

Recently, it was published in Circulation that empagliflozin inhibits H2O2-induced cardiac late sodium current (late INa).1 Using computational modeling and point mutagenic approaches, Philippaert et al. suggested a possible site of empagliflozin-binding within NaV1.5 similar to that of local anesthetics, supportive of direct drug binding to NaV1.5, although this remains to be determined conclusively and alternative mechanisms may exist.1 We have previously shown that Ca/calmodulin-dependent kinase II (CaMKII) binds to NaV1.5, stimulates late INa and affects its H2O2-dependent regulation.2,3 We also demonstrated that empagliflozin inhibits CaMKII in failing human and murine cardiomyocytes.4
Here we show that inhibition of H2O2-induced late INa by empagliflozin cannot solely be mediated via direct drug binding but depends on CaMKII-dependent phosphorylation of NaV1.5 at serine 571. We demonstrate that empagliflozin inhibits late INa in patients with aortic stenosis (AS) and phenotypic features of heart failure (HF) with preserved ejection fraction (HFpEF).
Raw data/analytic methods can be made available for purposes of reproducing results or replicating procedures. Human tissue/proprietary antibodies cannot be made available for legal constraints. Experiments conform to the Declaration of Helsinki. Human/murine studies were approved by institutional committee. Written informed consent was obtained from patients prior to tissue donation. LV samples were obtained from septal resections of 11 patients (8 male/ 3 female, aged 69.3±2.6) with AS undergoing valve replacement. Patients had a HFpEF-like phenotype with hypertrophy and preserved EF (59.4±1.7%). Murine models of CaMKIIδ knock-out (CaMKIIδ−/−)3, inhibition of CaMKII-dependent NaV1.5 phosphorylation at serine 571 (S571A), and with CaMKII phospho-mimetic NaV1.5 S571E mutation were tested for involvement of CaMKII-NaV1.5 phosphorylation. Isolated ventricular myocytes were incubated (30 min) with empagliflozin (1 μmol/l) or control (DMSO). Some cardiomyocytes were incubated with inhibitors of open-state Na channel inactivation (ATX-II or veratridine) or lidocaine (100 μmol/l, 30 min) for direct Na channel inhibition. H2O2 (100 μmol/l, 5 min) was used to induce reactive oxygen species, which stimulate late INa in HF via CaMKII3 (tested with CaMKII-inhibitor myristoylated-AiP; 2 μmol/l, 30 min). For some experiments, empagliflozin was washed-in to ATX-II or H2O2 pre-incubated myocytes.
Late INa was measured as described previously2,3. Resting membrane potential was held at -120 mV and INa elicited by depolarizing to -20 mV for 1000 ms, quantified by integrating from 100 to 500 ms of the start of depolarization (normalized to membrane capacitance). Western blots used human ventricular tissue exposed to empagliflozin/vehicle (30 min)4. Data were analyzed using mixed-effects analysis with Holm-Sidak, linear mixed model with random factor ‘individual’ and Sidak correction, or paired t-test (GraphPad Prism 9).
