

1. Introduction
There exist limited data to inform mechanism-based understanding and management of chronic pain, and thus there is a clear unmet need to better define the molecular mechanisms of pain in order to develop novel treatment strategies. Generating knowledge regarding molecular pathophysiology of pain states using human cohorts has an obvious advantage over animal models, and can be achieved using molecular and cellular genome-wide approaches in cohorts that have been characterized for different pain states and different pain-related intermediate phenotypes. Crucially, such approaches are hypothesis-free, and thus both heuristic and resulting in the unbiased interpretation of data, not restricted by or funnelled through specific hypotheses or gene candidates. There has been tremendous progress lately in the development of unbiased molecular screening approaches and analytic tools [67]. These multi-omics approaches include molecular assays of DNA, RNA, proteins, and small molecules in a high-throughput, comprehensive manner. Their use has advanced and even changed our understanding of the pathophysiology of many diseases including stroke, diabetes, and cancer [27]. Importantly, -omics approaches permit the systematic use and integration of multiple datasets, creating further dimensionality in the interpretation of the results that can lead to the development of new conceptual approaches and treatment strategies.
In this review, we will discuss these approaches for studying the molecular pathophysiology of pain states at molecular and genetic levels using three recent examples from our laboratories, describing both the -omic techniques identifying hypotheses and the animal model mechanistic studies that follow. Overall, these findings support the critical role of neuro-immune interactions in pain resolution processes that involve, over time, different subsets of immune cells.
2. Genome-Wide Association Analysis of Pain Phenotypes
At the level of DNA, whole-genome or exome sequencing [23], genome-wide genotyping [80], and DNA methylation assays [2; 89] have been rapidly developing. Genotyping platform development for genome-wide association studies (GWASs) was the first-developed high-throughput -omics approach, and as such today there exist a large number of published pain-relevant GWASs. The GWAS approach identifies genetic variants with relatively high frequency in the population and associates them statistically with either the presence or severity of a particular disease. The genetic variants, called alleles, are either single nucleotide polymorphisms (SNPs) or indels (insertion or deletion of nucleotides). Variants considered in GWAS studies generally appear in the study population at relatively high frequencies, typically >1%, whereas lower-frequency alleles would be termed rare variants, which are usually identified using direct DNA sequencing approaches. Functional annotation of the variants establishes the consequences of each allele at each variant that are found inside a gene’s locus. Examples of the impacts of variants in a protein-coding gene—which can be dramatic or subtle—include single amino acid substitution, insertion of a premature stop codon, or alteration in the expression levels (so-called expression quantitative trait loci, eQTLs).
The first pain GWAS was published in 2010, on migraineurs [1]. Multiple published migraine GWASs, each bigger than the last, have to date identified 38 genetic variants and 123 genetic susceptibility loci [14]. In the beginning, emphasis was placed on the genome-wide significance of the finding, and tying each genetic variant to a particular gene. Over time, the substantial polygenic nature of pain phenotypes (similar to other complex phenotypes) became apparent, undermining the importance of any individual genes and demanding new types of integrative analyses.
For migraine, a meta-analysis of 22 migraine GWASs [24] first allowed an unbiased reconstructing of the pathophysiology of migraine. Since strong arguments for both vascular and neuronal mechanisms of migraine existed [62], it was tempting to use GWAS results to try to establish which of these is the exclusive or at least major driver of migraine development. The first migraine-associated variants provided much stronger support for the involvement of vascular and smooth muscle dysfunction in migraine, with only 5 loci out of 38 containing neuronal (ion channel) genes. However, the latest even larger GWAS meta-analysis on 102,084 migraine cases and 771,257 controls [28] showed equal support for both vascular and neuronal contribution to the pathophysiology of migraine, suggesting that instead of contrasting these hypotheses we perhaps should embrace the possibility of their interacting contribution, or that migraine may have more substantial heterogeneity than previously thought.
Existing GWASs for other chronic pain syndromes include back pain [6; 20; 73], shoulder pain [10; 49], temporomandibular disorder (TMD) [66; 70], multisite pain [33–35], neuropathic pain [55; 64], and other chronic musculoskeletal pain conditions [61; 63; 72; 79]. Functional analyses of these GWASs has provided critical information on the pathophysiology of chronic pain. These functional analyses derive mostly from the annotating the corresponding genes [60; 88]; partitioning heritability [18; 19], a method which estimates enrichment of corresponding gene expression in specific tissues; and, pathway analysis, an approach which tests for the enrichment of the corresponding genes in predefined biological pathways like those in the Gene Ontology (GO) dataset [3]. Such functional analyses revealed the strong contribution of the central nervous system to the pathophysiology of back pain [20], neck/shoulder pain [49], and multisite pain [33]. Genes expressed in the muscle and skeletal tissues were found to contribute to the pathophysiology of back pain [6; 20] and chronic widespread musculoskeletal pain [63]. Finally, immune system genes were found to contribute to back pain [6; 57], TMD [57; 70], and shoulder impingement syndrome [10]. The most recent GWAS meta-analysis on seventeen pain susceptibility traits reveals equal neuronal and immunological etiology for pain susceptibility [51].
Further analyses of different sub-clusters of human pain conditions have provided additional critical information on the pathophysiology of chronic pain. It seems that chronic versus acute pain, as well as chronic overlapping pain conditions (COPC) versus single body site pain, are mediated by different biological pathways. For example, genes mapped to variants associated with chronic back pain, but not acute back pain, were found to be significantly enriched in the pathways for neurogenesis and synaptic plasticity. In contrast, connective tissue and bone remodeling pathways, cardiac muscle depolarization, and immune response via Th2-helper cells were enriched in acute back pain, but not at all in chronic pain back [6]. Whereas a report of chronic single-site pain did not identify enrichment in any GO biological process pathway, the GWAS on COPC identified a total of 60 pathways, with the overwhelming majority of the pathways involved in neural function and development [35]. The other significant pathways included the contribution of the immune system, such as regulation of monocyte differentiation, and vascular system development, such as aorta morphogenesis. Importantly, a further functional analysis pointed to axonogenesis in brain tissues as a major contributor to COPC. This observation was also supported by multimodal structural brain imaging, demonstrating that the top associated gene in this analysis, DCC netrin-1 receptor (DCC), is strongly expressed in subcortical limbic regions and is associated with alterations in the uncinate fasciculus microstructure, suggesting that DCC-dependent axonogenesis may contribute to COPC via cortico-limbic circuits (Figure 1) [35].
Figure 1. The contribution of axonogenesis to chronic overlapping pain conditions (COPCs) based on the results from genome-wide association studies (GWASs) and brain imaging data (based on Khoury etal. 2022).
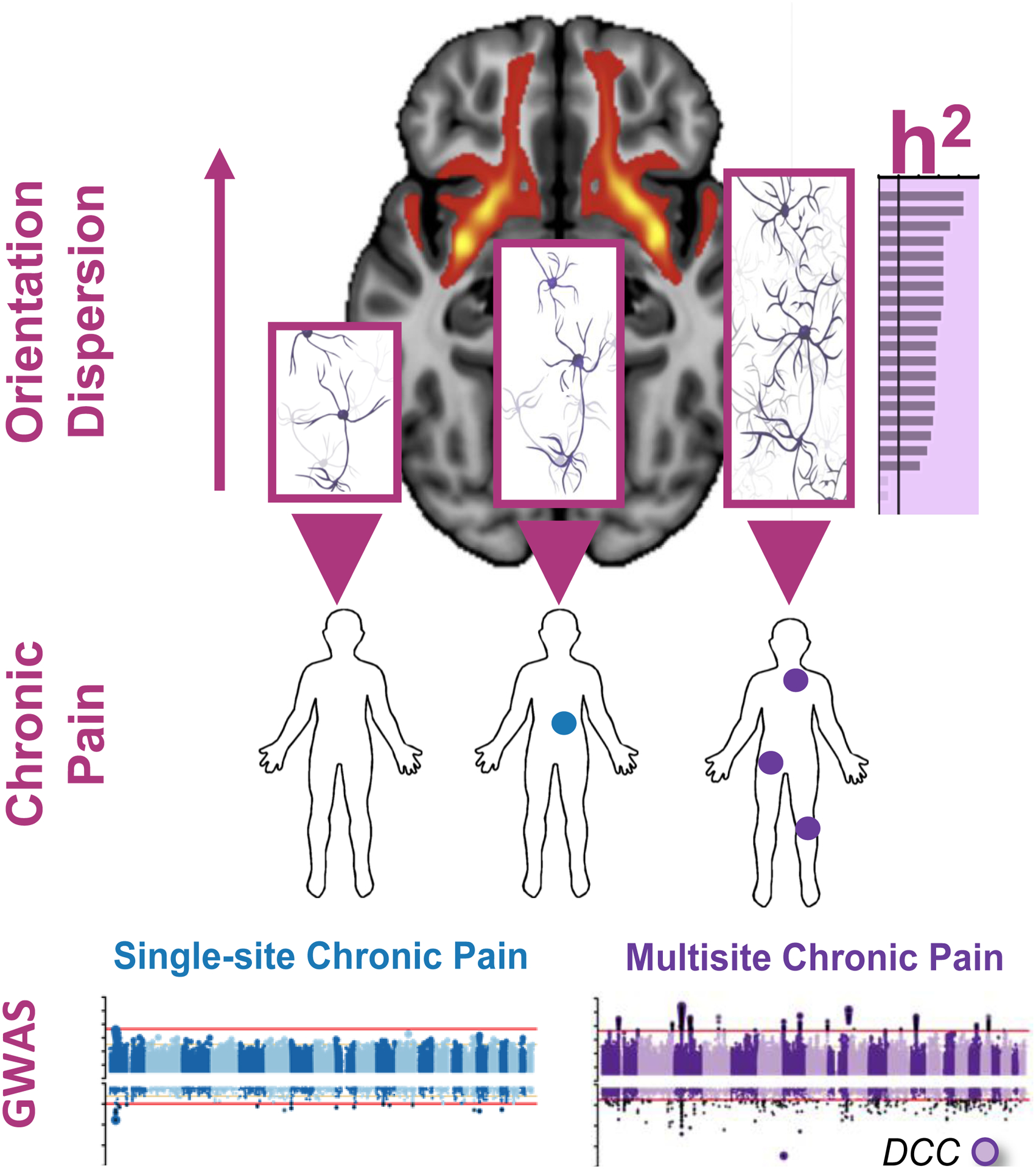
GWASs were conducted in human subjects reporting chronic pain at a single body site (N=1 site, blue) or multiple body sites (N≥2 sites, purple), compared with human subjects not reporting any pain (N=0). The DCC Netrin 1 Receptor (DCC) gene was found to be strongly associated with multisite chronic pain but not with single-site chronic pain. DCC is a gene that critically contributes to axonogenesis, the process of steered development of neuronal axons towards their synaptic targets. Consistent with the association of DCC gene with chronic multisite pain, the GWAS-based pathways analyses identified axonogenesis in brain tissues as the major contributing pathway to chronic multisite pain. Genetic partitioned heritability (h2) analysis indicated a significant enrichment of heritability in SNPs at loci of genes exclusively expressed in brain regions for multisite chronic pain but not for single-site chronic pain. Brain imaging data identified increased neurite orientation dispersion (OD) with an increased number of chronic pain sites. The OD of neurites can range from highly parallel (coherently oriented white matter structures) to highly dispersed (grey matter structures characterized by sprawling dendritic processes in all directions), as suggested by the neuronal networks inside the bar plot’s bars. OD is shown for the uncinate fasciculus (UF) brain sub-structure. The background brain image shows evidence for DCC expression in the UF. Together, our results suggest that genetically determined DCC-dependent disorganization in axonal tracks in patients with chronic overlapping pain conditions may contribute to pathgenesis of disease via corticolimbic circuits.
