl normalized intensity level. Error bars ± s.d. of 4 independent experiments. J, Relative proliferation measured at 7 d of TCC-Pan2 cells expressing sgControl or sgNCOA4-1 treated with or without ferric ammonium citrate (FAC) (50 μM) and ferrostatin-1 (500 nM, see methods). Values normalized to Day 0. Error bars ± s.d., triplicate wells of a representative experiment of 3 independent experiments. K, Relative proliferation of control versus NCOA4 clonal KO PaTu-8988T cells grown in media containing 10% FBS. Values normalized to Day 0. Error bars represent s.d. of 3 technical replicates (representative of 3 independent experiments). L, Relative proliferation of control versus NCOA4 KO PaTu-8988T cells grown in media containing 1% FBS as in K. M, Relative proliferation of control versus NCOA4 KO PaTu-8988T cells grown in media containing 1% FBS with FAC (100 μM) and ferrostatin-1 (1 μM). N, In vivo subcutaneous tumor growth in immunocompromised NCr-Foxn1nu mice of PaTu-8988T control versus NCOA4 KO cells as in K. Error bars ± s.e.m., n=14–15 mice. For panels G-N, significance determined with t-test. ^ p < 0.1, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
To evaluate whether NCOA4 is a cancer dependency, we queried the Cancer Dependency Map (DepMap), a library of genome-wide CRISPR loss-of-function screens in hundreds of cancer cell lines (17,18). NCOA4 scored as a cancer dependency in 335/777 cell lines, including PDAC cell lines (Fig. 1D). Among selective autophagy receptors enriched in PDAC autophagosomes (6), NCOA4 scores as the most dependent in PDAC cells (Fig. 1E). NCOA4 dependency was significantly and most highly correlated with ferritin heavy chain (FTH1) mRNA and protein expression (Fig. 1F, Supplementary Fig. S1B) suggesting that tumor cell lines with high basal levels of iron and thereby iron storage in ferritin are more dependent on ferritinophagy for proliferation. This correlation was specific for FTH1 and not FTL, which is consistent with NCOA4 directly interacting with FTH1 for autophagic targeting of FTH1/FTL ferritin complexes (Supplementary Fig. S1C) (14). Indeed, NCOA4 protein expression was highly correlated with FTH1 expression (Supplementary Fig. S1D)
To further validate the effects of NCOA4 depletion, we used CRISPR-SpCas9-mediated NCOA4 depletion (cell pools of >90% knockdown (‘KD’) or clonal knockout (‘KO’)) in established PDAC cell lines (Supplementary Fig. S1E-F, see methods). Cell growth and clonogenicity were significantly decreased in a panel of NCOA4 KD PDAC cells, as predicted by DepMap (Fig. 1G–H, Supplementary Fig. S1G-J). As NCOA4-mediated ferritinophagy is important for maintaining cytosolic labile iron levels, we measured labile iron levels in NCOA4 KD cells. NCOA4 depletion led to a significant decrease in ‘free iron’ (Fe2+), that was comparable to treatment of cells with deferoxamine (DFO), an iron chelator (Fig. 1I). Moreover, treatment of NCOA4 KD cells with iron in the form of ferric ammonium citrate (FAC) was sufficient to rescue cell growth confirming on-target effect of NCOA4 depletion (Fig. 1J). Across multiple additional PDAC cell lines, we noted variability in the degree of proliferative block following NCOA4 depletion, as predicted by dependency scores in DepMap (Fig. 1K, Supplementary Fig. S1G). We reasoned that the in vitro conditions under which NCOA4 depletion was tested in DepMap may not reflect in vivo conditions, in particular the amount of iron-bound transferrin (holo-transferrin) availability, a primary component of fetal bovine serum (FBS). Therefore, we evaluated a panel of PDAC cell lines cultured in media with lower percentage of FBS (1–5%) that reduced holo-transferrin availability but still supported proliferation of control cells. Under these conditions, PDAC cell lines with NCOA4 depletion (KD or clonal KO) demonstrated more marked growth delay and reduced clonogenicity compared to cells grown in 10% FBS (Fig. 1L, Supplementary Fig. S1H-J). To evaluate the specificity of this reduced growth to iron availability, we rescued NCOA4 clonal KO PaTu-8988T cells with FAC (Fig. 1M). We next determined the in vivo tumor growth of xenografted PaTu-8988T NCOA4 ‘wild type’ versus NCOA4 KO cells to determine the relevance of reduced serum cell growth conditions to in vivo growth. Growth of NCOA4 KO xenografts demonstrated marked tumor growth delay (Fig. 1N). These results demonstrate that NCOA4 is important for proliferation of PDAC cells and xenograft tumors.
Ncoa4 ablation improves survival in a genetically engineered mouse model of PDAC
Next, we used autochthonous genetically engineered PDAC mouse models to study the role of Ncoa4 in the context of a model that more closely recapitulates the human PDAC condition (19). We and others previously demonstrated that systemic ablation of Ncoa4 is not embryonic lethal or lethal in the context of acute inducible ablation in an adult mouse (9,10). Under basal conditions, Ncoa4-null mice develop tissue ferritin accumulation, decreased serum iron availability, and a mild microcytic hypochromic anemia but no overt pancreatic pathology (10). To investigate the role of Ncoa4 loss in PDAC pathogenesis, we crossed a conditional Ncoa4fl/fl allele with a genetically engineered PDAC mouse model that uses Pft1a-Cre to drive pancreas expression of mutant KRAS (KrasLSL-G12D/+) with loss of p53 (Trp53fl/+) (KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre mice (KPC)). These crosses generated a KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre; Ncoa4fl/fl (KPCN) mouse model. As adequate immunohistochemistry (IHC) reagents for Ncoa4 are lacking, we evaluated Ncoa4 expression in tumor-derived cell lines. Ncoa4 expression was ablated in KPCN-derived tumor cell lines (Fig. 2A) and no tumor lines examined demonstrated re-expression of Ncoa4. Ncoa4 ablation significantly delayed appearance of tumors as measured by high-resolution ultrasound (Fig. 2B) (median age of diagnosis KPC: 8.86 weeks (95% confidence interval 8, 14.57 weeks); KPCN: 12.57 weeks (11.57, 14.29 weeks)). There was no significant difference in size of tumor detected in KPC versus KPCN mice at time of diagnosis (Fig. 2C). In a cohort of mice evaluated at time of diagnosis, histologic analysis demonstrated no significant difference in area of pancreas affected by invasive PDAC or PanIN (Fig. 2D).
Figure 2. Loss of NCOA4 extends murine PDAC survival.
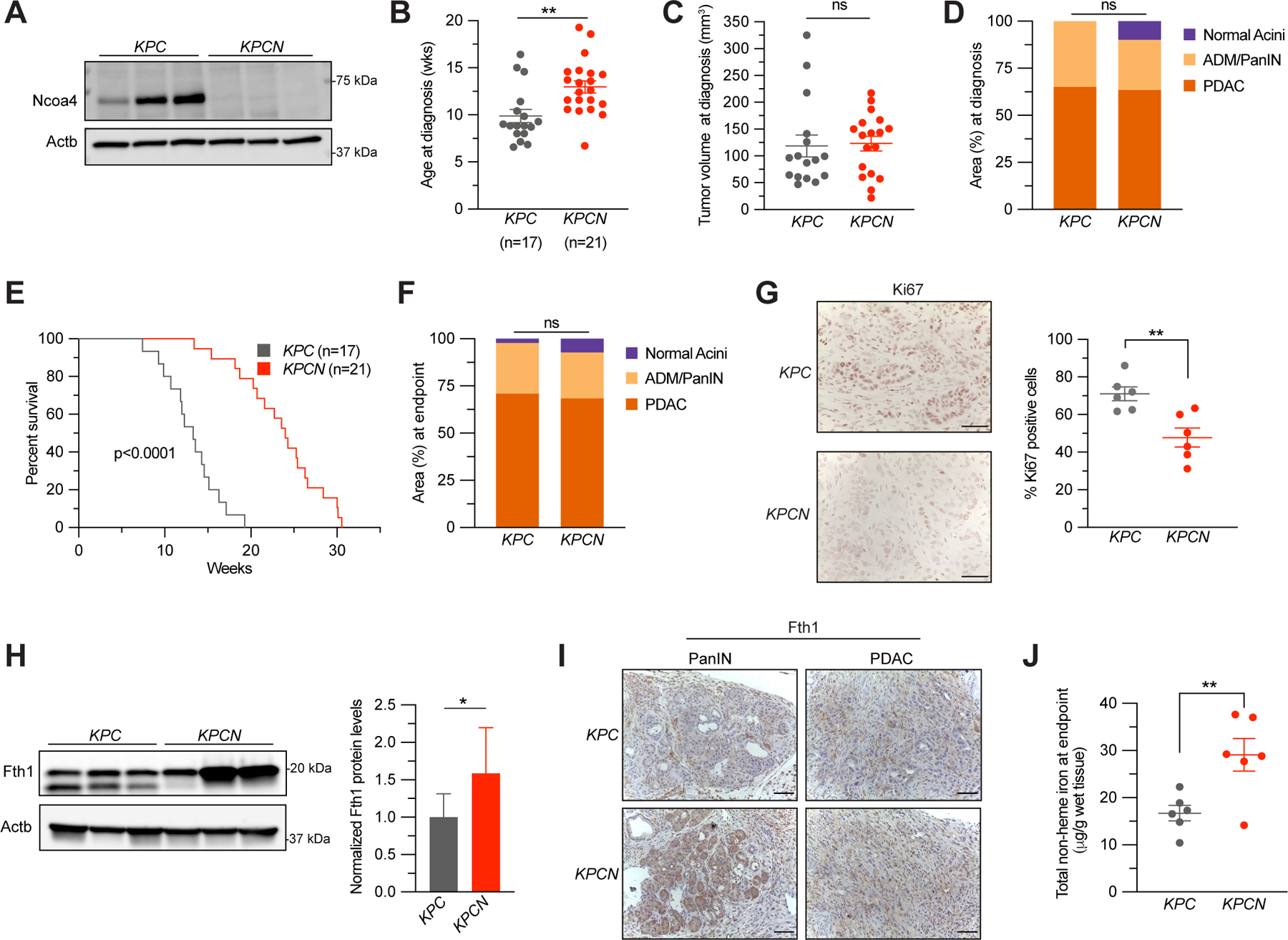
A, Immunoblot showing Ncoa4 protein levels in lysates from tumor-derived cell lines from KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre (KPC) and KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre; Ncoa4fl/fl (KPCN) mice (n=3 independent tumor-derived cell lines per genotype). B, Age at pancreatic tumor diagnosis, as determined by high-resolution ultrasound. C, Tumor volume at time of diagnosis, as measured by ultrasound, KPC, n=16, KPCN, n=18. D, Quantification of normal acinar, acinar to ductal metaplasia (ADM)/pancreatic intraepithelial neoplasia (PanIN), and PDAC area in KPC (n=4) versus KPCN (n=3) pancreata at time of diagnosis as measured on H&E staining. E, Kaplan-Meier analysis comparing overall survival of KPC (n=17) versus KPCN (n=21) mice. Log-rank test, p < 0.0001. F, Quantification of normal acinar, ADM/PanIN, and PDAC area in KPC (n=16) versus KPCN (n=16) pancreata at endpoint as measured on H&E staining. G, Pancreatic tumors from KPC and KPCN tumors at endpoint stained with Ki67 antibody, representative field displayed. The percentage of Ki67 positive cells was counted in 5 fields from each tumor, n=6 tumors per genotype, scale bar = 50 μm. H, Immunoblot showing Fth1 protein levels in lysates from tumor-derived cell lines from KPC and KPCN mice (n=3 independent tumor-derived cell lines per genotype, quantification is of 3 KPC and 3 KPCN cells with 3 replicates). Lower band in Fth1 blot is indicative of a lysosomal processed form of Fth1. Error bars ± s.d. I, Fth1 immunostaining from PanIN and PDAC areas from representative KPC and KPCN pancreata, scale bar = 50 μm. J, Tumor non-heme iron levels in KPC versus KPCN tumors at endpoint, n=6 tumors per genotype. For panels B-D and F-J, significance determined with t-test. ns = not significant: p > 0.05, * p<0.05, ** p < 0.01. Error bars ± s.e.m.