We demonstrate that late INa can be reduced by empagliflozin in ventricular myocytes from patients with AS similar to CaMKII-inhibitor AiP (figure 1A). ATX-II-dependent (figure 1B) enhancement of late INa in murine wildtype cardiomyocytes was not affected by empagliflozin (not even at 10 and 100 μmol/l), nor after wash-in (at 1 μM) to ATX-II pre-incubated myocytes, which would be expected if empagliflozin were a direct NaV1.5 inhibitor. Moreover, wash-in of empagliflozin (up to 10 μM) also did not inhibit late INa in myocytes preincubated with a moderate concentration of veratridine (16 nM, experimentally determined as EC50 by dose-response, data not shown). In sharp contrast, both veratridine and ATX-II-enhanced late INa were blocked by lidocaine (not shown). Empagliflozin robustly inhibited H2O2-induced late INa, (figure 1C), with maximal efficacy at 6 min but not already at 2 min after onset of exposure (late INa integral during wash-in: 0 min −50.8±4.3 A*F−1*ms, 2 min: −39.9±4.6 A*F−1*ms, p=0.0934 vs. 0 min; 4 min: −24.2±4.6 A*F−1*ms, p=0.0007 vs. 0 min; 6 min: −17.2±4.0 A*F−1*ms, p<0.0001 vs. 0 min). No additional effect of empagliflozin on late INa was observed upon AiP (not shown), or in myocytes lacking either CaMKIIδ (CaMKIIδ−/−) or CaMKII-dependent NaV1.5 phosphorylation at serine 571 (S571A, figure 1C). Accordingly, the enhanced late INa in mice with CaMKII phospho-mimetic NaV1.5 S571E was neither blocked by empagliflozin nor AiP (figure 1D). In contrast, lidocaine inhibited late INa in S571E cells, underscoring that empagliflozin primarily acts via CaMKII-NaV1.5 phosphorylation. Empagliflozin dose-response revealed an IC50 for inhibition of H2O2-dependent late INa of 0.086 μmol/l in murine myocytes (not shown). Empagliflozin inhibited CaMKII-autophosphorylation and CaMKII-dependent phosphorylation of NaV1.5 in AS and HF (figure 1E+F).
Figure 1:
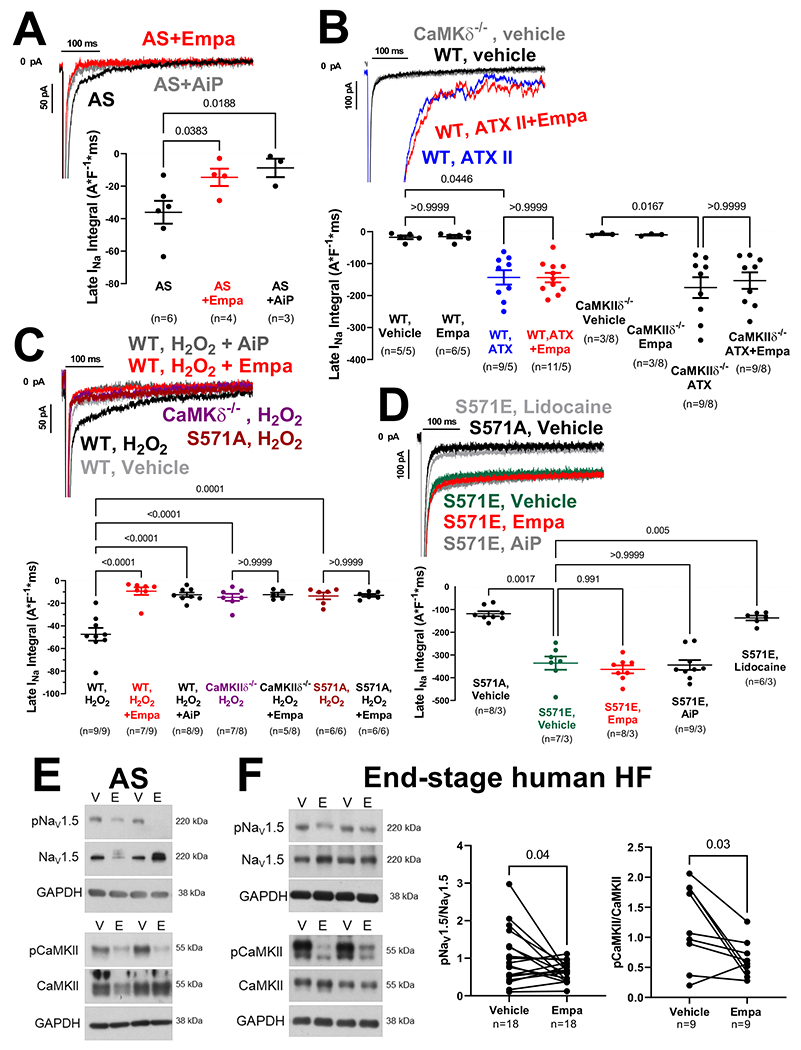
A) Original recordings and mean data of empagliflozin- or AiP-mediated inhibition of late INa in human ventricular cardiomyocytes from patients with AS (n=patients). B) Original recordings and mean data of late INa in murine cardiomyocytes from WT or CaMKIIδ−/− mice (n=cells/mice). The ATX-dependent enhancement of late INa could not be blocked by empagliflozin. C) In contrast, the H2O2-dependent stimulation of late INa was blocked by either CaMKII inhibition (AiP, CaMKII−/−), transgenic inhibition of CaMKII-dependent NaV1.5 phosphorylation (S571A) or in the presence of empagliflozin. D) In contrast to local anesthetic lidocaine, neither empagliflozin nor AiP could block enhanced late INa in mice with phosphomimetic substitution of glutamic acid for serine at 571 (S571E). E+F) Western blots of cardiomyocytes upon empagliflozin shows reduced CaMKII-autophosphorylation (T287) and reduced CaMKII-dependent NaV1.5 phosphorylation.