In addition to understanding biological pathways contributing to pain states, GWAS-derived heritability estimates—the proportion of variation in pain phenotypes due to genes versus the environment—has provided valuable information. Contrasting chronic and acute back pain [6] demonstrates that the heritability of chronic back pain (4.6%) is much higher than for acute back pain (0.8%), and, similarly, the heritability of COPC (19%) is much higher than the heritability of pain in any single chronic body site (1–10%) [35].
Among other insights provided by the analyses of human GWASs to the pathophysiology of chronic pain is a strong genetic correlation between different chronic pain conditions. In line with existing epidemiological data on the comorbidity between different pain conditions [69], this genetic correlation was the strongest for the physically proximal pain sites [35]. Also, in line with previous observations in twin studies [82], headaches demonstrated the smallest genetic correlations with any other chronic pain site, suggestive of distinct pathophysiology relative to other chronic pain conditions. Furthermore, genetic correlations between chronic pain and other comorbid conditions have been reported. Psychiatric phenotypes showed the strongest genetic correlations, including depressive symptoms, neuroticism, anxiety, schizophrenia, and post-traumatic stress disorder [6; 20; 33; 35]. Importantly, GWAS-based Mendelian Randomization, an analysis that investigates causal relationships between phenotypes, identified an effect of chronic multisite pain on major depressive disorders, but not the other way around [33]. Other reported genetic correlations with chronic pain include immune and sleep disorders [6; 20; 33]. These results provide a molecular genetic basis for pain-related comorbidities previously reported by epidemiological studies.
Sex-specific analyses of GWASs of pain conditions have also provided important insights. For both back pain [21] and multisite pain [34], the genetic correlation between male- and female-specific association results were very high: 0.84 and 0.92, respectively. Nonetheless, the exact list of statistically significant SNPs looked quite different between males and females. On the one hand, these results suggest that the genetic mechanisms contributing to chronic pain susceptibility are largely shared by men and women. On the other hand, the results suggest that different pathways contribute to pain with different relative strengths in a sex-specific fashion. Limited efforts have been made in these two studies to elaborate on the true sex-specificity of identified SNPs beyond measuring the relative strength of association. As sex-specific differences in pain mechanisms continue to be identified in animal models, further efforts in identifying genetic sex differences are required. In particular, it will be interesting to highlight differences not only at the SNP-level, but also at the gene-, pathway-, and even at the tissue-level as well. Furthermore, since sex chromosomes have been excluded from the reported analyses due to technical difficulties with analyses of the sex chromosomes in GWASs [36], their inclusion in future analyses may provide more insights into sex-specific pain mechanisms
Finally, many more genetic avenues are left to be explored for a complete understanding of pain. Of these, polygenic risk scores have become increasingly popular to summarize the effects at hundreds or thousands of genetic loci into a single score [78]. The scores are not only useful for diagnostic and prognostic purposes, but also to highlight the shared genetic burden of painful conditions [40; 42; 81]. Alongside genes, the environment also has a great deal of say in the variability of pain sensitivity as well as in the development of painful conditions. Moreover, there are also interactions between genetics and environmental factors (termed GxE), which are now possible to investigate using genome-wide by environment interaction studies (GWEIS) [90]. For pain, exposure to chemicals known to affect the nervous or immune systems and traumas of any kind are of particular interest. At the level of the single gene, even a gene-sex-environment interaction for stress response was reported for COMT [48] and vasopressin [53]. Most large genetics cohorts have focused on populations of European ancestry, but arguments for inclusion of diverse racial/ethnic groups have been proposed [5], despite challenges in conducting GWAS in diverse or admixed populations (e.g. [47]), prompting renewed interest in methodological developments (e.g. [41; 59]).
3. Transcriptome Profiling of Pain Phenotypes
High-throughput measurement of gene expression at the transcriptome-wide level (i.e., measurement of all mRNAs expressed in a tissue) was first demonstrated in 1996 by Derisi et. al. [13] using a method that allowed very high-density cDNA arrays on glass substrates, although cDNA microarray technologies date back to the 1980s [8; 71]. Fueled by the fast development of human genome sequencing technologies at the turn of the 21st century, an approach to sequence short nucleic acid fragments (DNA or RNA) in a high-throughput fashion was developed: so-called next-generation deep-sequencing, which is usually referred to as RNA-seq for transcriptomics [23; 45]. Even though both technologies quantify gene the RNA-seq approach has many advantages. Analysis of publicly available gene expression data from pain phenotypes in the Gene Expression Omnibus [16] indicated the popularity of cDNA microarrays in early pain-relevant studies, with the focus shifted now towards RNA-seq as costs diminish. Now, gene expression at the level of the single cell in a particular tissue (e.g., dorsal root ganglia [45; 86] called single cell RNA-seq (scRNA-seq) yields even more detailed results [37; 74]). Noteworthy is the fact that RNA-seq not only permits gene quantification at the level of transcription (transcriptomics), but also at the translation level (translatomics) via sequencing of ribosome-protected fragments or Ribo-seq [30]. The validity of transcriptional analyses as a substitute for direct protein quantification or indirect ribosomal footprints is suggested by their highly correlated measurements [87], although the method is blind to post-transcriptional and post-translational gene regulation.
Fewer studies have tested human samples characterized for pain conditions at the transcriptome level (i.e., RNA expression) in comparison with the genome level (i.e., DNA variants). A concern remains that the only tissue readily available for human transcriptomics studies – peripheral blood – is not fully relevant to pain conditions. However, increasing evidence suggests that the pathophysiology of chronic pain involves an interplay between the nervous and immune systems [31]. For example, when eQTL analysis of human dorsal root ganglia were mapped to human pain GWASs [57], the strongest evidence for overlap was the human leukocyte antigen (HLA) locus containing MHCII genes. This newly identified role of MHCII genes in the development and maintenance of pain states was validated in a mouse pain model [57], suggesting that the MHCII complex contributes to successful pain resolution after injury, and highlighting the critical role of the immune system in the pathophysiology of pain states. In general, neuro-immune interactions are thought to contribute to the development of pain states and are possibly especially important in the first stages of the acute-to-chronic pain transition[9; 32; 50].
A number of transcriptome profiling studies have been performed in humans to help decode the molecular pathways responsible for pain states. In particular, Guo and colleagues studied the stages in rheumatoid arthritis to understand the disease’s progression, and more specifically inflammation markers [26]. Changes in the transcriptomes between osteoarthritis patients with and without pain were tracked by Bratus-Neuenschwander and co-workers [7]. In this study, proteins promoting neuronal cell survival under stress were found to be enriched in top differentially expressed genes, alongside other genes modulating GABAergic activity. Theken and collaborators [77] investigated Ibuprofen’s efficacy in humans in relation to transcriptional changes in third molar extraction setting, highlighting the role of cyclooxygenase activation. Held and colleagues looked at differences in immune-related expression patterns in patients with sustained peripheral nerve lesions that did or didn’t feature neuropathic pain [29]. They found that neuropathic pain patients exhibited a systemic proinflammatory gene expression pattern. Finally, whole blood transcriptomes of healthy subjects were compared with those with acute or chronic back pain [15]. It was found that genes of the extracellular matrix, from the major histocompatibility complex (MHC) locus, and those participating in mitochondria’s oxidative phosphorylation pathway defined a unique genomic signature in the risk for chronic back pain.