In a survival analysis cohort, remarkably, KPCN mice had statistically improved survival compared to KPC mice (Fig. 2E) (median survival KPC: 13.57 weeks (95% C.I. 11.9, 14.6 weeks); KPCN: 23.57 weeks (20.7, 25.3 weeks)). This improved survival was not reflected in any obvious difference in amount of invasive cancer (Fig. 2F), size of tumor, differentiation status, or extent of desmoplasia in KPCN versus KPC tumors at endpoint, or metastases in KPCN versus KPC mice at endpoint (of note, in this particular KPC model, there is a low frequency of metastases) (Supplementary Fig. S2A-D). However, KPCN tumors at endpoint demonstrated a significant decrease in Ki67 staining consistent with a lower proliferative rate (Fig. 2G). Consistent with a block in ferritinophagic flux, Fth1 accumulated in KPCN versus KPC tumor-derived cell lines with absence of the faster migrating band indicative of lysosomal processing (Fig. 2H) (7). To evaluate Fth1 levels in vivo, we used Fth1 IHC (Fig. 2I). Normal acini, PanIN, and tumor regions demonstrated Fth1 accumulation, consistent with a block in Ncoa4-mediated ferritinophagy. Finally, total non-heme iron levels in KPCN tumors were significantly elevated suggesting accumulation of ferritin-bound iron (Fig. 2J, Supplementary Fig. S2E). Among a panel of metals measured by inductively coupled plasma-mass spectrometry (ICP-MS), iron was the only metal increased (Supplementary Fig. S2F-G). Altogether, loss of Ncoa4 in a KPC mouse model delays progression of tumors and improves survival through impairment of ferritinophagy, thus supporting a role for this process in PDAC pathogenesis.
Enhanced ferritinophagy accelerates PDAC initiation and worsens survival
If Ncoa4 and iron availability are necessary for tumor initiation and progression, we hypothesized that enhanced ferritinophagy via Ncoa4 over-expression would accelerate tumor progression and decrease survival. On the other hand, NCOA4 over-expression promotes sensitivity to ferroptosis, which is a vulnerability in PDAC (12,20). To address these questions, we generated a conditional transgenic mouse model of Ncoa4 over-expression by knocking in a murine Ncoa4 open reading frame into the Rosa26 locus using CRISPR-spCas9 (hereafter: Ncoa4Tg/+) (Supplementary Fig. S3A). To validate the allele, we used a CMV-Cre driven systemic model of over-expression (Ncoa4Tg/+; CMV-Cre). Mice with the target genotypes were born at expected frequency (Supplementary Fig. S3B). Ncoa4 protein level was elevated and Fth1 decreased in the liver (Supplementary Fig. S3C-D). Ncoa4 was similarly elevated in pancreata with decreased Fth1 consistent with increased ferritinophagic flux (Supplementary Fig. S3E-F). Pancreatic acinar and ductal morphology was normal (Supplementary Fig. S3G) and there was no significant difference in body weight, suggesting intact pancreatic exocrine and endocrine function (Supplementary Fig. S3H). Likewise, there was no difference in survival in mice monitored to 38 weeks of age (Supplementary Fig. S3I).
To evaluate the effect of enhanced ferritinophagy on PDAC, we crossed KrasLSL-G12D; Trp53L/+; Pft1a-Cre mice with the Ncoa4Tg/+ allele to generate KPC-Ncoa4Tg/+ mice (Supplementary Fig. S3J). Among 200 mice from consecutive litters, the KPC-Ncoa4Tg/+ genotype frequency was significantly reduced compared to the expected number (Fig. 3A). Of four viable KPC-Ncoa4Tg/+ mice, one died at 2.1 weeks of age (unable to harvest tissues) and three others were euthanized at three weeks of age due to morbidity. KPC-Ncoa4Tg/+ mice had significantly reduced body weight (Fig. 3B) and a dramatic decrease in survival compared to KPC mice (Fig. 3C). Pancreata from KPC-Ncoa4Tg/+ mice were grossly abnormal with significantly increased pancreas-body weight ratio (Fig. 3D). Histologic evaluation of KPC-Ncoa4Tg/+ mice demonstrated total replacement of pancreata by invasive adenocarcinoma (Fig. 3E–F). This suggests that the reduced body weight phenotype was a consequence of pancreatic insufficiency due to a lack of normal functioning pancreas in KPC-Ncoa4Tg/+ mice. Age-matched KPC mice had minimal acinar-to-ductal metaplasia or early PanIN and pancreata from PC-Ncoa4Tg/+ mice were histologically normal. Tumors from KPC-Ncoa4Tg/+ were histologically similar to tumors from KPC mice that developed at a later age (Fig. 3G). Fth1 levels in PC-Ncoa4Tg/+ acinar regions of pancreata were reduced in comparison to KPC mice and significantly reduced compared to tumor-bearing KPC mice (Fig. 3H). There was a trend towards decreased ferritin levels in tumor-bearing KPC-Ncoa4Tg/+ versus tumor-bearing KPC mice.
Figure 3. Enhanced NCOA4-mediated ferritinophagy accelerates PDAC initiation and worsens murine survival.
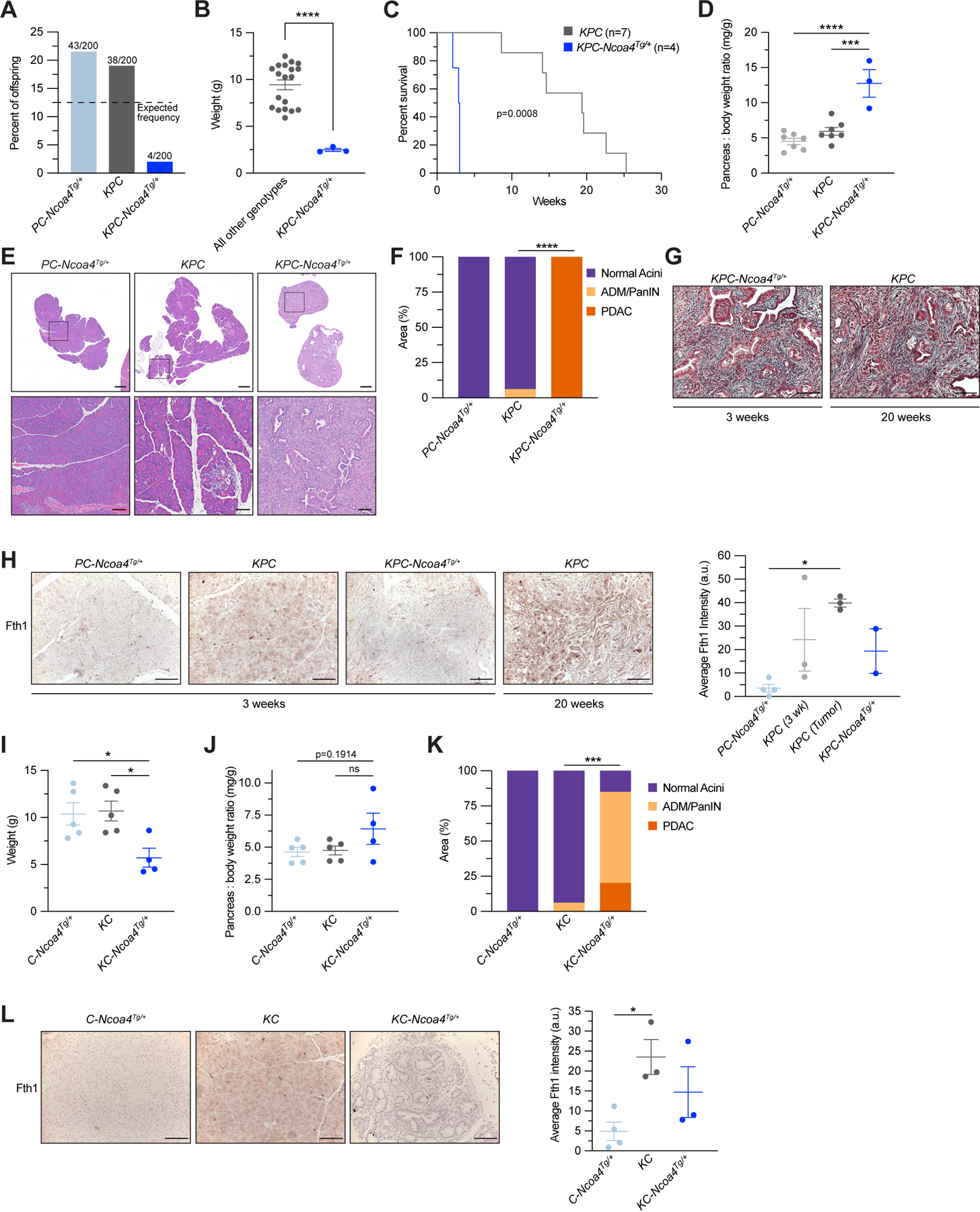
A, Genotype frequency of Trp53fl/+; Pft1a-Cre; Ncoa4Tg/+ (PC-Ncoa4Tg/+), KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre; (KPC) and KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre; Ncoa4Tg/+ (KPC-Ncoa4Tg/+) among 200 genotyped offspring mice, dotted line represents expected frequency of genotypes (12.5%). B, Body weight of KPC-Ncoa4Tg/+ mice versus mice of all other genotypes, measured at 3 weeks of age. C, Kaplan-Meier analysis comparing overall survival of KPC-Ncoa4Tg/+ (n=4) versus KPC (n=7) mice. Log-rank test, p=0.0008. D, Pancreas to body weight ratio of PC-Ncoa4Tg/+, KPC, and KPC-Ncoa4Tg/+ mice. E, H&E staining of representative pancreata from PC-Ncoa4Tg/+, KPC, and KPC-Ncoa4Tg/+ mice harvested at 3 weeks of age. Bottom row panels are magnifications of top row images. Top row scale bar = 500 μm. Bottom row scale bar = 100 μm. F, Quantification of normal acinar, acinar to ductal metaplasia (ADM)/pancreatic intraepithelial neoplasia (PanIN), and PDAC area in PC-Ncoa4Tg/+ (n=2), KPC (n=4), and KPC-Ncoa4Tg/+ (n=2) mice harvested at 3–4 weeks of age. G, Trichrome staining of representative PDAC tumors from a 3-week-old KPC-Ncoa4Tg/+ mouse and a 20-week-old tumor-bearing KPC mouse. H, Fth1 immunostaining of pancreata from PC-Ncoa4Tg/+, KPC, and KPC-Ncoa4Tg/+ mice at 3–4 weeks of age and tumor-bearing KPC mice. Scale bar = 50 μm. Quantification of average intensity in arbitrary units (5 random fields per sample, n=2–4 mice/group, error bars represent s.e.m.). I, Body weight of C-Ncoa4Tg/+ (n=5), KC (n=5), and KC-Ncoa4Tg/+ (n=4) mice, measured at 3 weeks of age. J, Pancreas to body weight ratio of C-Ncoa4Tg/+ (n=5), KC (n=5), and KC-Ncoa4Tg/+ (n=4) mice measured at 3 weeks of age. K, Quantification of normal acinar, ADM/PanIN, and PDAC area in C-Ncoa4Tg/+ (n=5), KC (n=6), and KC-Ncoa4Tg/+ (n=3) mice harvested at 3–4 weeks of age. L, Fth1 immunostaining of pancreata from C-Ncoa4Tg/+, KC, and KC-Ncoa4Tg/+ mice at 3–4 weeks of age. Scale bar = 50 μm. Quantification of average intensity in arbitrary units (5 random fields per sample, n=3–4 mice/group, error bars represent s.e.m.). For panels B, F, and K significance determined with t-test. For panels D, H, I, J, and L, significance determined using ANOVA. ns = not significant: p > 0.05, ** p < 0.01. Error bars ± s.e.m.