For comparison of multiple groups, mixed-effects analysis+Holm-Sidak (1A) or linear mixed model+Sidak were performed. For comparison of two groups, paired t-test was done (1F).
In conclusion, inhibition of late INa by empagliflozin is at least in part due to inhibition of CaMKII-dependent regulation of NaV1.52,4. If cardiac Na channels were solely directly inhibited, empagliflozin, like local anesthetics, should have blocked ATX-II/veratridine-stimulated late INa, but it did not. Nevertheless, the target of empagliflozin in the heart remains unclear5 and further research is needed to better understand direct vs. indirect effects on late INa. We demonstrate that empagliflozin also inhibits late INa in patients with AS and features of HFpEF, which may reduce the propensity for arrhythmias and contribute to the positive results of the EMPEROR-Preserved trial.
Funding sources
JM is funded by the German Cardiac Society Clinician Scientist program and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) grant MU 4555/2-1. LSM is funded by DFG grant MA1982/5-1. SW and LSM are funded by SFB 1350 TPA6 and the University of Regensburg ReForM C program. SW is funded by DFG grants WA 2539/4-1, 5-1, 7-1, 8-1. MJB is supported by German Heart Foundation/German Foundation of Heart Research – F/50/20. SP, TS, and MT are funded by the Else-Kröner-Fresenius Stiftung (EKFS, 2019_A84). SS is funded by DFG (SO 1223/4-1) and EKFS (2017_A137). TJH is funded by NIH grants R01 HL156652 and R01 HL135096. PJM is funded by NIH grant R35 HL135754 and Leducq Foundation. The authors have declared that no conflict of interest exists.
Footnotes
Conflict of Interest Disclosures
None.
References
- 1.Philippaert K, Kalyaanamoorthy S, Fatehi M, Long W, Soni S, Byrne NJ, Barr A, Singh J, Wong J, Palechuk T The Cardiac Late Sodium Channel Current is a Molecular Target for the Sodium-Glucose Co-Transporter 2 Inhibitor Empagliflozin. Circulation [Internet]. 2021. [cited 2021 May 21];0. Available from: 10.1161/CIRCULATIONAHA.121.053350 [DOI] [PMC free article] [PubMed]
- 2.Wagner S, Dybkova N, Rasenack E, Jacobshagen C, Fabritz L, Kirchhof P, Maier S, Zhang T, Hasenfuss G, Brown JH Ca/Calmodulin-dependent protein kinase II regulates cardiac Na channels. Journal of Clinical Investigation. 2006;116(12): 3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J Reactive Oxygen Species–Activated Ca/Calmodulin Kinase IIδ Is Required for Late INa Augmentation Leading to Cellular Na and Ca Overload. Circulation Research. 2011;108:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mustroph J, Wagemann O, Lücht CM, Trum M, Hammer KP, Sag CM, Lebek S, Tarnowski D, Reinders J, Perbellini F Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Failure. 2018;5:642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopaschuk Gary D, Verma Subodh. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors. JACC: Basic to Translational Science. 2020;5:632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]