Our recent prospective whole-transcriptome analysis of peripheral immune blood cells of patients with acute low back pain (LBP) provided new and unexpected insights into the pathophysiology of the acute-to-chronic pain transition [58]. We found that patients who resolve their acute back pain within three months after the acute episode have thousands of genes that change their expression levels over this time period. However, no statistically significant transcriptional changes were identified in the blood of the patients whose pain persisted at three months. These transcriptional change dynamics were mirrored by identified changes in white blood cell composition, and only in the blood of the patients with resolved pain: their neutrophils and mast cells precursors were decreased in number, and T- and natural killer (NK) cells were increased over the 3-month observation period.
We next found that one of the strongest biological processes that drive these dynamic changes is the transient neutrophil-dependent upregulation of inflammatory responses. There was a strong correlation between transcriptional processes in the subjects who resolve their pain and those who don’t; however, in patients who resolve their pain these processes are about 30-fold more active. We validated these results in an independent cohort of temporomandibular disorder (TMD) patients. Unlike the LBP cohort, the TMD cohort also contained healthy controls. Comparing the transcriptomics of the recent onset TMD, chronic TMD and healthy controls, as well with existing data of inflammatory response at different stages of chronic pain development [68; 83], we concluded that although it is the patients with resolved pain that have robust upregulation of the inflammatory response at acute pain stage, the patients who will not resolve their pain display a higher inflammatory state prior to tissue damage. Furthermore, patients with persistent pain continue to increase their inflammatory state over time (Figure 2). Overall, our analysis suggested that there is an active, adaptive component of the immune system that protects against a transition to chronic pain. Inefficiency in these processes—either intrinsic or drug-induced—and, in particular, in the transient inflammatory response in humans with physical injury may lead to pain chronification.
Figure 2. Inflammatory response contribution to acute pain resolution based on the results from transcriptomics-wide profiling of a prospective cohort of acute low back pain (LBP) patients (based on Parisien et al. 2022).
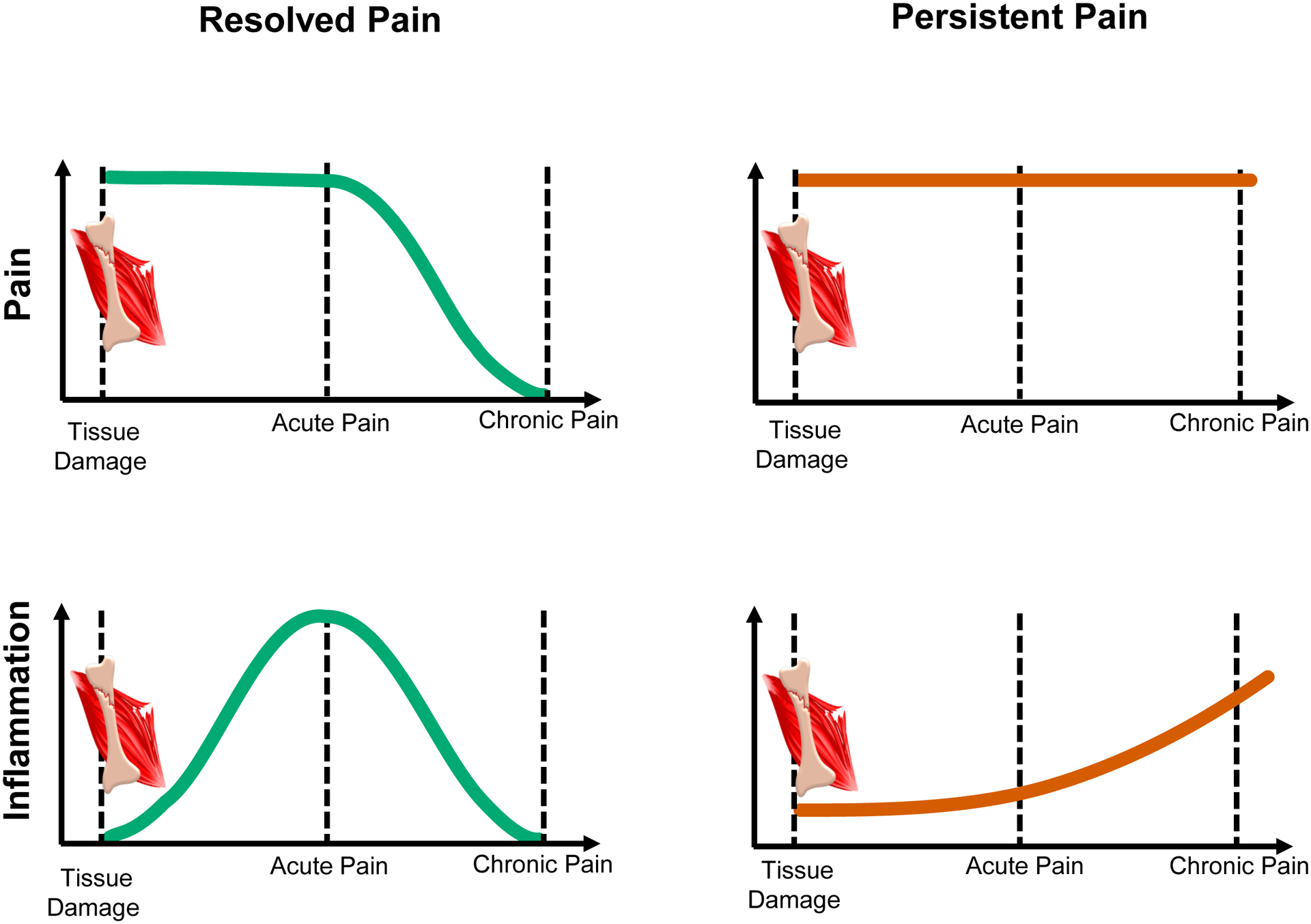
Human patients incur tissue damage eliciting back pain. After an acute pain period, pain in some patients resolves, the “resolved pain” group (left, green), whereas for others the pain persists, the “persistent pain” group (right, red). Evidence from transcriptomics studies indicates different time trajectories for the inflammatory response in the two groups (bottom row). During the acute pain stage, the inflammatory response in the resolved pain group is stronger than for the persistent pain group. With time, the inflammatory response substantially lessened in patients with resolved pain (left). In patients with persistent pain, the inflammatory response is already elevated at the time of injury but does not rise during the acute pain stage, however transitions to a low-grade inflammatory state over time (right). The inflammatory response seems to coincide with neutrophil count and activation. Together, the acute-to-chronic pain transition features the absence of active processes of upregulation and subsequent resolution of inflammation.
We tested this counterintuitive hypothesis in mouse pain assays. We found that early treatment with the steroid drug, dexamethasone, or a non-steroidal anti-inflammatory drug (NSAID), but not other analgesics, greatly prolongs the duration of mechanical allodynia caused by complete Freund’s adjuvant (CFA), chronic constriction injury (CCI) of the sciatic nerve, or injection of nerve growth factor (NGF) into the muscles of the low back. Experimental depletion of neutrophils also greatly delayed the resolution of pain behavior, whereas peripheral injection of neutrophils (or the S100A8 and S100A9 “alarmin” compounds released by neutrophils) prevented the development of long-lasting pain.
Finally, we analyzed the reported back pain trajectories in the large UK Biobank cohort. We found that human subjects reporting acute back pain at the baseline data collection time point were much more likely to report having chronic back pain 2–6 years later if they were taking NSAIDs in comparison with those who were not. This relationship was not observed for back pain sufferers taking antidepressants or acetaminophen (paracetamol).
In summary, our transcriptome-wide analysis of longitudinal data of the blood of human patients with acute back pain provided important insight into the pathophysiology of the acute-to-chronic pain transition. A heuristic model based on our findings is presented in Figure 2. Overall, our results have several translational ramifications, the most immediate of which is that despite high analgesic efficacy at early time points, the inhibition of acute inflammatory response in pain management may be counterproductive for long-term outcomes of pain patients.
The results of these studies let us generate new hypotheses regarding the pathophysiology of chronic pain and provide new frameworks to think about chronic pain development. That is, although we generally think about chronic pain as an active pathological process, it might be better conceptualized as resulting from the absence of an active adaptive process. We generally try to find the ”pain genes”, when it might be more accurate to think in terms of “pain pathways”. We generally measure and value pain intensity, and forget about the equal or greater importance of pain duration. We generally think about pain resolution as a linear process, when it might be better conceptualized as an inverted U-shaped process, in which one biological process can go through a full temporal cycle resulting in the initiation of the next one. These results also pose a new set of questions. What are the dynamics of this cascade of immune responses? When does the anti-inflammatory response start to take place? What are the triggers for the adaptive immune response? Which pharmacological target(s) with analgesic effects can be beneficial and safe for people with a painful injury?
4. Unbiased Immune Profiling of Pain Phenotypes
The final example presented in this review is based on the results of unbiased immune profiling of peripheral blood mononuclear cells (PBMCs) of fibromyalgia patients compared to healthy controls [84]. Although there is solid evidence regarding the contribution of the immune system to fibromyalgia development, existing studies have typically focused on a specific immune cell subset [4; 11]. This is true for other chronic pain conditions as well, with the largest focus overall on macrophages/monocytes, T-cells, and mast cells [39; 43; 56]. Technological limitations are perhaps an important reason for this cell type-focused approach. Although multicolor fluorescence-based flow cytometry was introduced approximately four decades ago and has proven to be an extremely effective tool for the phenotypic characterization and functional analysis of immune cells, it still cannot capture all immune cells subtypes in one run [80]. The number of parameters that can be measured simultaneously has steadily increased, resulting in higher throughput and possibility of capturing more and more immune cell subtypes. However, the integration of other molecular approaches, such as RNA-seq and multiplexed functional assays with higher high-throughput capacities, has been proposed as a method of further expansion [80].