To evaluate the effects of Ncoa4 over-expression in a less aggressive PDAC model, we crossed the Ncoa4Tg/+ allele with KC mice where tumors typically appear after 30 weeks of age. Remarkably, at 3 weeks of age KC-Ncoa4Tg/+ mice were also underweight, likely due to pancreatic insufficiency (Fig. 3I, Supplementary Fig. S3K), and there was a trend towards increased pancreas-body weight ratio (Fig. 3J). Pancreata evaluated at 3 weeks of age demonstrated near complete replacement with ADM/PanIN or PDAC (Fig. 3K, Supplementary Fig. S3L). Fth1 was significantly decreased in C-Ncoa4Tg/+ acini in comparison to KC mice with a trend towards a decrease in KC-Ncoa4Tg/+ mice when compared to age-matched KC mice (Fig. 3L). Taken together, mouse models with Ncoa4 ablation or over-expression in the context of PDAC mouse models identify a role for Ncoa4-mediated ferritinophagy in modulating PDAC progression and survival.
Ferritinophagy supports iron-sulfur cluster protein levels and activity
To identify the PDAC tumor cell autonomous pathways supported by NCOA4, we first evaluated genes with parallel patterns of essentiality to NCOA4 in the Cancer Dependency Map (18). Clustering analysis using STRING (21) identified functional clusters related to autophagy, as expected (Fig. 4A). Specifically, NCOA4 dependency correlated with TAX1BP1, RB1CC1 (FIP200), and ATG9A dependencies (Fig. 4A), which are upstream autophagic regulators of NCOA4 trafficking via an ATG8-independent autophagic pathway (22). Remarkably, the remainder of the top NCOA4-correlated dependencies cluster in iron metabolism-related functional groups spanning the cycle of Transferrin receptor-dependent iron import, intracellular trafficking (lysosomal iron export e.g. SLC11A2, mitochondrial import, e.g. SLC25A28), iron utilization, and cellular iron metabolism regulatory machinery (Fig. 4A–B). Clusters related to functional use of iron include iron-sulfur cluster (ISC) protein synthesis (23), DNA replication, and RNA metabolism. Multiple genes that require ISCs directly or in complex were correlated (e.g. ACO1, POLE3, POLE4). Finally, IREB2, the master post-transcriptional regulator of iron response element (IRE)-containing genes was correlated and its negative regulator, FBXL5, was inversely correlated. NCOA4 dependency was similarly associated with iron metabolism genes in an independent dataset (Supplementary Fig. S4A) (24). STRING-based mapping of TAX1BP1 and RB1CC1-correlated dependencies also demonstrated enrichment for iron metabolism genes (Supplementary Fig. S4B-C) supporting the importance of these upstream regulators of NCOA4 in iron metabolism.
Figure 4. NCOA4-mediated ferritinophagy supports iron-sulfur cluster protein levels and activity in PDAC cells.
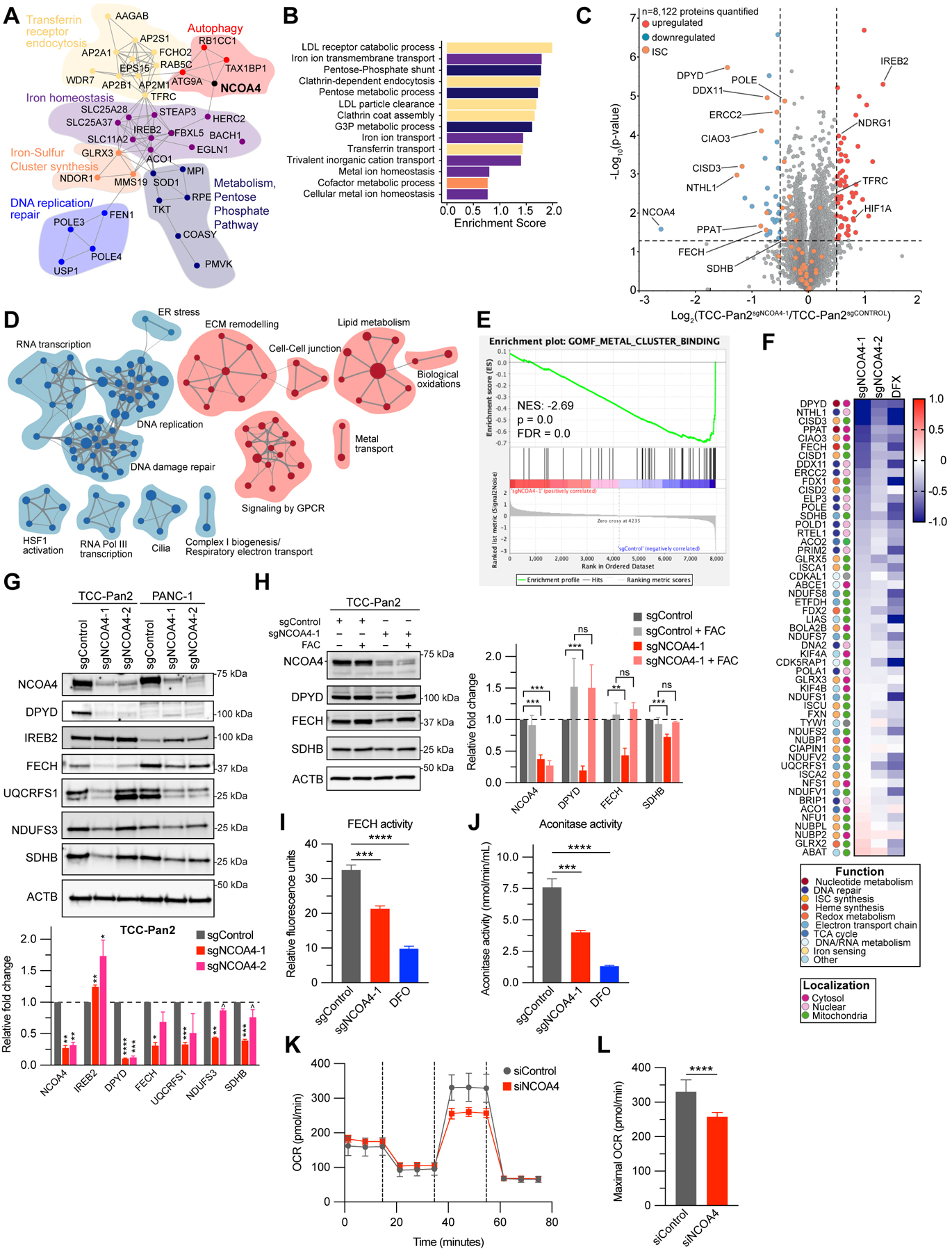
A, Network map showing gene dependencies most highly correlated with NCOA4 dependency in the Cancer Dependency Map (21Q1). Grey connections indicate interaction between genes as predicted by STRING. Functional clusters manually annotated based on Gene Ontology Biological Process analysis as in B. FBXL5 is denoted by a hashed circle as it is anti-correlated to NCOA4 dependency. B, Gene Ontology Biological Process enrichment analysis of gene dependencies most highly correlated with NCOA4 dependency in the Cancer Dependency Map (21Q1). C, Volcano plot illustrates statistically significant protein abundance differences in TCC-Pan2 cells lentivirally transduced with sgNCOA4-1-Cas9 versus sgControl-Cas9. Volcano plots display the −log10 (p value) versus the log2 of the relative protein abundance of mean sgNCOA4-1 to mean sgControl samples. Orange circles represent iron-sulfur cluster (ISC) proteins identified in the dataset (n=53). Red circles represent significantly upregulated (log2 fold change>0.5) while blue circles represent significantly down-regulated proteins (log2 fold change<−0.5) (Data from four sgControl or three sgNCOA4-1 independent plates). D, Enrichment map of gene set enrichment analysis (GSEA-Reactome) of sgNCOA4-1 compared to sgControl TCC-Pan2 proteome. (p < 0.01, FDR q value <0.1; Jaccard coefficient >0.5). Node size is related to the number of components identified within a gene set, and the width of the line is proportional to the overlap between related gene sets. GSEA terms associated with upregulated (red) and downregulated (blue) proteins are colored accordingly and grouped into nodes with associated terms. E, Enrichment plot for Gene Ontology (Molecular Function) Metal Cluster Binding gene set of sgNCOA4-1 compared to sgControl TCC-Pan2 proteome. F, Log2 (Fold change) heatmap for iron-sulfur cluster containing proteins and iron-sulfur cluster synthesis proteins as measured from TCC-Pan2 cells (comparisons are: (sgNCOA4-1 / sgControl), (sgNCOA4-2 / sgControl), (sgControl + Deferasirox (DFX) / sgControl)). Values presented are the mean of three independent plates. Legend: color scheme for functional and localization categories of ISCs. G, Immunoblot showing ISC protein levels in lysates from TCC-Pan2 and PANC-1 cell lines lentivirally transduced with sgControl, sgNCOA4-1, or sgNCOA4-2 (NCOA4 knockdown). Relative fold-change quantification is of 2–3 replicates. Increased IREB2 levels serve as a positive control indicative of decreased free iron. H, Immunoblot showing ISC protein levels in lysates from TCC-Pan2 cell lines lentivirally transduced with sgControl or sgNCOA4-1 with or without ferric ammonium citrate (FAC, 100 μM). Relative fold-change quantification is of 4 replicates. I, FECH activity from TCC-Pan2 cells lentivirally transduced with sgControl, sgNCOA4-1, or sgControl cells treated with DFO (12 h, 1 mm) as measured by formation of zinc-mesoporphyrin (monitored at 400 nm). J, Cytosolic aconitase activity from TCC-Pan2 cells lentivirally transduced with sgControl, sgNCOA4-1, or sgControl cells treated with DFO (12 h, 1 mm). K, Oxygen consumption rate (OCR) of mitochondrial respiratory complex using Seahorse in TCC-Pan2 cells transduced with siControl or siNCOA4. L, Maximal OCR in TCC-Pan2 cells transduced with siControl or siNCOA4. For panels G-J, and L significance determined with t-test. ns non-significant, ^ p<0.1, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Error bars are ± s.d.