In our recent study, we tested PBMCs of fibromyalgia patients via multicolor flow cytometry in a case-control design. Our analysis used markers for cytokine production, chemokine receptor expression, and cell surface markers that allowed us to distinguish all major blood cell types and many of their subtypes [84]. In this hypothesis-free comparison, we found that fibromyalgia patients have fewer circulating natural killer (NK) cells, and increased numbers of circulating B cells. As the changes in the NK cells were stronger, we concentrated our study on NK cells. It is interesting to note, though, that neither NK nor B cells have been considered “major suspects” in the pathophysiology of fibromyalgia or other chronic pain conditions.
To characterize circulating NK cells from fibromyalgia patients, we tested them via a set of known surface markers and cytokines. To our surprise, we found that even though the NK cells are depleted in the blood of fibromyalgia patients; they are hyperactivated, yet exhausted. These conclusions are based on the decreased surface expression of resting NK cell markers, namely, CD16, CD96, and CD226, and increased surface expression of degranulation marker, CD107a, and cell exhaustion marker, TIGIT. The hyperactivation of NK cells in fibromyalgia patients was also confirmed through the functional in-vitro activation assays. When co-cultured with HLA-null target cells, fibromyalgic NK cells showed increased production of the chemokine CCL4 and increased expression of CD107a. Following our immunophenotyping experiments, we also performed GWAS and RNA-seq analyses of whole blood samples of both fibromyalgia patients and healthy controls. Our genetic and transcriptomic pathway analyses confirmed the immune profiling conclusions.
Looking for an explanation for the observed cellular phenotype of NK cells in fibromyalgia patients, we hypothesized that they may be redistributed somewhere else in the body (i.e., out of the bloodstream) where they are chronically activated. Since infiltration of cytotoxic NK cells into the sciatic nerve following peripheral nerve injury has been recently reported in mice [12], we explored if the depletion of circulating NK cells in the blood of fibromyalgia patients may be associated with their recruitment to and consequent degeneration of peripheral nerves. Such a hypothesis would be also in line with the previously observed presence of small-fiber neuropathy in fibromyalgia patients [17; 25; 46].
We tested this hypothesis in an independent cohort of fibromyalgia patients. Indeed, relative to healthy controls, the skin biopsies of fibromyalgia patients showed increased expression of the NK cell activation ligand, UL16-binding protein (ULBP), on the subepidermal nerves. This elevated expression of ULBP was correlated with an increased number of NK cells near the peripheral nerves, and reduced epidermal nerve density. Usually, the expression of these ligands is restricted in normal tissues, but they are known to be expressed in infected, malignant, or stressed cells, marking those cells for removal through the NK cell cytotoxic mechanism [22]. Importantly, in the mouse model, an endogenous ligand for NK cells was shown to be expressed in DRG neurons following peripheral nerve injury, triggering selective degeneration of injured axons [12]. It remains a question why DRG neurons in fibromyalgia patients marked themselves, via ULBP, as damaged and needing to be removed. Moreover, it is unclear if ULBP expression is a cause or consequence of the nerve damage in fibromyalgia patients. Based on our results, we propose a new heuristic model of the neuro-immune interaction pathogenesis of fibromyalgia (Figure 3).
Figure 3. The contribution of Natural Killer cells to the pathogenesis of fibromyalgia based on the results of unbiased immune profiling of fibromyalgia cases and controls (based on Verma et al. 2021).
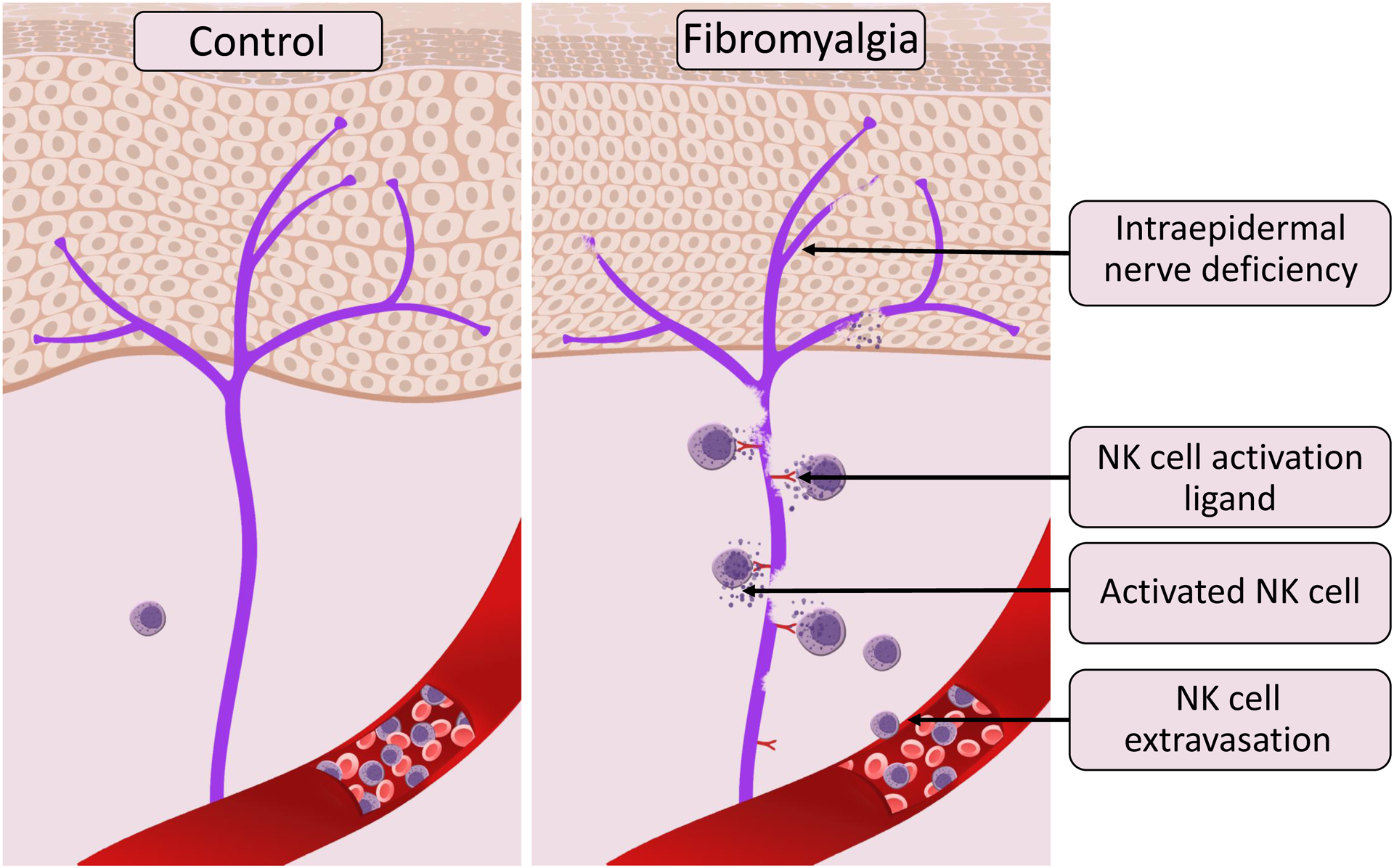
Panels show intraepidermal innervation (purple) in control human subjects (left) and patients with fibromyalgia (right). In those with fibromyalgia, but not in the controls, intraepidermal nerves express a ligand for NK cell activation, which in turn coaxes NK cells to extravasate from the bloodstream and follow the ligand’s gradient. Thus, depletion of NK cells can be observed in blood samples of patients with fibromyalgia compared to controls. The NK cells recruited to the damaged nerve are activated through the immune synapse formation leading to peripheral nerve degeneration through NK cell cytotoxic functions. Thus, the chronic expression of an NK activation ligand(s) stimulates chronic extravasation, recruitment, and activation of circulating NK cells, resulting in persistent peripheral nerve degeneration, and chronic activation of NK cells.
The contribution of NK cells to the pathogenesis of chronic pain has just started to be understood, and NK cells are clearly not the “usual suspects” among immune cell types. However, we note that another unbiased immune profiling experiment in the cerebrospinal fluid of patients with ostherpetic neuralgia, and patients with polyneuropathy in the ERA-Net NEURON study, also identified the reduction of NK cells as a major change in the immune cell composition associated with clinical pain [38].
5. Discussion
-Omics analyses provide a new and critical tool for pain research, contributing new pieces of the puzzle of the pathophysiology of chronic pain. Our GWAS, transcriptomics, and immune profiling studies provided separate examples of these approaches. In each case, the study led to an independent novel insight, and it is our vision that with the increased numbers of such studies being conducted the gaps between seemingly unrelated findings will close. Needless to say, the list of -omics approaches to study pain is growing, and many studies are analyzing human samples at the levels of methylomics, metabolomics, lipidomics, and proteomics, promising new and exciting discoveries. These -omics studies will be also combined with other techniques in the wider context of pain research, including use of demographic data, health data, psycho-social data, qualitative research, and contributions from people living with pain who can ensure that relevant phenotypes are researched.
Importantly, each -omics approach has its own strengths and limitations, and in the long term, a wide variety of -omics analyses should be used. The integration between different -omics approaches is also important, and the genetics field offers more and more tools allowing the integration of -omics approaches in an efficient and meaningful manner, such as in our example with unbiased immune profiling [84].
We note that although the fact that -omics approaches can be performed in human pain patients represents an obvious advantage compared to prior approaches using animal models, animal models are still critical to the overall strategy. There is very limited value in simply identifying genetic, cellular, or pathway “targets” without the ability to define the mechanism by which those targets cause or maintain pain. Animal models remain critical to this aim, and we note that modern animal model implementation, with its increased focus on spontaneous, non-reflexive, and integrative behaviors [52; 65], features much more translational relevance than has been true previously. In addition, more attention is starting to be paid to duration of behaviors in these assays as well as their peak intensity [54; 57; 70; 75]
It is important to note that some biological processes can be captured well by one -omics method but not others. The genes of the inflammatory response represent one example. Because of the inverted U-shaped time-dependent contribution of inflammatory processes to pain resolution, and the gradual increase of a pro-inflammatory state in chronic pain development, inflammatory genes relevant to pain can be captured well only by -omics methods measuring dynamic responses, such as transcriptomics and, probably, metabolomics and methylomics. In contrast, genetic variation in the inflammatory genes themselves, which is a static, inherited molecular feature, may not easily be found among the genome-wide significant SNPs in a GWAS. The same genetic variant that, for example, increases the intensity of inflammatory response, will be protective for the transition from acute to chronic pain but if a patient nevertheless develops chronic pain, this same variant will be a risk factor for chronic pain. Similarly, the same genetic variant could be associated with the intensity of acute pain in an opposite manner to its association with chronic pain. In general, acute pain measures will be difficult for genetic association analyses, as the results will always depend on the degree of injury and degree of inflammation-dependent pain. This may be one reason why post-GWAS functional analyses for pain phenotypes identify more robust evidence for brain-expressed genes among top associated SNPs [6; 20; 33; 35; 49], but less evidence for immune cell-specific SNPs [6; 10; 35; 70]. The enrichment for brain-expressed genes in the GWAS results may be a reflection of the linear dose-dependent and time-dependent relationship between neuronal processes and chronic pain development, in contrast to the curvilinear features of immune cell processes.
We observed a similar phenomenon for loss-of-function genetic variants of the gene encoding epiregulin (EREG) [85]. A strong association with lower chronic pain intensity in facial pain (TMD) patients was observed for an alternative (minor) allele of the EREG-adjacent SNP rs6836436. However, the same allele was associated with higher facial pain intensity among recent-onset patients. These results were validated in an independent cohort, the UK Biobank, where this minor allele was associated with a higher number of reported acute pain sites but a lower number of chronic pain sites. Mouse models of chronic and acute pain confirmed the dichotomous role of epiregulin in pain states [85].
The -omics studies results suggest that chronic pain is a multi-stage process developing over a substantial period of time. For example, our study on LBP patients showed different blood cell types dynamically changed their levels; neutrophils and mast cells were increased and activated in the blood at the acute pain stage of those who resolved their pain, and then were substantially reduced over 3 months, whereas NK and T cells, in contrast, increased over the three months of pain resolution [58]. In the blood of chronic fibromyalgia patients, we found decreased levels of NK cells and increased B cell levels [84]. Even though a particular -omics analysis might identify the association with a specific immune cell type (or specific gene, or specific biological process), the overall process of pain development or resolution most likely includes multiple immune cell types. These cells might contribute to the process in a sequential manner, or they might be complementary to each other.
Furthermore, it will be useful to further our understanding of downstream neuro-immune interactions using human -omics approaches. Neuro-immune interactions are obviously more difficult to capture in humans in large datasets due to highly limited access to relevant nervous system tissues (e.g., peripheral nerve, DRG, spinal cord, cortex) in comparison with blood, although single-cell RNA-seq has started to be applied to human and primate DRGs [37; 76]. The integration of the results of multiple -omics approaches will be the next step in building a holistic picture of the pathophysiology of chronic pain through the generation of testable hypotheses. For example, GWAS association of chronic pain with netrin-driven axonal guidance [35] can be related to the proposed NK-dependent reduction in epidermal nerve density in fibromyalgia patients [84], as the regrowth of the peripheral nerves removed by activated NK cells will rely on the efficiency of axonal guidance pathways.
The -omics approaches are well-known to be prone to Type I errors; that is, false positives, and an increasing sample size, is the most robust way to mitigate these risks. -Omics integration approaches would require even further increases in required sample sizes. There is a clear need to consolidate pain cohorts to facilitate further studies and summarize current results, and now is the time to start to create a pain genetics consortium(s) to combine cohorts across the field. A recent effort using the UK Biobank in which a pain questionnaire was assembled by experts and filled online by more than 157,000 participants is highly commendable (https://biobank.ndph.ox.ac.uk/showcase/label.cgi?id=154), and should prove very useful despite limitations [44].
In summary, human -omics approaches provide a useful avenue to study the pathophysiology of human chronic pain, and for the first time enable the full complexity of various contributing biological processes to be considered. We have started to identify a mosaic of “pain genes” and biological pathways contributing to human chronic pain, which we believe will result in new targets for more effective therapeutic interventions.
Funding:
This work was funded by the Canadian Excellence Research Chairs (CERC09) to LD.
Footnotes
Declaration of conflict of interests: The authors have no conflict of interests to declare.
REFFERENCES
- [1].Anttila V, Stefansson H, Kallela M, Todt U, Terwindt GM, Calafato MS, Nyholt DR, Dimas AS, Freilinger T, Muller-Myhsok B, Artto V, Inouye M, Alakurtti K, Kaunisto MA, Hamalainen E, de Vries B, Stam AH, Weller CM, Heinze A, Heinze-Kuhn K, Goebel I, Borck G, Gobel H, Steinberg S, Wolf C, Bjornsson A, Gudmundsson G, Kirchmann M, Hauge A, Werge T, Schoenen J, Eriksson JG, Hagen K, Stovner L, Wichmann HE, Meitinger T, Alexander M, Moebus S, Schreiber S, Aulchenko YS, Breteler MM, Uitterlinden AG, Hofman A, van Duijn CM, Tikka-Kleemola P, Vepsalainen S, Lucae S, Tozzi F, Muglia P, Barrett J, Kaprio J, Farkkila M, Peltonen L, Stefansson K, Zwart JA, Ferrari MD, Olesen J, Daly M, Wessman M, van den Maagdenberg AM, Dichgans M, Kubisch C, Dermitzakis ET, Frants RR, Palotie A, International Headache Genetics C. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet 2010;42(10):869–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Arora I, Tollefsbol TO. Computational methods and next-generation sequencing approaches to analyze epigenetics data: Profiling of methods and applications. Methods 2021;187:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25(1):25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Banfi G, Diani M, Pigatto PD, Reali E. T Cell Subpopulations in the Physiopathology of Fibromyalgia: Evidence and Perspectives. Int J Mol Sci 2020;21(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ben-Eghan C, Sun R, Hleap JS, Diaz-Papkovich A, Munter HM, Grant AV, Dupras C, Gravel S. Don’t ignore genetic data from minority populations. Nature 2020;585(7824):184–186. [DOI] [PubMed] [Google Scholar]
- [6].Bortsov AV, Parisien M, Khoury S, Zaykin DV, Martinsen AE, Lie MU, Heuch I, Pain HA-I, Hveem K, Zwart J-A, Winsvold BS, Diatchenko L. Genome-wide analysis identifies significant contribution of brain-expressed genes in chronic, but not acute, back pain. PAIN Rep 2022:in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bratus-Neuenschwander A, Castro-Giner F, Frank-Bertoncelj M, Aluri S, Fucentese SF, Schlapbach R, Sprott H. Pain-Associated Transcriptome Changes in Synovium of Knee Osteoarthritis Patients. Genes (Basel) 2018;9(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bumgarner R Overview of DNA microarrays: types, applications, and their future. Curr Protoc Mol Biol 2013;Chapter 22:Unit 22 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chapman CR, Vierck CJ. The Transition of Acute Postoperative Pain to Chronic Pain: An Integrative Overview of Research on Mechanisms. J Pain 2017;18(4):359 e351–359 e338. [DOI] [PubMed] [Google Scholar]