We similarly analyzed for gene expression inversely correlated to NCOA4 dependency where high expression predicts sensitivity to NCOA4 depletion. In addition to FTH1 (Fig. 1F), among the highest anti-correlated genes were those that promote increased intracellular iron levels: HMOX1 which degrades heme to release iron and IL6, a cytokine that can induce HAMP (Hepcidin) mRNA expression thereby reducing SLC40A1 (FPN)-mediated iron export (Supplementary Fig. S4D) (25). Genes associated with autophagic and lysosomal activity (CTSZ, SQSTM1, BHLHE40, EVA1A) were inversely correlated with NCOA4 dependency suggesting cell lines with increased autophagic/lysosomal activity are sensitive to NCOA4 depletion. Among the highest positively correlated genes was SLC25A28, a mitochondrial iron importer, suggesting cell lines with robust mitochondrial iron import may be relatively resistant to NCOA4 depletion (Supplementary Fig. S4D). Collectively, these observations suggest that expression of NCOA4 and other genes related to iron metabolism or lysosomal activity may serve as biomarkers of NCOA4 dependency.
Iron is critical for a host of metabolic and enzymatic cellular processes active in both non-tumor and tumor cells. In cellular and zebrafish models of erythropoiesis, we and others showed NCOA4-mediated ferritinophagy was necessary for mitochondrial heme synthesis (14,26). Furthermore, in HeLa and SH-SY5Y (neuronal) cells, ferritinophagy maintains mitochondrial iron levels and respiratory activity (27). The relevance of these findings to PDAC are unclear. Therefore, we evaluated the global proteomic response to NCOA4 depletion in a NCOA4-dependent PDAC cell line using multiplexed isobaric tag-based quantitative mass spectrometry to systematically identify NCOA4-dependent pathways (Fig. 4C, Supplementary Fig. S4E-G, Supplementary Table 1) (28,29). Overall, we observed proteomic alterations in NCOA4-depleted cells consistent with 1) a block in ferritinophagic flux; 2) upregulation of iron transport; and 3) downregulation of cellular processes dependent on ISC-containing proteins. We identified and quantified 8,122 proteins with an expected significant decrease in NCOA4 levels in NCOA4 KD conditions. Consistent with a decrease in bioavailable iron due to a block in ferritinophagic flux, we saw an increase in IREB2, which is stabilized from FBXL5-mediated proteasomal degradation during iron deprivation. HIF1A protein levels were also elevated, likely due to decreased activity of iron cofactor-dependent prolyl hydroxylases.
To gain a comprehensive understanding of the pathways altered in response to NCOA4 depletion, we performed gene set enrichment analysis (GSEA) on the proteome (Supplementary Table 2) (30). Reactome enrichment map nodes associated with upregulated proteins included metal transport consistent with a compensatory increase in iron import via TFRC (Fig. 4D). Enrichment map nodes associated with downregulated proteins included mitochondrial complex I biogenesis and electron transport chain (Fig. 4D, Supplementary Fig. S4H), DNA damage repair, and RNA/DNA replication (31). Gene Ontology molecular function GSEA revealed ‘Metal Cluster Binding’, ‘2 Iron 2 sulfur cluster binding’ and ‘4 iron 4 sulfur cluster binding’ as three of the four most downregulated molecular function pathways (Fig. 4E, Supplementary Table 2). Indeed, the most downregulated proteins were ISC-containing proteins, or those proteins involved in ISC synthesis with a broad downregulation of this protein class across functional and localization groupings (Fig. 4F).
We validated by immunoblot decreases in ISC-containing mitochondrial electron transport proteins including NADH:Ubiquinone Oxidoreductase Core Subunit S3 (NDUFS3, Complex I component), Succinate Dehydrogenase Complex Iron Sulfur Subunit B (SDHB, Complex II), and Ubiquinol-Cytochrome C Reductase, Rieske Iron-Sulfur Polypeptide 1 (UQCRFS1, Complex III) (Fig. 4G, Supplementary Fig. S4I). Likewise, we validated decreases in Dihydropyrimidine dehydrogenase (DPYD, pyrimidine catabolism pathway) and Ferrochelatase (FECH, heme synthesis pathway) ISC-containing proteins (Fig. 4G). ISC-protein decreases in NCOA4 KD PDAC cells are presumably a consequence of limited cytosolic and mitochondrial iron availability (Fig. 1I, Supplementary Fig. S4J) due to decreased ferritin degradation. To test this, we evaluated levels of ISC proteins in response to addition of iron (FAC). FAC addition rescued decreased levels of ISCs in NCOA4 KD cells as did expression of a sgNCOA4-resistant murine Ncoa4 rescue plasmid (Fig. 4H, Supplementary Fig. 4K). Finally, we demonstrated decreased ISC-protein activity in response to NCOA4 depletion, including ferrochelatase activity (FECH) (Fig. 4I) and cytosolic aconitase activity (ACO1) (Fig. 4J). Electron transport chain capacity, as measured by maximal oxygen consumption rate (OCR), was decreased with RNAi-mediated NCOA4 KD (Fig. 4K, Supplementary Fig. S4L) consistent with previously reported effects of NCOA4 depletion on OCR (27). These findings demonstrate a critical role for NCOA4 in regulating ISC-protein level and activity with broad implications for not only mitochondrial function but also DNA repair and DNA/RNA metabolism.
Ferritinophagy supports mitochondrial iron-sulfur cluster protein levels and mitochondria structure in murine PDAC tumors
To translate these findings to in vivo PDAC models, we similarly used mass spectrometry-based quantitative proteomics of KPC and KPCN tumors to identify proteomic alterations that correlate with iron metabolic pathways fueled by NCOA4-mediated ferritinophagy in vivo (Supplementary Fig. S5A, Supplementary Table 1). Here, we identified and quantified 9,259 proteins (Fig. 5A) with an expected enrichment in Fth1 and Ftl1 proteins consistent with a block in ferritinophagy. Enrichment map nodes associated with downregulated proteins included electron transport chain and RNA metabolism suggesting an in vivo reliance of Ncoa4-mediated ferritinophagy to fuel ISC-related processes (Fig. 5B, Supplementary Table 2). To correlate these findings linking Ncoa4 to maintenance of processes that require ISC proteins in vivo, we evaluated expression levels of mitochondrial ISCs in KPC and KPCN tumors. KPCN tumors demonstrated significantly lower immunostaining for Sdhb and Ndufs3 (Fig. 5C–D). We next evaluated number of cristae per mitochondria by transmission electron microscopy in KPC versus KPCN mice. Here, we identified a significant decrease in cristae per mitochondria in KPCN mice suggesting defective mitochondrial health (Fig. 5E, Supplementary Fig. S5B). To determine if tumors with Ncoa4 over-expression had increased ISCs, we evaluated tumor Sdhb levels by immunostaining; however, there were no discernible alterations in tumors from KPC-Ncoa4Tg/+ and KC-Ncoa4Tg/+ mice in comparison with KPC tumors (Supplementary Fig. S5C). As discussed below, we considered alternative explanations for the accelerated tumor phenotype in the Ncoa4 over-expression model. These data demonstrate that NCOA4-mediated ferritinophagy is important for maintenance of ISC-related iron homeostasis in PDAC in vivo.
Figure 5. NCOA4-mediated ferritinophagy supports mitochondrial iron-sulfur cluster protein levels and mitochondria structure in murine PDAC tumors.
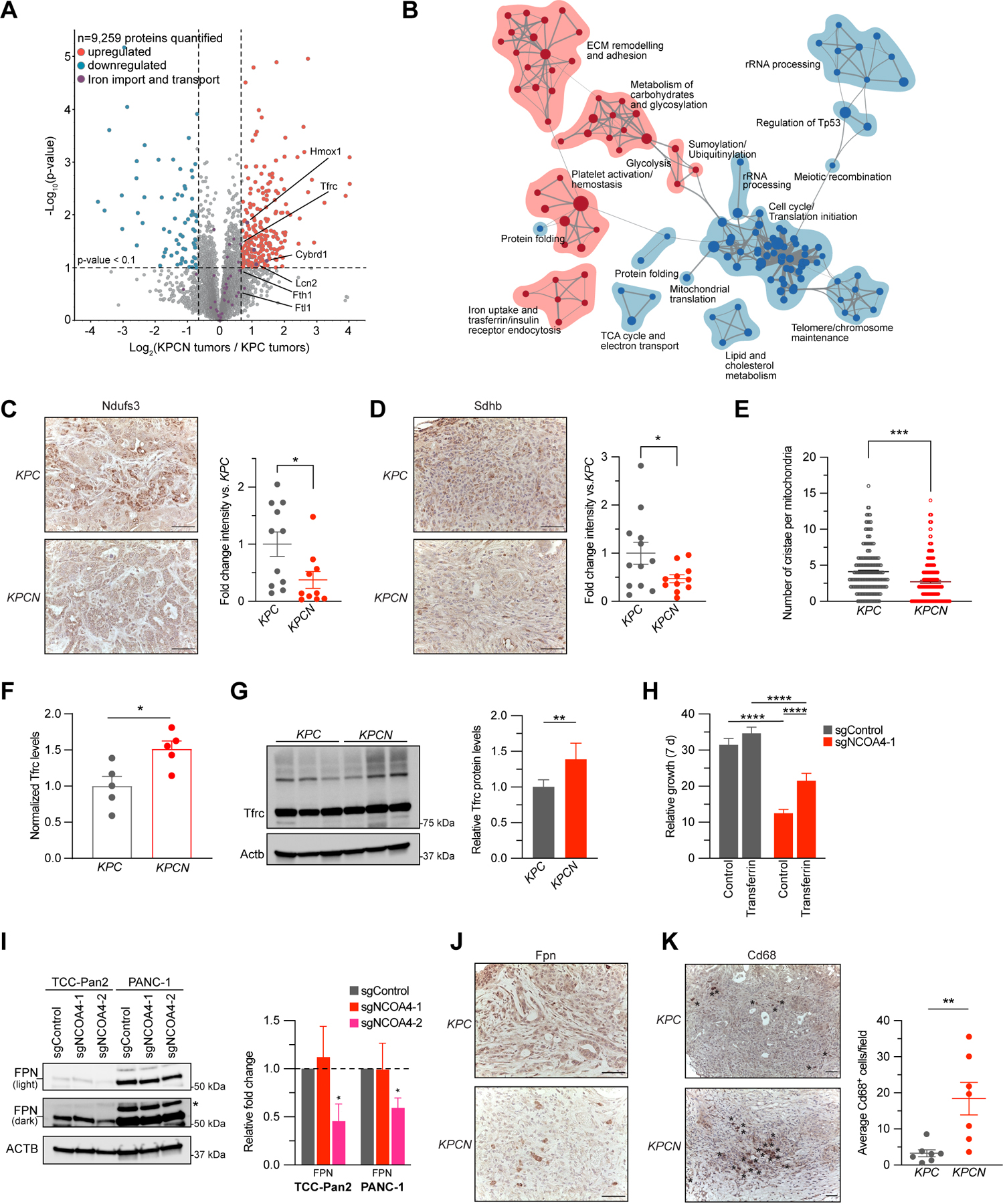
A, Volcano plot illustrates statistically significant protein abundance differences in KPCN versus KPC tumors. Volcano plots display the −log10 (p value) versus the log2 of the relative protein abundance of mean KPCN to KPC tumors (n=5 tumors per genotype, proteins with log2 fold change between −4 to 4 are graphed). Purple circles represent iron transport proteins identified in the dataset. Red circles represent significantly upregulated (log2 fold change>0.7) while blue circles represent significantly down-regulated proteins (log2 fold change<−0.7). B, Enrichment map of gene set enrichment analysis (GSEA-Reactome) of KPCN compared to KPC tumor proteomes. (p < 0.005, FDR q value <0.05; Jaccard coefficient >0.5). Node size is related to the number of components identified within a gene set, and the width of the line is proportional to the overlap between related gene sets. GSEA terms associated with upregulated (red) and downregulated (blue) proteins are colored accordingly and grouped into nodes with associated terms. C, Ndufs3 immunostaining of tumors from KPC and KPCN tumors at endpoint, representative image displayed. Scale bar = 50 μm. Quantification of average intensity presented in relative fold change (5 random fields per sample, n=10–11 mice/group, error bars represent s.e.m.). D, Sdhb immunostaining of tumors from KPC and KPCN tumors at endpoint, representative image displayed. Scale bar = 50 μm. Quantification of average intensity presented in relative fold change (5 random fields per sample, n=11–12 mice/group, error bars represent s.e.m.). E, Quantification of mitochondrial cristae number from electron microscopy images of pancreata from KPC and KPCN mice (n=3 mice/group, 40–100 mitochondria quantified for each mouse). F, Relative protein levels of Tfrc as measured in global KPC and KPCN proteome as in A, normalized to KPC Tfrc protein levels. G, Immunoblot showing Tfrc protein levels in lysates from tumor-derived cell lines from KPC and KPCN mice (n=3 independent tumor-derived cell lines per genotype, quantification is of 3 KPC and 3 KPCN cells with 2 replicates). H, Relative proliferation measured at 7 d of TCC-Pan2 cells expressing sgControl versus sgNCOA4-1 treated with or without Transferrin, 10 μM. Values normalized to Day 0. Error bars ± s.d., n=6 wells of a representative experiment of 3 independent experiments. I, Immunoblot showing Ferroportin (SLC40A1/FPN) protein levels in lysates from TCC-Pan2 and PANC-1 cell lines lentivirally transduced with sgControl, sgNCOA4-1, or sgNCOA4-2. Relative fold-change quantification is of 3 replicates. J, Fpn immunostaining of tumors from KPC and KPCN tumors at endpoint, representative image displayed, Scale bar = 50 μm. K, Cd68 immunostaining of tumors from KPC and KPCN mice at endpoint. Representative field, Scale bar = 50 μm, Cd68 positive cells indicated by asterisks. Quantification (at least 5 fields per sample) of average Cd68+ cells per field (n=7 mice/group, error bars represent s.e.m.). For panels C-I and K significance determined with t-test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Error bars for C-F and K ± s.e.m. for G-I ± s.d.