- [10].Cheng B, Ning Y, Liang C, Li P, Liu L, Cheng S, Ma M, Zhang L, Qi X, Wen Y, Zhang F. Genome-Wide Association Analysis Identified ANXA1 Associated with Shoulder Impingement Syndrome in UK Biobank Samples. G3 (Bethesda) 2020;10(9):3279–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Conti P, Gallenga CE, Caraffa A, Ronconi G, Kritas SK. Impact of mast cells in fibromyalgia and low-grade chronic inflammation: Can IL-37 play a role? Dermatol Ther 2020;33(1):e13191. [DOI] [PubMed] [Google Scholar]
- [12].Davies AJ, Kim HW, Gonzalez-Cano R, Choi J, Back SK, Roh SE, Johnson E, Gabriac M, Kim MS, Lee J, Lee JE, Kim YS, Bae YC, Kim SJ, Lee KM, Na HS, Riva P, Latremoliere A, Rinaldi S, Ugolini S, Costigan M, Oh SB. Natural Killer Cells Degenerate Intact Sensory Afferents following Nerve Injury. Cell 2019;176(4):716–728 e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet 1996;14(4):457–460. [DOI] [PubMed] [Google Scholar]
- [14].Dias A, Mariz T, Sousa A, Lemos C, Alves-Ferreira M. A review of migraine genetics: gathering genomic and transcriptomic factors. Hum Genet 2022;141(1):1–14. [DOI] [PubMed] [Google Scholar]
- [15].Dorsey SG, Renn CL, Griffioen M, Lassiter CB, Zhu S, Huot-Creasy H, McCracken C, Mahurkar A, Shetty AC, Jackson-Cook CK, Kim H, Henderson WA, Saligan L, Gill J, Colloca L, Lyon DE, Starkweather AR. Whole blood transcriptomic profiles can differentiate vulnerability to chronic low back pain. PLoS One 2019;14(5):e0216539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30(1):207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Evdokimov D, Frank J, Klitsch A, Unterecker S, Warrings B, Serra J, Papagianni A, Saffer N, Meyer Zu Altenschildesche C, Kampik D, Malik RA, Sommer C, Uceyler N. Reduction of skin innervation is associated with a severe fibromyalgia phenotype. Ann Neurol 2019;86(4):504–516. [DOI] [PubMed] [Google Scholar]
- [18].Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, Anttila V, Xu H, Zang C, Farh K, Ripke S, Day FR, Purcell S, Stahl E, Lindstrom S, Perry JRB, Okada Y, Raychaudhuri S, Daly MJ, Patterson N, Neale BM, Price AL, ReproGen C, Schizophrenia Working Group of the Psychiatric Genomics C, The RC. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nature Genetics 2015;47(11):1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, Gazal S, Loh PR, Lareau C, Shoresh N, Genovese G, Saunders A, Macosko E, Pollack S, Brainstorm C, Perry JRB, Buenrostro JD, Bernstein BE, Raychaudhuri S, McCarroll S, Neale BM, Price AL. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 2018;50(4):621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Freidin MB, Tsepilov YA, Palmer M, Karssen LC, Suri P, Aulchenko YS, Williams FMK, Group CMW. Insight into the genetic architecture of back pain and its risk factors from a study of 509,000 individuals. Pain 2019;160(6):1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Freidin MB, Tsepilov YA, Stanaway IB, Meng W, Hayward C, Smith BH, Khoury S, Parisien M, Bortsov A, Diatchenko L, Borte S, Winsvold BS, Brumpton BM, Zwart JA, Aulchenko YS, Suri P, Williams FMK, Pain HA-I. Sex- and age-specific genetic analysis of chronic back pain. Pain 2021;162(4):1176–1187. [DOI] [PubMed] [Google Scholar]
- [22].Gonzalez S, Lopez-Soto A, Suarez-Alvarez B, Lopez-Vazquez A, Lopez-Larrea C. NKG2D ligands: key targets of the immune response. Trends Immunol 2008;29(8):397–403. [DOI] [PubMed] [Google Scholar]
- [23].Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 2016;17(6):333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gormley P, Anttila V, Winsvold BS, Palta P, Esko T, Pers TH, Farh KH, Cuenca-Leon E, Muona M, Furlotte NA, Kurth T, Ingason A, McMahon G, Ligthart L, Terwindt GM, Kallela M, Freilinger TM, Ran C, Gordon SG, Stam AH, Steinberg S, Borck G, Koiranen M, Quaye L, Adams HH, Lehtimaki T, Sarin AP, Wedenoja J, Hinds DA, Buring JE, Schurks M, Ridker PM, Hrafnsdottir MG, Stefansson H, Ring SM, Hottenga JJ, Penninx BW, Farkkila M, Artto V, Kaunisto M, Vepsalainen S, Malik R, Heath AC, Madden PA, Martin NG, Montgomery GW, Kurki MI, Kals M, Magi R, Parn K, Hamalainen E, Huang H, Byrnes AE, Franke L, Huang J, Stergiakouli E, Lee PH, Sandor C, Webber C, Cader Z, Muller-Myhsok B, Schreiber S, Meitinger T, Eriksson JG, Salomaa V, Heikkila K, Loehrer E, Uitterlinden AG, Hofman A, van Duijn CM, Cherkas L, Pedersen LM, Stubhaug A, Nielsen CS, Mannikko M, Mihailov E, Milani L, Gobel H, Esserlind AL, Christensen AF, Hansen TF, Werge T, International Headache Genetics C, Kaprio J, Aromaa AJ, Raitakari O, Ikram MA, Spector T, Jarvelin MR, Metspalu A, Kubisch C, Strachan DP, Ferrari MD, Belin AC, Dichgans M, Wessman M, van den Maagdenberg AM, Zwart JA, Boomsma DI, Smith GD, Stefansson K, Eriksson N, Daly MJ, Neale BM, Olesen J, Chasman DI, Nyholt DR, Palotie A. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 2016;48(8):856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Grayston R, Czanner G, Elhadd K, Goebel A, Frank B, Uceyler N, Malik RA, Alam U. A systematic review and meta-analysis of the prevalence of small fiber pathology in fibromyalgia: Implications for a new paradigm in fibromyalgia etiopathogenesis. Semin Arthritis Rheum 2019;48(5):933–940. [DOI] [PubMed] [Google Scholar]
- [26].Guo Y, Walsh AM, Fearon U, Smith MD, Wechalekar MD, Yin X, Cole S, Orr C, McGarry T, Canavan M, Kelly S, Lin TA, Liu X, Proudman SM, Veale DJ, Pitzalis C, Nagpal S. CD40L-Dependent Pathway Is Active at Various Stages of Rheumatoid Arthritis Disease Progression. J Immunol 2017;198(11):4490–4501. [DOI] [PubMed] [Google Scholar]
- [27].Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol 2017;18(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hautakangas H, Winsvold BS, Ruotsalainen SE, Bjornsdottir G, Harder AVE, Kogelman LJA, Thomas LF, Noordam R, Benner C, Gormley P, Artto V, Banasik K, Bjornsdottir A, Boomsma DI, Brumpton BM, Burgdorf KS, Buring JE, Chalmer MA, de Boer I, Dichgans M, Erikstrup C, Farkkila M, Garbrielsen ME, Ghanbari M, Hagen K, Happola P, Hottenga JJ, Hrafnsdottir MG, Hveem K, Johnsen MB, Kahonen M, Kristoffersen ES, Kurth T, Lehtimaki T, Lighart L, Magnusson SH, Malik R, Pedersen OB, Pelzer N, Penninx B, Ran C, Ridker PM, Rosendaal FR, Sigurdardottir GR, Skogholt AH, Sveinsson OA, Thorgeirsson TE, Ullum H, Vijfhuizen LS, Widen E, van Dijk KW, International Headache Genetics C, Headache HA-i, Danish Blood Donor Study Genomic C, Aromaa A, Belin AC, Freilinger T, Ikram MA, Jarvelin MR, Raitakari OT, Terwindt GM, Kallela M, Wessman M, Olesen J, Chasman DI, Nyholt DR, Stefansson, Stefansson K, van den Maagdenberg A, Hansen TF, Ripatti S, Zwart JA, Palotie A, Pirinen M. Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat Genet 2022;54(2):152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Held M, Karl F, Vlckova E, Rajdova A, Escolano-Lozano F, Stetter C, Bharti R, Forstner KU, Leinders M, Dusek L, Birklein F, Bednarik J, Sommer C, Uceyler N. Sensory profiles and immune-related expression patterns of patients with and without neuropathic pain after peripheral nerve lesion. Pain 2019;160(10):2316–2327. [DOI] [PubMed] [Google Scholar]
- [30].Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet 2014;15(3):205–213. [DOI] [PubMed] [Google Scholar]
- [31].Ji R-R, Chamessian A, Zhang Y-Q. Pain regulation by non-neuronal cells and inflammation. Science 2016;354(6312):572–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology 2018;129(2):343–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Johnston KJA, Adams MJ, Nicholl BI, Ward J, Strawbridge RJ, Ferguson A, McIntosh AM, Bailey MES, Smith DJ. Genome-wide association study of multisite chronic pain in UK Biobank. PLoS Genet 2019;15(6):e1008164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Johnston KJA, Ward J, Ray PR, Adams MJ, McIntosh AM, Smith BH, Strawbridge RJ, Price TJ, Smith DJ, Nicholl BI, Bailey MES. Sex-stratified genome-wide association study of multisite chronic pain in UK Biobank. PLoS Genet 2021;17(4):e1009428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Khoury S, Parisien M, Thompson SJ, Vachon-Presseau E, Roy M, Martinsen AE, Winsvold BS, Pain HA-I, Mundal IP, Zwart JA, Kania A, Mogil JS, Diatchenko L. Genome-wide analysis identifies impaired axonogenesis in chronic overlapping pain conditions. Brain 2021. [DOI] [PubMed] [Google Scholar]
- [36].Konig IR, Loley C, Erdmann J, Ziegler A. How to include chromosome X in your genome-wide association study. Genet Epidemiol 2014;38(2):97–103. [DOI] [PubMed] [Google Scholar]
- [37].Kupari J, Usoskin D, Parisien M, Lou D, Hu Y, Fatt M, Lonnerberg P, Spangberg M, Eriksson B, Barkas N, Kharchenko PV, Lore K, Khoury S, Diatchenko L, Ernfors P. Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain. Nat Commun 2021;12(1):1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lassen J, Sturner KH, Gierthmuhlen J, Dargvainiene J, Kixmuller D, Leypoldt F, Baron R, Hullemann P. Protective role of natural killer cells in neuropathic pain conditions. Pain 2021;162(9):2366–2375. [DOI] [PubMed] [Google Scholar]
- [39].Laumet G, Ma J, Robison AJ, Kumari S, Heijnen CJ, Kavelaars A. T Cells as an Emerging Target for Chronic Pain Therapy. Front Mol Neurosci 2019;12:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li Z, Wu X, Leo PJ, De Guzman E, Akkoc N, Breban M, Macfarlane GJ, Mahmoudi M, Marzo-Ortega H, Anderson LK, Wheeler L, Chou CT, Harrison AA, Stebbings S, Jones GT, Bang SY, Wang G, Jamshidi A, Farhadi E, Song J, Lin L, Li M, Wei JC, Martin NG, Wright MJ, Lee M, Wang Y, Zhan J, Zhang JS, Wang X, Jin ZB, Weisman MH, Gensler LS, Ward MM, Rahbar MH, Diekman L, Kim TH, Reveille JD, Wordsworth BP, Xu H, Brown MA, Group TA. Polygenic Risk Scores have high diagnostic capacity in ankylosing spondylitis. Ann Rheum Dis 2021;80(9):1168–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lin M, Park DS, Zaitlen NA, Henn BM, Gignoux CR. Admixed Populations Improve Power for Variant Discovery and Portability in Genome-Wide Association Studies. Front Genet 2021;12:673167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lobo JJ, McLean SA, Tungate AS, Peak DA, Swor RA, Rathlev NK, Hendry PL, Linnstaedt SD. Polygenic risk scoring to assess genetic overlap and protective factors influencing posttraumatic stress, depression, and chronic pain after motor vehicle collision trauma. Transl Psychiatry 2021;11(1):359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Luchting B, Rachinger-Adam B, Heyn J, Hinske LC, Kreth S, Azad SC. Anti-inflammatory T-cell shift in neuropathic pain. J Neuroinflammation 2015;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Macfarlane GJ, Beasley M, Smith BH, Jones GT, Macfarlane TV. Can large surveys conducted on highly selected populations provide valid information on the epidemiology of common health conditions? An analysis of UK Biobank data on musculoskeletal pain. Br J Pain 2015;9(4):203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mardis ER. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif) 2013;6:287–303. [DOI] [PubMed] [Google Scholar]
- [46].Martinez-Lavin M. Fibromyalgia and small fiber neuropathy: the plot thickens! Clin Rheumatol 2018;37(12):3167–3171. [DOI] [PubMed] [Google Scholar]
- [47].Medina-Gomez C, Felix JF, Estrada K, Peters MJ, Herrera L, Kruithof CJ, Duijts L, Hofman A, van Duijn CM, Uitterlinden AG, Jaddoe VW, Rivadeneira F. Challenges in conducting genome-wide association studies in highly admixed multi-ethnic populations: the Generation R Study. Eur J Epidemiol 2015;30(4):317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Meloto CB, Bortsov AV, Bair E, Helgeson E, Ostrom C, Smith SB, Dubner R, Slade GD, Fillingim RB, Greenspan JD, Ohrbach R, Maixner W, McLean SA, Diatchenko L. Modification of COMT-dependent pain sensitivity by psychological stress and sex. Pain 2016;157(4):858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Meng W, Chan BW, Harris C, Freidin MB, Hebert HL, Adams MJ, Campbell A, Hayward C, Zheng H, Zhang X, Colvin LA, Hales TG, Palmer CNA, Williams FMK, McIntosh A, Smith BH. A genome-wide association study finds genetic variants associated with neck or shoulder pain in UK Biobank. Hum Mol Genet 2020;29(8):1396–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mifflin KA, Kerr BJ. The transition from acute to chronic pain: understanding how different biological systems interact. Can J Anaesth 2014;61(2):112–122. [DOI] [PubMed] [Google Scholar]
- [51].Mocci E, Ward K, Dorsey SG, Ament SA. GWAS meta-analysis reveals dual neuronal and immunological etiology for pain susceptibility. medRxiv 2021:2021.2008.2023.21262510. [Google Scholar]
- [52].Mogil JS. Animal models of pain: progress and challenges. Nat Rev Neurosci 2009;10(4):283–294. [DOI] [PubMed] [Google Scholar]
- [53].Mogil JS, Sorge RE, LaCroix-Fralish ML, Smith SB, Fortin A, Sotocinal SG, Ritchie J, Austin JS, Schorscher-Petcu A, Melmed K, Czerminski J, Bittong RA, Mokris JB, Neubert JK, Campbell CM, Edwards RR, Campbell JN, Crawley JN, Lariviere WR, Wallace MR, Sternberg WF, Balaban CD, Belfer I, Fillingim RB. Pain sensitivity and vasopressin analgesia are mediated by a gene-sex-environment interaction. Nat Neurosci 2011;14(12):1569–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Muralidharan A, Sotocinal SG, Yousefpour N, Akkurt N, Lima LV, Tansley S, Parisien M, Wang C, Austin JS, Ham B, Dutra GM, Rousseau P, Maldonado-Bouchard S, Clark T, Rosen SF, Majeed MR, Silva O, Nejade R, Li X, Donayre Pimentel S, Nielsen CS, Neely GG, Autexier C, Diatchenko L, Ribeiro-da-Silva A, Mogil JS. Long-term male-specific chronic pain via telomere- and p53mediated spinal cord cellular senescence. J Clin Invest 2022;132(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Nishizawa D, Iseki M, Arita H, Hanaoka K, Yajima C, Kato J, Ogawa S, Hiranuma A, Kasai S, Hasegawa J, Hayashida M, Ikeda K. Genome-wide association study identifies candidate loci associated with chronic pain and postherpetic neuralgia. Mol Pain 2021;17:1744806921999924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].O’Brien JA, McGuire HM, Shinko D, Fazekas de St Groth B, Russo MA, Bailey D, Santarelli DM, Wynne K, Austin PJ. T lymphocyte and monocyte subsets are dysregulated in type 1 diabetes patients with peripheral neuropathic pain. Brain Behav Immun Health 2021;15:100283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Parisien M, Khoury S, Chabot-Dore AJ, Sotocinal SG, Slade GD, Smith SB, Fillingim RB, Ohrbach R, Greenspan JD, Maixner W, Mogil JS, Belfer I, Diatchenko L. Effect of human genetic variability on gene expression in dorsal root ganglia and association with pain phenotypes. Cell Rep 2017;19(9):1940–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Parisien M, Lima LV, Dagostino C, El-Hachem N, Drury GL, Grant AV, Huising J, Verma V, Meloto CB, Silva JR, Dutra GGS, Markova T, Dang H, Tessier PA, Slade GD, Nackley AG, Ghasemlou N, Mogil JS, Allegri M, Diatchenko L. Acute Inflammatory Response via Neutrophil Activation Protects Against the Development of Chronic Pain. Science Translational Medicine 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pasaniuc B, Zaitlen N, Lettre G, Chen GK, Tandon A, Kao WH, Ruczinski I, Fornage M, Siscovick DS, Zhu X, Larkin E, Lange LA, Cupples LA, Yang Q, Akylbekova EL, Musani SK, Divers J, Mychaleckyj J, Li M, Papanicolaou GJ, Millikan RC, Ambrosone CB, John EM, Bernstein L, Zheng W, Hu JJ, Ziegler RG, Nyante SJ, Bandera EV, Ingles SA, Press MF, Chanock SJ, Deming SL, Rodriguez-Gil JL, Palmer CD, Buxbaum S, Ekunwe L, Hirschhorn JN, Henderson BE, Myers S, Haiman CA, Reich D, Patterson N, Wilson JG, Price AL. Enhanced statistical tests for GWAS in admixed populations: assessment using African Americans from CARe and a Breast Cancer Consortium. PLoS Genet 2011;7(4):e1001371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pers TH, Karjalainen JM, Chan Y, Westra HJ, Wood AR, Yang J, Lui JC, Vedantam S, Gustafsson S, Esko T, Frayling T, Speliotes EK, Genetic Investigation of ATC, Boehnke M, Raychaudhuri S, Fehrmann RS, Hirschhorn JN, Franke L. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun 2015;6:5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Peters MJ, Broer L, Willemen HL, Eiriksdottir G, Hocking LJ, Holliday KL, Horan MA, Meulenbelt I, Neogi T, Popham M, Schmidt CO, Soni A, Valdes AM, Amin N, Dennison EM, Eijkelkamp N, Harris TB, Hart DJ, Hofman A, Huygen FJ, Jameson KA, Jones GT, Launer LJ, Kerkhof HJ, de Kruijf M, McBeth J, Kloppenburg M, Ollier WE, Oostra B, Payton A, Rivadeneira F, Smith BH, Smith AV, Stolk L, Teumer A, Thomson W, Uitterlinden AG, Wang K, van Wingerden SH, Arden NK, Cooper C, Felson D, Gudnason V, Macfarlane GJ, Pendleton N, Slagboom PE, Spector TD, Volzke H, Kavelaars A, van Duijn CM, Williams FM, van Meurs JB. Genome-wide association study meta-analysis of chronic widespread pain: evidence for involvement of the 5p15.2 region. Ann Rheum Dis 2013;72(3):427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pietrobon D, Striessnig J. Neurobiology of migraine. Nat Rev Neurosci 2003;4(5):386–398. [DOI] [PubMed] [Google Scholar]