As KPCN tumors ultimately progressed, we evaluated the KPCN proteome to identify compensatory pathways that circumvent loss of ferritinophagy. Enrichment map nodes associated with upregulated proteins included iron uptake and transferrin receptor (Tfrc)-mediated endocytosis (Fig. 5B, Supplementary Fig. S5D). NCOA4 depletion has previously been shown to decrease bioavailable iron in cellular models leading to increased IREB2 levels thereby stabilizing TFRC mRNA for increased TFRC translation (6). Here, we show this compensatory response to Ncoa4 depletion is conserved in an in vivo cancer model with a significant 1.5-fold elevation of Tfrc levels in bulk tumor proteome and in KPCN tumor-derived cell lines (Fig. 5F, G). Likewise, IRP2 and TFRC levels were elevated in NCOA4-depleted human PDAC cell lines (Fig. 4C). To evaluate the potential for compensation of transferrin-mediated iron uptake to rescue proliferation, we tested TCC-Pan2 NCOA4 KD cells for rescue by holo-transferrin demonstrating a partial rescue (Fig. 5H).
Given the partial rescue phenotype, we evaluated the KPCN proteome to identify additional compensatory mechanisms supporting progression of KPCN tumors in vivo. First, we identified upregulation of Cybrd1 (Dcytb), a plasma membrane ferrireductase involved in uptake of non-transferrin bound iron (Fig. 5A). Hmox1 was similarly upregulated in KPCN tumors suggesting heme degradation may compensate for decreased ferritinophagy (Fig. 5A). We evaluated levels of Slc11a2 (Dmt1), an endosomal/lysosomal iron exporter, to determine if there was an upregulation of iron export from endosomes/lysosomes but saw no increase (Supplementary Table 1). Lipocalin-2 (Lcn2) levels were also elevated consistent with upregulation of extracellular iron scavenging by KPCN tumors (Fig. 5A) (32). Intriguingly, LCN2 levels were also significantly elevated in TCC-Pan2 NCOA4 KD proteomes (Supplementary Fig. S5E). SLC40A1 (FPN) is the primary cellular iron export mechanism, and its modulation can affect cellular iron levels. Therefore, we evaluated FPN expression by immunoblot (not detected in proteome) and demonstrated a decrease in expression in TCC-Pan2 and PANC-1 NCOA4 KD cells (Fig. 5I). Fpn immunostaining similarly showed a decrease in tumor-associated staining in KPCN tumors (Fig. 5J). Interestingly, in KPCN tumors, we saw an increase in Fpn staining in cells that appeared to be non-tumor in origin. Tumor associated macrophages (TAMs) are known to affect tumor cell iron metabolism in cancers via elevated Fpn levels to export iron (33,34). Therefore, we evaluated tumors for Cd68+ macrophage immunostaining identifying a significant upregulation of Cd68+ cells in KPCN tumors (Fig. 5K). Taken together, these findings suggest multi-factorial tumor cell autonomous and non-autonomous mechanisms of adaptation to Ncoa4 tumor cell loss to support tumor cell iron metabolism.
A ferritinophagy expression signature is a prognostic marker in PDAC
As few targeted therapies exist for autophagy or iron metabolism, we evaluated the consequences of acute Ncoa4 ablation in formed tumors as opposed to genetic ablation prior to initiation or implantation. We first used inducible RNAi-mediated Ncoa4 knockdown in a syngeneic ‘KPCY’ PDAC tumor cell line model (35) (Fig. 6A, Supplementary Fig. S6A) demonstrating a significant decrease in clonogenicity. We subsequently evaluated this model in the context of an in vivo syngeneic transplant tumor experiment demonstrating a significant decrease in relative growth of Ncoa4 KD tumors in comparison to a non-induced control (Fig. 6B, see methods). Of note, there was no increase in apoptosis in Ncoa4 knockdown tumors (Supplementary Fig. S6B) suggesting the effect of Ncoa4 knockdown was due to a decrease in proliferation, in line with decreased Ki67 staining in KPCN tumors (Fig. 2G).
Figure 6. NCOA4-mediated ferritinophagy expression signature is a prognostic marker in human PDAC.
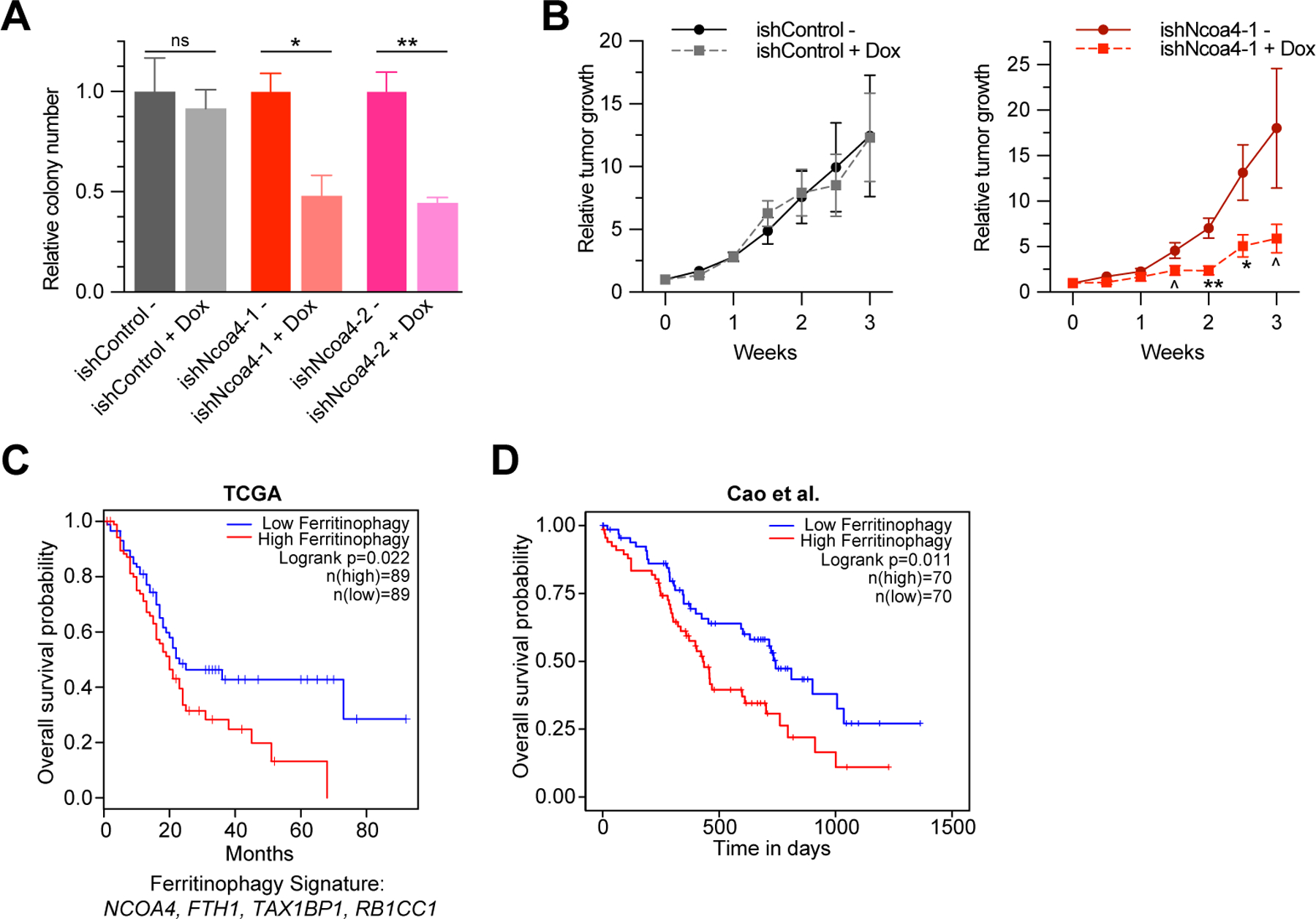
A, Relative clonogenic growth of KPCY (KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre; Rosa26YFP/YFP) murine PDAC cells expressing doxycycline-inducible lentiviral shRNAs: ishControl, ishNcoa4-1, and ishNcoa4-2 treated with or without doxycycline (100 ng/mL). Error bars ± s.d., triplicate wells of a representative experiment of 3 independent experiments, significance determined by two-sided t test. B, In vivo subcutaneous tumor growth in C57BL/6 mice of KPCY ishControl versus ishNcoa4-1 cells with and without doxycycline containing food. Doxycycline was initiated after tumors reached an average of 75 mm3 (Day 0 on graph). Error bars ± s.e.m., n=6 mice per group. Significance determined by one-sided t test. ^ p < 0.1, * p < 0.05, ** p < 0.01. C, Survival analysis with log-rank test of a ferritinophagy gene expression signature (NCOA4, FTH1, TAX1BP1, RB1CC1) in PDAC using TCGA dataset. D, Survival analysis with log-rank test of a ferritinophagy gene expression signature (NCOA4, FTH1, TAX1BP1, RB1CC1) in PDAC using Cao et al. 2021 dataset (37).
To identify the relevance of ferritinophagic flux in human PDAC, we evaluated a ferritinophagy mRNA expression signature derived from the overlap of upstream regulators (RB1CC1, TAX1BP1) and downstream targets (FTH1) with correlated DepMap data (NCOA4, TAX1BP1, RB1CC1, and FTH1 expression levels) in the Cancer Genome Atlas (TCGA) PDAC database (16,36). Low expression of the ferritinophagy signature was significantly correlated with improved overall and disease-free survival in human PDAC (Fig. 6C, Supplementary Fig. S6C). We similarly evaluated the ferritinophagy mRNA signature in PDAC transcriptome data from the Cancer Clinical Proteomics Research (CPTAC) dataset confirming improved survival in patients with lower expression of the signature (Fig. 6D, Supplementary Fig. S6D) (37). Examination of patient outcome based on individual genes in the ferritinophagy signature revealed that the difference in outcome was not driven solely by RB1CC1 expression that may have suggested global autophagy dependence as opposed to ferritinophagy dependence (Supplementary Fig. S6E-L). As NCOA4 was not measured in the proteome in all CPTAC tumors, we evaluated the correlation of FTH1 protein levels with survival demonstrating improved survival of patients with lower FTH1 tumor expression (Supplementary Fig. S6M). Together, these data nominate NCOA4-mediated ferritinophagy as critical for fueling PDAC iron metabolism and tumor progression and suggest a targeted approach for dual selective autophagy and iron metabolism inhibition in PDAC.
DISCUSSION
In this study we used orthogonal approaches to demonstrate that disruption of NCOA4-mediated ferritinophagy impairs PDAC tumor progression and is a potential PDAC therapeutic target. While the role of autophagy in PDAC is well-established, an understanding of which autophagic substrates and corresponding breakdown products are critical for the cell autonomous role of autophagy in supporting PDAC proliferation have remained unclear. A major consequence of autophagy/lysosomal inhibition in PDAC with hydroxychloroquine or by genetic silencing of ATG genes is a defect in mitochondrial oxidative phosphorylation (3). Prior studies have implicated various metabolites derived from autophagic degradation (38) as well as selective mitochondrial degradation (mitophagy) as supportive of mitochondrial oxidative phosphorylation. We propose that iron released via ferritinophagy is a critical component of PDAC autophagy that supports ISC proteins and ultimately mitochondrial function. While the relative contribution of different selective autophagic pathways and breakdown products in PDAC is not evaluated here, studies demonstrate that iron homeostasis is the critical function of lysosomal acidification to support proliferation (39). However, these studies did not evaluate the relative contribution to proliferation of NCOA4-mediated ferritinophagy versus TFRC-dependent transferrin endocytosis, which also requires an acidified endolysosomal environment for iron release.
In loss-of-function cell culture-based screens, cancer cells are more dependent on TFRC than NCOA4 (Supplementary Fig. S6N, Fig. 1E). We speculate that iron endocytosed and released from holo-transferrin is apportioned to at least two pools, a ferritin-bound pool, and the cytosolic labile iron pool. As TFRC loss affects both pools, this may explain the differential yet correlated dependency of TFRC and NCOA4 (Supplementary Fig. S6O). Still, there remain a subset of cells that are highly dependent on NCOA4 and furthermore, in holo-TF limiting culture conditions, an additional dependency on NCOA4 is clear. The relevance of these findings to in vivo tumors where TF may be limited has been unclear. We now demonstrate an in vivo PDAC dependency of the NCOA4-mediated ferritinophagy arm of iron availability. Undoubtedly, TFRC-mediated iron acquisition is critical in vivo for all organs and higher expression is correlated with worse survival in PDAC. However, ablation of TFRC is embryonic lethal (40) and TFRC-targeted therapies have deleterious effects on erythropoiesis. Therefore, identifying cancers with in vivo reliance on NCOA4 could reveal a targeted and tolerable strategy for disrupting tumor iron metabolism given systemic ablation of NCOA4 has minimal effects on physiologic iron metabolism. Our analyses of murine PDAC tumors with Ncoa4 ablation identify a role for NCOA4-mediated ferritinophagy in PDAC progression and credential NCOA4 as a therapeutic target. Our results also show that compensatory upregulation of Tfrc partially rescues iron availability, and we note multiple other putative tumor cell autonomous and non-autonomous compensatory mechanisms that support PDAC iron metabolism in the absence of NCOA4. Future work will define the relative contributions of tumor cell autonomous and non-autonomous iron regulation and may provide additional strategies for targeting iron metabolism in the tumor cell and the tumor microenvironment.
We note that our study represents the first evaluation of NCOA4 ablation in the context of an autochthonous GEMM. Prior studies have evaluated the role of NCOA4 in breast (41), prostate (42), and ovarian cancers (43) but with unclear relevance to in vivo tumor microenvironments. Interestingly, different tumor contexts may have opposite dependencies on ferritinophagy. For instance, a low ferritinophagy signature in renal clear cell carcinoma (RCC) correlated with worse survival (Supplementary Fig. S6P) (44). This suggests that highly ferroptosis sensitive tumors such as RCC have an inverse survival relationship to ferritinophagy levels with high ferritinophagy levels predisposing to increased sensitivity to tumor ferroptosis and thereby better patient survival.
As NCOA4 ablation decreases sensitivity to ferroptosis induction in cell culture models of PDAC, one potential limitation of a strategy targeting NCOA4 is that it may reduce sensitivity to ferroptosis-inducing therapies or more broadly therapies that induce oxidative stress. However, we did not observe any correlation between ferritinophagy signature levels and responsiveness to components of standard of care chemotherapy (gemcitabine/5-FU), as assessed in the Cancer Therapeutics Response Portal (Supplementary Table 3) (45). Further evaluation in in vivo PDAC models with NCOA4 ablation are required to fully address this question. On the other hand, PDAC cell lines transduced with a NCOA4 over-expression plasmid are more sensitive to ferroptosis induction (12). While our murine models of Ncoa4 over-expression highlight the role of Ncoa4 in tumor initiation and progression, a question remains as to the consequences of Ncoa4 over-expression in an established PDAC tumor. Whether strategies to increase NCOA4 levels (such as HERC2 inhibition, Supplementary Fig. S6Q) accelerate tumor progression or induce ferroptosis may depend on the level of NCOA4 induction and presence of additional ferroptotic stimuli. We note that systemic Ncoa4 over-expression is tolerable and does not induce ferroptosis under basal conditions. Similar strategies aimed at inducing increased iron levels to precipitate sensitivity to ferroptosis are being evaluated (46).
The dramatic acceleration in PDAC initiation in KPC-Ncoa4Tg/+ mice suggests a potent interaction between ‘free iron’ and oncogenic KRas. Indeed, a recent study of oncogenic KRas-driven PDAC mouse models fed a high iron diet demonstrated accelerated pancreatic tumorigenesis via a proposed ferroptotic damage mechanism (47). Given the limited survival of KPC-Ncoa4Tg/+ mice, evaluating whether the effects of Ncoa4 over-expression could be rescued with iron chelation was not possible. Future work will evaluate the consequences of increasing ferritinophagic flux in an established tumor in vivo to determine whether upregulated ferritinophagy enhances tumor growth and/or sensitizes to ferroptosis.
Finally, NCOA4-mediated ferritinophagy is further activated under various conditions and suggests there are likely situations where ferritinophagy is further upregulated in PDAC. Our study of temporal proteomic adaptations to oncogenic KRASG12C inhibition identified NCOA4 as highly upregulated suggesting extra-reliance on NCOA4 in response to KRAS pathway inhibition (29). KRAS pathway inhibition and NCOA4 inhibition may therefore represent a potential combination therapy with future development of NCOA4-targeted inhibitors. We note the recent development of a compound reported to bind NCOA4 and inhibit ferritinophagy suggesting proof-of-principle that NCOA4 can be targeted (48). Together, our data identify NCOA4-mediated ferritinophagy as a selective autophagy PDAC dependency that warrants further evaluation as an anti-PDAC therapeutic target.
METHODS
Cell culture
PaTu-8988T, PaTu-8902, JoPaca-1 and YAPC cells were obtained from DSMZ, PANC-1 and HEK-293T cells from ATCC, and TCC-Pan2 cells from JCRB (RRIDs: CVCL_1847, CVCL_1845, CVCL_S507, CVCL_1794, CVCL_0480, CVCL_0063, CVCL_3178). KPCY-2838c3 cell line was a gift from Dr. Stanger (35), now available via Kerafast (EUP013-FP, RRID:CVCL_YM18). KPC/KPCN murine PDAC lines were harvested from a section of tumor, minced and digested in 4% collagenase. Cell lines were maintained in a centralized cell bank, authenticated by assessment of cell morphology as well as STR fingerprinting, and routinely inspected for mycoplasma contamination using PCR (most recently March 2022). After thawing, cell lines were cultured for no longer than 30 days. Cell lines were maintained at 37°C with 5% CO2 and grown in DMEM or RPMI 1640, supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin/Streptomycin, unless otherwise specified. HPDE cells were cultured as previously described (3).
Western blotting
Cells were lysed in RIPA buffer with protease inhibitors, centrifuged and supernatants were collected. 60 μg of protein was resolved on 4–12% SDS-PAGE gels and transferred to nitrocellulose or PVDF membranes. Membranes were blocked in 5% milk, incubated with primary antibodies and then with peroxidase-conjugated secondary antibody. Membranes were developed using the ECL Detection System (Thermo, 32209). Antibodies: NCOA4 (Santa Cruz Biotechnology sc-373739, RRID:AB_10915585, 1:100), FTH1 (Cell Signaling (CS) #4393, RRID:AB_11217441, 1:2000), ACTB (Sigma A5441, RRID:AB_476744, 1:5000), TFRC (Abcam ab84036, RRID:AB_10673794, 1:1000), FPN/SLC40A1 (NovusBio, NBP1-21502SS, RRID:AB_1660489, 1:1000), IREB2/IRP2 (CS #37135, RRID:AB_2799110, 1:100), SDHB (Abcam ab14714, RRID:AB_301432, 1:1000), FECH (Proteintech 14466-1-AP, RRID:AB_2231579, 1:1000), NDUFS3 (Proteintech 15066-1-AP, RRID:AB_2151109, 1:1000), UQCRFS1 (Abcam ab14746, RRID:AB_301445, 1:1000), DPYD (NovusBio, NBP2-92308, RRID:AB_2910205, 1:1000), MAP1LC3B (CS #3868, RRID:AB_2137707, 1:1000), Anti-rabbit IgG (H1L) HRP conjugate (Thermo 31460, 1:3000); Anti-mouse IgG(H1L) HRP conjugate (Promega W4021, 1:7000). Quantification performed using ImageJ (RRID:SCR_003070).
Analysis of TCGA/GTEx data
NCOA4 PDAC and normal pancreas expression levels were obtained from the Cancer Genome Atlas and GTEx databases via the Gene Expression Profiling Interactive Analysis server (16) (RRID:SCR_018294).
Analysis of Cancer Dependency Map
To identify NCOA4 and other gene dependencies across cancer cell lines, we analyzed data from pooled, genome-scale CRISPR-SpCas9 loss-of-function data within the Broad Institute’s Cancer Dependency Map Public 21Q1 release (17,18) (RRID:SCR_017655). NCOA4 dependency scores across 777 cell lines were correlated with gene expression, proteomics, and other CRISPR dependency scores. Pearson’s correlations were performed in R using the cor.test function. P-values were corrected for false discovery using the Benjamini-Hochberg method of the p.adjust function in R and q-values were -log10-normalized. For correlation analyses, the top 100 most correlated genes were analyzed and top positively correlated genes were clustered and visualized using functional associations predicted with STRING (21) (RRID:SCR_005223).