- [63].Rahman MS, Winsvold BS, Chavez Chavez SO, Borte S, Tsepilov YA, Sharapov SZ, Pain HA-I, Aulchenko YS, Hagen K, Fors EA, Hveem K, Zwart JA, van Meurs JB, Freidin MB, Williams FM. Genome-wide association study identifies RNF123 locus as associated with chronic widespread musculoskeletal pain. Ann Rheum Dis 2021;80(9):1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Reyes-Gibby CC, Wang J, Yeung SJ, Chaftari P, Yu RK, Hanna EY, Shete S. Genome-wide association study identifies genes associated with neuropathy in patients with head and neck cancer. Sci Rep 2018;8(1):8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sadler KE, Mogil JS, Stucky CL. Innovations and advances in modelling and measuring pain in animals. Nat Rev Neurosci 2022;23(2):70–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sanders AE, Jain D, Sofer T, Kerr KF, Laurie CC, Shaffer JR, Marazita ML, Kaste LM, Slade GD, Fillingim RB, Ohrbach R, Maixner W, Kocher T, Bernhardt O, Teumer A, Schwahn C, Sipila K, Lahdesmaki R, Mannikko M, Pesonen P, Jarvelin M, Rizzatti-Barbosa CM, Meloto CB, Ribeiro-Dasilva M, Diatchenko L, Serrano P, Smith SB. GWAS Identifies New Loci for Painful Temporomandibular Disorder: Hispanic Community Health Study/Study of Latinos. J Dent Res 2017;96(3):277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sharma J, Balakrishnan L, Kaushik S, Kashyap MK. Editorial: Multi-Omics Approaches to Study Signaling Pathways. Front Bioeng Biotechnol 2020;8:829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Slade GD, Conrad MS, Diatchenko L, Rashid NU, Zhong S, Smith S, Rhodes J, Medvedev A, Makarov S, Maixner W, Nackley AG. Cytokine biomarkers and chronic pain: association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain 2011;152(12):2802–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Slade GD, Rosen JD, Ohrbach R, Greenspan JD, Fillingim RB, Parisien M, Khoury S, Diatchenko L, Maixner W, Bair E. Anatomical selectivity in overlap of chronic facial and bodily pain. Pain Rep 2019;4(3):e729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Smith SB, Parisien M, Bair E, Belfer I, Chabot-Dore AJ, Gris P, Khoury S, Tansley S, Torosyan Y, Zaykin DV, Bernhardt O, de Oliveira Serrano P, Gracely RH, Jain D, Jarvelin MR, Kaste LM, Kerr KF, Kocher T, Lahdesmaki R, Laniado N, Laurie CC, Laurie CA, Mannikko M, Meloto CB, Nackley AG, Nelson SC, Pesonen P, Ribeiro-Dasilva MC, Rizzatti-Barbosa CM, Sanders AE, Schwahn C, Sipila K, Sofer T, Teumer A, Mogil JS, Fillingim RB, Greenspan JD, Ohrbach R, Slade GD, Maixner W, Diatchenko L. Genome-wide association reveals contribution of MRAS to painful temporomandibular disorder in males. Pain 2019;160(3):579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Southern EM. DNA microarrays. History and overview. Methods Mol Biol 2001;170:1–15. [DOI] [PubMed] [Google Scholar]
- [72].Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, Guiducci C, Chen R, Alfredsson L, Amos CI, Ardlie KG, Consortium B, Barton A, Bowes J, Brouwer E, Burtt NP, Catanese JJ, Coblyn J, Coenen MJ, Costenbader KH, Criswell LA, Crusius JB, Cui J, de Bakker PI, De Jager PL, Ding B, Emery P, Flynn E, Harrison P, Hocking LJ, Huizinga TW, Kastner DL, Ke X, Lee AT, Liu X, Martin P, Morgan AW, Padyukov L, Posthumus MD, Radstake TR, Reid DM, Seielstad M, Seldin MF, Shadick NA, Steer S, Tak PP, Thomson W, van der Helm-van Mil AH, van der Horst-Bruinsma IE, van der Schoot CE, van Riel PL, Weinblatt ME, Wilson AG, Wolbink GJ, Wordsworth BP, Consortium Y, Wijmenga C, Karlson EW, Toes RE, de Vries N, Begovich AB, Worthington J, Siminovitch KA, Gregersen PK, Klareskog L, Plenge RM. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 2010;42(6):508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Suri P, Palmer MR, Tsepilov YA, Freidin MB, Boer CG, Yau MS, Evans DS, Gelemanovic A, Bartz TM, Nethander M, Arbeeva L, Karssen L, Neogi T, Campbell A, Mellstrom D, Ohlsson C, Marshall LM, Orwoll E, Uitterlinden A, Rotter JI, Lauc G, Psaty BM, Karlsson MK, Lane NE, Jarvik GP, Polasek O, Hochberg M, Jordan JM, Van Meurs JBJ, Jackson R, Nielson CM, Mitchell BD, Smith BH, Hayward C, Smith NL, Aulchenko YS, Williams FMK. Genome-wide meta-analysis of 158,000 individuals of European ancestry identifies three loci associated with chronic back pain. PLoS Genet 2018;14(9):e1007601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Tansley S, Uttam S, Urena Guzman A, Yaqubi M, Pacis A, Parisien M, Deamond H, Wong C, Rabau O, Brown N, Haglund L, Ouellet J, Santaguida C, Ribeiro-da-Silva A, Tahmasebi S, Prager-Khoutorsky M, Ragoussis J, Zhang J, Salter MW, Diatchenko L, Healy LM, Mogil JS, Khoutorsky A. Single-cell RNA sequencing reveals time- and sex-specific responses of mouse spinal cord microglia to peripheral nerve injury and links ApoE to chronic pain. Nat Commun 2022;13(1):843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tao X, Lee MS, Donnelly CR, Ji RR. Neuromodulation, Specialized Proresolving Mediators, and Resolution of Pain. Neurotherapeutics 2020;17(3):886–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tavares-Ferreira D, Shiers S, Ray PR, Wangzhou A, Jeevakumar V, Sankaranarayanan I, Cervantes AM, Reese JC, Chamessian A, Copits BA, Dougherty PM, Gereau RWt, Burton MD, Dussor G, Price TJ. Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci Transl Med 2022;14(632):eabj8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Theken KN, Hersh EV, Lahens NF, Lee HM, Li X, Granquist EJ, Giannakopoulos HE, Levin LM, Secreto SA, Grant GR, Detre JA, FitzGerald GA, Grosser T, Farrar JT. Variability in the Analgesic Response to Ibuprofen Is Associated With Cyclooxygenase Activation in Inflammatory Pain. Clin Pharmacol Ther 2019;106(3):632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet 2018;19(9):581–590. [DOI] [PubMed] [Google Scholar]
- [79].Tsepilov YA, Freidin MB, Shadrina AS, Sharapov SZ, Elgaeva EE, Zundert JV, Karssen Lcapital Es C, Suri P, Williams FMK, Aulchenko YS. Analysis of genetically independent phenotypes identifies shared genetic factors associated with chronic musculoskeletal pain conditions. Commun Biol 2020;3(1):329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Uffelmann E, Posthuma D. Emerging Methods and Resources for Biological Interrogation of Neuropsychiatric Polygenic Signal. Biol Psychiatry 2021;89(1):41–53. [DOI] [PubMed] [Google Scholar]
- [81].van Reij RRI, Voncken JW, Joosten EAJ, van den Hoogen NJ. Polygenic risk scores indicates genetic overlap between peripheral pain syndromes and chronic postsurgical pain. Neurogenetics 2020;21(3):205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Vehof J, Zavos HMS, Lachance G, Hammond CJ, Williams FMK. Shared genetic factors underlie chronic pain syndromes. Pain 2014;155(8):1562–1568. [DOI] [PubMed] [Google Scholar]
- [83].Verdonk F, Einhaus J, Tsai AS, Hedou J, Choisy B, Gaudilliere D, Kin C, Aghaeepour N, Angst MS, Gaudilliere B. Measuring the human immune response to surgery: multiomics for the prediction of postoperative outcomes. Curr Opin Crit Care 2021;27(6):717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Verma V, Drury GL, Parisien M, Ozdag Acarli AN, Al-Aubodah TA, Nijnik A, Wen X, Tugarinov N, Verner M, Klares R 3rd, Linton A, Krock E, Morado Urbina CE, Winsvold B, Fritsche LG, Fors EA, Piccirillo C, Khoutorsky A, Svensson CI, Fitzcharles MA, Ingelmo PM, Bernard NF, Dupuy FP, Uceyler N, Sommer C, King IL, Meloto CB, Diatchenko L, Pain HU-AI. Unbiased immune profiling reveals a natural killer cell-peripheral nerve axis in fibromyalgia. Pain 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Verma V, Khoury S, Parisien M, Cho C, Maixner W, Martin LJ, Diatchenko L. The dichotomous role of epiregulin in pain. Pain 2020;161(5):1052–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wang K, Wang S, Chen Y, Wu D, Hu X, Lu Y, Wang L, Bao L, Li C, Zhang X. Single-cell transcriptomic analysis of somatosensory neurons uncovers temporal development of neuropathic pain. Cell Res 2021;31(8):904–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang ZY, Leushkin E, Liechti A, Ovchinnikova S, Mossinger K, Bruning T, Rummel C, Grutzner F, Cardoso-Moreira M, Janich P, Gatfield D, Diagouraga B, de Massy B, Gill ME, Peters A, Anders S, Kaessmann H. Transcriptome and translatome co-evolution in mammals. Nature 2020;588(7839):642–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017;8(1):1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wilhelm-Benartzi CS, Koestler DC, Karagas MR, Flanagan JM, Christensen BC, Kelsey KT, Marsit CJ, Houseman EA, Brown R. Review of processing and analysis methods for DNA methylation array data. Br J Cancer 2013;109(6):1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Winham SJ, Biernacka JM. Gene-environment interactions in genome-wide association studies: current approaches and new directions. J Child Psychol Psychiatry 2013;54(10):1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]