CRISPR-SpCas9 genome editing in cell lines
SgRNA oligonucleotides were cloned into lentiCRISPRv2 (RRID:Addgene_52961). Sequences can be found in the Supplementary Methods. Lentiviral particles were generated by co-transfection into HEK-293T cells with a packaging (psPAX2; RRID:Addgene_12260) and VSV-G envelope plasmid (pMD2.G; RRID:Addgene_12259). Target cells were transduced with lentiviruses. After puromycin selection, pools of cells were used in assays. The pool level depletion of NCOA4 was >90%. To distinguish from clonally selected knockout cells, these pools are referred to in the text as ‘NCOA4 knockdown (KD)’ or ‘pools of cells transduced with lentivirally encoded sgNCOA4’. Rescue was accomplished using lentiviral-based expression of murine Ncoa4 (NM_019744.4) with a ‘pLV-3xFLAG-mNcoa4’ sgNCOA4-resistant plasmid or an empty vector negative control (‘pLV-Empty’) with blasticidin selection. PaTu-8988T NCOA4 knockout (KO) clones were generated as previously described (14) and are referred to as ‘NCOA4 knockout (KO) cells’.
Cell proliferation assay
Cells were plated at 3000–4000 cells/well. For rescue experiments, ferric ammonium citrate (FAC) was added at 50–100 μM, holo-transferrin at 10 μM, and ferrostatin-1 (0.5–1 μM to alleviate deleterious effects of excess free iron, as described (39)). Cells were fixed in 10% formalin and stained with 0.1% crystal violet. Dye was extracted with 10% acetic acid, and relative proliferation was determined by measuring OD at 595 nm.
Clonogenic assay
For clonogenic assays, 400–800 cells/well were plated. After 7–10 days, cells were fixed in 80% methanol, stained with 0.2% crystal violet, and colonies were counted.
Iron quantification in cells
For cytosolic labile iron measurements, 1×105 cells were washed with PBS, stained with Ferroorange in DMEM without serum (1 μM, 30 minutes), washed with PBS, trypsinized and diluted in PBS for fluorescence reading (PE filter) using a Beckman Coulter Cytoflex (RRID:SCR_019627). For mitochondrial iron measurements, cells were washed with HBSS, stained with Mito-FerroGreen (5 μM, 30 minutes), washed with HBSS, and analyzed by flow cytometry.
Subcutaneous mouse xenograft and allograft studies
Animal studies were performed in accordance with Dana-Farber Cancer Institute’s IACUC-approved protocols (10–055, 15–020). Mice were housed in pathogen-free animal facilities at DFCI. For PaTu-8988T NCOA4 KO subcutaneous xenograft experiments, 1×106 cells were suspended in PBS and mixed with Matrigel 1:1 in 100 μL and injected in the flank of female CrTac:NCr-Foxn1nu mice (Taconic:NCRNU-F RRID:IMSR_TAC:ncrnu). For inducible ‘ishNcoa4’ allograft experiments, 4×105 stably transduced ‘KPCY-2838c3’ cells were suspended in PBS and mixed with Matrigel 1:1 in 100 μL and injected in the flank of C57BL/6NCrl female mice (Charles River Laboratories, RRID:IMSR_CRL:027). When tumors reached 75 mm3, mice were randomly assigned to control or doxycycline-containing diets (625 ppm) (n=6/group). Tumor volume was measured twice a week with calipers: volume=(length×width2)/2.
Genetically engineered mouse models
KrasLSL-G12D/+, Trp53fl/+, Pft1a-Cre strains were generated as previously described (49–52) (RRID:MGI:3836620, RRID:IMSR_JAX:008462). Ncoa4fl/fl mice were generated as previously described (10) (RRID:IMSR_JAX:033295). KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre (KPC) and KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre; Ncoa4fl/fl (KPCN) mice were obtained by interbreeding Ncoa4fl/fl mice to the KPC PDAC model maintained on a mixed background. Pft1a-Cre transmission was from the maternal lineage for all crossings. A Ncoa4 overexpression allele was generated by Cyagen in a C57BL/6N and C57BL/6J mixed background. A targeting vector containing a “CAG-loxP-Stop-loxP-mouse Ncoa4 open reading frame-polyA” cassette was cloned into intron 1 of the Rosa26 safe locus in reverse direction using CRISPR-SpCas9-mediated genome engineering (hereafter: Ncoa4Tg/+). The Ncoa4Tg/+ allele was crossed to the CMV-Cre allele (B6.C-Tg(CMV-Cre)1Cgn/J, RRID:IMSR_JAX:006054) to generate Ncoa4Tg/+; CMV-Cre mice on a mixed background. Similarly, the Ncoa4Tg/+ allele was crossed to the KPC and KC models to generate KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre; Ncoa4Tg/+ (KPC-Ncoa4Tg/+) and KrasLSL-G12D/+; Pft1a-Cre; Ncoa4Tg/+ (KC-Ncoa4Tg/+ ) mice on a mixed background. Male and female animals were utilized for experiments with genetically engineered mice and the number of animals used in each experiment is stated in the figure legends.
Tumor identification
Genetically engineered PDAC mice were screened for tumors beginning at 6 weeks of age using abdominal palpation and high-resolution ultrasound (Vevo 3100, RRID: SCR_022152). For ultrasound measurements, tumor volumes were measured by 3D-ultrasound imaging (53). Mice included in the survival analysis were euthanized when endpoint criteria were met, including severe cachexia, ascites, weight loss >15% of initial weight, or extreme weakness/inactivity.
Histology
Hematoxylin and eosin-stained slides were scored in a blinded manner. Percentage area of normal pancreatic parenchyma/acinar structures, regions of acinar-to-ductal metaplasia or pancreatic intraepithelial neoplasia, and pancreatic ductal adenocarcinoma was scored by a gastrointestinal pathologist across the total area of the pancreas. Tumor differentiation was categorized based upon degree of gland formation as moderately, moderately-poorly, or poorly differentiated.
Immunohistochemistry
Tissues were processed as previously described (53). Briefly, tissues were fixed in formalin and paraffin embedded. After deparaffinization, primary antibody was incubated followed by secondary antibody and then developed by DAB. Antibodies: Fth1 (CS #4393, RRID:AB_11217441, 1:500), Cd68 (Abcam ab31630, RRID:AB_1141557, 1:200), Tfrc (Abcam ab84036, RRID:AB_10673794, 1:1000), Fpn (Alpha Diagnostic International MTP11-A, RRID:AB_1619475, 1:200), Ndufs3 (Proteintech 15066-1-AP, RRID:AB_2151109, 1:100), Sdhb (Abcam ab14714, RRID:AB_301432, 1:600), Dpyd (NovusBio NBP2-92308, RRID:AB_2910205, 1:300), Ki67 (Abcam ab15580, RRID:AB_443209, 1:700), Cleaved Caspase-3 (Asp175) (CS #9661, RRID:AB_2341188, 1:400). For quantification of IHC intensity staining more than 5 fields (40x or 20x) per mouse were analyzed.
Iron quantification in tissues
Total non-heme iron levels in tissues were quantified using an iron assay kit (Sigma MAK025) following the manufacturer’s instructions.
Inductively coupled plasma mass spectrometry (ICP-MS)
Metal quantifications by ICP-MS were performed as previously described (54). Tissue samples were digested with 2 mL/g total weight nitric acid for 24h and then digested with 1 mL/g total weight hydrogen peroxide for 24h. Samples were analyzed using a Perkin-Elmer NexION 2000 ICP-MS (RRID:SCR_022153).
Quantitative Proteomics
Mass spectrometry-based proteomics was performed as previously described (29). Cells or tumors were lysed using 8 M urea, 200 mM EPPS, pH 8.5 with protease inhibitors. 50–100 μg of protein extracts were reduced using TCEP and alkylated with 10 mM iodoacetamide followed by chloroform/methanol precipitation. Protein pellets were digested overnight with Lys-C and trypsin digested the next day. 50 μg of peptides were labeled using 100 μg of TMT reagent. To equalize protein loading, a ratio check was performed by pooling 2 μg of each TMT-labeled sample. Pooled TMT-labeled peptide samples were fractionated with basic-pH reverse-phase HPLC. Samples were desalted using StageTips prior to analyses using LC-MS/MS/MS. All MS data was acquired using an Orbitrap Lumos mass spectrometer in-line with a Proxeon NanoLC-1200 UHPLC system (RRID:SCR_020562). Machine settings are noted in the Supplementary Methods section. All acquired data were processed using Comet (55) and a previously described informatics pipeline (28). Spectral searches were done using fasta-formatted databases (Uniprot Human, 2020 or Uniprot Mouse, 2020). Protein quantitative values were normalized so that the sum of the signal for all protein in each channel was equal to account for sample loading.
Bioinformatic analysis
Protein quantification values were processed using Perseus (56) (RRID:SCR_015753) to calculate Log2-fold changes and p-values. Briefly, two-way Welch’s t-test analysis was performed to compare two datasets and correction for multiple comparisons was achieved by the permutation-based FDR method. Proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier: PXD033356 (RRID:SCR_003411) (57). For principal component analysis (PCA), the fold changes of proteins were calculated and were used as inputs. Gene set enrichment analysis (GSEA) was performed with GSEA v3.0 (47) (RRID:SCR_003199) and visualized with EnrichmentMap (3.2.1) (31) (RRID:SCR_016052) in Cytoscape (3.7.2) (58) (RRID:SCR_003032). Gene Ontology (GO) enrichment analyses were performed by using two unranked lists of genes, target list and background lists.
Iron sulfur-cluster activity assays
Ferrochelatase (FECH) activity was performed as previously described (59). Briefly, cell lysates were incubated with mesoporphyrin and zinc acetate at 37°C for 60 minutes. The reaction was terminated, samples were centrifuged at 14,000 × g, and the supernatant was fractionated on a HPLC in tandem with a fluorescence spectrophotometer to measure zinc-mesoporphyrin (400 nm). Cytosolic aconitase activity was quantified using an aconitase assay kit (Cayman 705502) following the manufacturer’s instructions. Mitochondrial respiration was measured using a Seahorse Metabolic Flux Analyzer e96 XF instrument (Agilent). 20,000 cells/well were seeded in the culture media the day prior to the assay. The next day media was exchanged to Seahorse assay media containing 25 mM glucose and 300 mg/L glutamine, adjusted to pH 7.4. The plate was equilibrated for 1 hour in a non-CO2, 37°C incubator and measurements for basal respiration were followed by a mitostress assay with sequential injection of 1μM oligomycin, 2 μM FCCP, and 0.5 μM rotenone/0.5 μM antimycin A.
Transmission Electron Microscopy
Tissue was fixed in 2% Paraformaldehyde/2.5% Glutaraldehyde in 0.1M Sodium Cacodylate Buffer, pH 7.4, washed in 0.1M cacodylate buffer and osmicated in 1% osmium tetroxide/1.5% potassium ferrocyanide, followed by washes of dH2O. 1% uranyl acetate in maleate buffer was added, washed with maleate buffer (pH 5.2), graded cold ethanol series up to 100%, and propylene oxide was added. Tissue was placed in 50:50 propylene oxide:Taab resin including catalyst and embedded in Taab resin mixture. After incubation at 60°C, 80 nm sections were cut with a microtome, picked up on formvar-carbon coated slot Cu grids, stained with 0.2% Lead Citrate, and imaged under a JEOL 1200EX 80 kV Transmission Electron Microscope. Cristae were counted for each mitochondrion (40–100 mitochondria quantified for each mouse, biological triplicates).
RNAi-mediated NCOA4 knockdown
Lentiviral-mediated shRNA knockdown was performed using the ‘Tet-pLKO-puro’ plasmid (RRID:Addgene_21915). siRNAs were transfected using a reverse transfection protocol and Lipofectamine RNAiMAX. Sequences are in the Supplementary Methods.
Chemicals
Deferoxamine mesylate (BioVision, 1883-1000), Ammonium ferric citrate (I72–500; Fisher Scientific), holo-transferrin human (Sigma, T0665), deferasirox (DFX, Selleckchem, S1712), ferrostatin-1 (Cayman, 17729), CQ (Sigma C6628), Ferroorange (Sigma, SCT210), Mito-FerroGreen (Dojindo Molecular Technologies, M489), Vectastain ABC-HRP kit (PK-4001; Vector Labs), MOM Mouse Elite detection kit (PK-2200; Vector labs), DAB Substrate kit (SK-4100; Vector labs).
Statistical Analysis
Differences in survival were assessed using the log-rank test (Mantel-Cox method). No statistical methods were used to predetermine sample size. For comparisons between two groups, Student’s t-test (unpaired, two-tailed or unpaired, one-tailed) was performed. Groups were considered different when p<0.05 or p<0.1, as indicated. For multiple comparisons, one way ANOVA with Tukey’s Statistical analyses were performed using Prism 8 (GraphPad Software, RRID:SCR_002798). R-based analyses and figure generation were performed in R v4.0.3 (RRID:SCR_001905).
Illustrations and diagrams
Drawings detailing experimental set-up were created in Adobe Illustrator (v24.1.2, RRID:SCR_010279) and utilized BioRender.com (RRID:SCR_018361) for portions of the figures.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Autophagy and iron metabolism are metabolic dependencies in PDAC. However, targeted therapies for these pathways are lacking. We identify NCOA4-mediated selective autophagy of ferritin (‘ferritinophagy’) as upregulated in PDAC. Ferritinophagy supports PDAC iron metabolism and thereby tumor progression and represents a new therapeutic target in PDAC.
Acknowledgements
This research was supported by NIH grants R01 DK124384 (J.D.M.), R01 CA148828, R01 CA245546, R01 DK095201 (Y.M.S.), K08 CA218420-02, P50 CA127003, U01 CA224146, and U01 CA250549 (A.J.A.), a Burroughs Wellcome Fund Career Award for Medical Scientists, the Sidney Kimmel Foundation Kimmel Scholar Program, the Manley Family Fund (to J.D.M.), the Lustgarten Foundation, the Doris Duke Charitable Foundation and Pancreatic Cancer Action Network (A.J.A.), and the Hale Family Center for Pancreatic Cancer Research (J.D.M., A.J.A, J.A.N., A.Y.). We acknowledge Xiaoxu Wang for assistance with generating PaTu-8988T NCOA4 knockout clones, Dr. Steven Gygi for use of CORE for MS data analysis software, Dr. Benjamin Stanger for the generous gift of the KPCY-2838c3 cell line, and the Harvard Medical School Electron Microscopy Centers for imaging support. We acknowledge the Perera Lab for fruitful conversations regarding the role of NCOA4-mediated ferritinophagy in PDAC.
Disclosures of Potential Conflicts of Interest:
J.D.M. is an inventor on a patent pertaining to the autophagic control of iron metabolism. A.J.A. has consulted for Oncorus, Inc., Arrakis Therapeutics, Syros Pharmaceuticals, Boehringer Ingelheim, T-knife Therapeutics, and Merck & Co., Inc, and has research funding from Mirati Therapeutics, Syros Pharmaceuticals, Bristol Myers Squibb, Revolution Medicines, Novo Ventures and Deerfield, Inc. that is unrelated to this work.
Data availability
The data generated in this study are available within the article and its supplementary data files. All genome-scale dependency data used in this analysis are publicly available for download at depmap.org. All tumor and normal expression and survival data are publicly available via the Gene Expression Profiling Interactive Analysis server: gepia2.cancer-pku.cn/#index. Raw proteomics data generated in this study have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier: PXD033356 (RRID:SCR_003411). Requests for resources and reagents should be directed to Dr. Joseph Mancias (joseph_mancias@dfci.harvard.edu).
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019 (US statistics). CA-Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Jiang H, Muir RK, Gonciarz RL, Olshen AB, Yeh I, Hann BC, et al. Ferrous iron-activatable drug conjugate achieves potent MAPK blockade in KRAS-driven tumors. J Exp Med 2022;219:e20210739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Gene Dev 2011;25:717–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santana-Codina N, Mancias JD, Kimmelman AC. The role of autophagy in cancer. Annu Rev Cancer Biol 2017;1:19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discov 2014;4:905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014;509:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 2014;16:1069–79. [DOI] [PubMed] [Google Scholar]
- 8.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis : An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellelli R, Federico G, Matte A, Colecchia D, Iolascon A, Chiariello M, et al. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep 2016;14:411–21. [DOI] [PubMed] [Google Scholar]
- 10.Santana-Codina N, Gableske S, Quiles del Rey M, Małachowska B, Jedrychowski MP, Biancur DE, et al. NCOA4 maintains murine erythropoiesis via cell autonomous and non-autonomous mechanisms. Haematologica 2019;104:1342–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das NK, Jain C, Sankar A, Schwartz AJ, Santana-Codina N, Solanki S, et al. Modulation of the HIF2α-NCOA4 axis in enterocytes attenuates iron loading in a mouse model of hemochromatosis. Blood 2022;139:2547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016;12:1425–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Sun G, Chen B, Xu L, Ye Y, He J, et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol Res 2021;174:105933. [DOI] [PubMed] [Google Scholar]
- 14.Mancias JD, Vaites LP, Nissim S, Biancur DE, Kim AJ, Wang X, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015;4:e10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paulo JA, Mancias JD, Gygi SP. Proteome-Wide Protein Expression Profiling Across Five Pancreatic Cell Lines. Pancreas 2017;46:690–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 2019;47:W556–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell 2017;170:564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bardeesy N, Aguirre AJ, Chu GC, Cheng K-H, Lopez LV, Hezel AF, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA 2006;103:5947–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee H-J, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020;368:85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodwin JM, Dowdle WE, DeJesus R, Wang Z, Bergman P, Kobylarz M, et al. Autophagy-Independent Lysosomal Targeting Regulated by ULK1/2-FIP200 and ATG9. Cell Rep 2017;20:2341–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maio N, Rouault TA. Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis. Trends Biochem Sci 2020;45:411–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill-Burger J, et al. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat Commun 2019;10:5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004;113:1271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J Clin Invest 2017;127:1786–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujimaki M, Furuya N, Saiki S, Amo T, Imamichi Y, Hattori N. Iron Supply via NCOA4-Mediated Ferritin Degradation Maintains Mitochondrial Functions. Mol Cell Biol 2019;39:e000010–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mcalister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin L, Erickson BK, et al. MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal Chem 2014;86:7150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santana-Codina N, Chandhoke AS, Yu Q, Małachowska B, Kuljanin M, Gikandi A, et al. Defining and Targeting Adaptations to Oncogenic KRASG12C Inhibition Using Quantitative Temporal Proteomics. Cell Rep 2020;30:4584–4599. [DOI] [PubMed] [Google Scholar]
- 30.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merico D, Isserlin R, Stueker O, Emili A, Bader G. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 2010;5:e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santiago-Sánchez GS, Pita-Grisanti V, Quiñones-Díaz B, Gumpper K, Cruz-Monserrate Z, Vivas-Mejía PE. Biological Functions and Therapeutic Potential of Lipocalin 2 in Cancer. Int J Mol Sci 2020;21:4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marques O, Porto G, Rêma A, Faria F, Cruz Paula A, Gomez-Lazaro M, et al. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer 2016;16:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Torti SV, Torti FM. Iron and Cancer: 2020 Vision. Cancer Res 2020;80:5435–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018;49:178–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao L, Huang C, Cui Zhou D, Hu Y, Lih TM, Savage SR, et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 2021;184:5031–5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 2016;30:1704–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber RA, Yen FS, Nicholson SPV, Alwaseem H, Bayraktar EC, Alam M, et al. Maintaining Iron Homeostasis Is the Key Role of Lysosomal Acidity for Cell Proliferation. Mol Cell 2020;77:645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet 1999;21:396–9. [DOI] [PubMed] [Google Scholar]
- 41.Wu X, Chen F, Sahin A, Albarracin C, Pei Z, Zou X, et al. Distinct function of androgen receptor coactivator ARA70alpha and ARA70beta in mammary gland development, and in breast cancer. Breast Cancer Res Treat 2011;128:391–400. [DOI] [PubMed] [Google Scholar]
- 42.Ligr M, Li Y, Zou X, Daniels G, Melamed J, Peng Y, et al. Tumor suppressor function of androgen receptor coactivator ARA70alpha in prostate cancer. Am J Pathol 2010;176:1891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rockfield S, Flores I, Nanjundan M. Expression and function of nuclear receptor coactivator 4 isoforms in transformed endometriotic and malignant ovarian cells. Oncotarget 2018;9:5344–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mou Y, Wu J, Zhang Y, Abdihamid O, Duan C, Li B. Low expression of ferritinophagy-related NCOA4 gene in relation to unfavorable outcome and defective immune cells infiltration in clear cell renal carcinoma. BMC Cancer 2021;21:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol 2016;12:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 2021;22:266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai E, Han L, Liu J, Xie Y, Zeh HJ, Kang R, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun 2020;11:6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang Y, Chen X, Tan Q, Zhou H, Xu J, Gu Q. Inhibiting Ferroptosis through Disrupting the NCOA4-FTH1 Interaction: A New Mechanism of Action. ACS Cent Sci 2021;7:980–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001;15:3243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 2001;29:418–25. [DOI] [PubMed] [Google Scholar]
- 51.Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CVE. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet 2002;32:128–34. [DOI] [PubMed] [Google Scholar]
- 52.Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov 2018;8:276–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biancur DE, Paulo JA, Małachowska B, Del Rey MQ, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun 2017;8:15965–15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi E-K, Aring L, Das NK, Solanki S, Inohara N, Iwase S, et al. Impact of dietary manganese on experimental colitis in mice. FASEB J. 2020;34:2929–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eng JK, Hoopmann MR, Jahan TA, Egertson JD, Noble WS, MacCoss MJ. A deeper look into Comet--implementation and features. J Am Soc Mass Spectrom 2015;26:1865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 2016;13:731–40. [DOI] [PubMed] [Google Scholar]
- 57.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019;47:D442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shannon P, Markiel A, Ozier O, Baliga N, Wang J, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taketani S Measurement of ferrochelatase activity. Curr Protoc Toxicol. 2001;Chapter 8:Unit 8.7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files. All genome-scale dependency data used in this analysis are publicly available for download at depmap.org. All tumor and normal expression and survival data are publicly available via the Gene Expression Profiling Interactive Analysis server: gepia2.cancer-pku.cn/#index. Raw proteomics data generated in this study have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier: PXD033356 (RRID:SCR_003411). Requests for resources and reagents should be directed to Dr. Joseph Mancias (joseph_mancias@dfci.harvard.edu).