

Abstract
In acute myeloid leukemia (AML), SWI/SNF chromatin remodeling complexes sustain leukemic identity by driving high levels of MYC. Previous studies have implicated the hematopoietic transcription factor PU.1 (SPI1) as an important target of SWI/SNF inhibition, but PU.1 is widely regarded to have pioneer-like activity. As a result, many questions have remained regarding the interplay between PU.1 and SWI/SNF in AML as well as normal hematopoiesis. Here we found that PU.1 binds to most of its targets in a SWI/SNF-independent manner and recruits SWI/SNF to promote accessibility for other AML core regulatory factors, including RUNX1, LMO2, and MEIS1. SWI/SNF inhibition in AML cells reduced DNA accessibility and binding of these factors at PU.1 sites and redistributed PU.1 to promoters. Analysis of non-tumor hematopoietic cells revealed that similar effects also impair PU.1-dependent B cell and monocyte populations. Nevertheless, SWI/SNF inhibition induced profound therapeutic response in an immunocompetent AML mouse model as well as in primary human AML samples. In vivo, SWI/SNF inhibition promoted leukemic differentiation and reduced the leukemic stem cell burden in bone marrow but also induced leukopenia. These results reveal a variable therapeutic window for SWI/SNF blockade in AML and highlight important off-tumor effects of such therapies in immunocompetent settings.
Keywords: BAF, CRC, BENC, immune cells, BRM014
Introduction
Acute myeloid leukemia (AML) is an aggressive malignancy associated with poor outcomes (1–3). An important hallmark of AML cell identity is a deregulated hematopoietic transcriptional program that promotes malignant self-renewal and impairs differentiation (4). Altered gene expression in AML is supported by a core regulatory circuitry (CRC) (5) composed of transcription factors (TFs) and other regulators, including RUNX1 (6), MEIS1 (7), LMO2 and several others (8,9), which form an autoregulatory circuit that maintains leukemic cell identity (10). Targeting the chromatin and transcriptional dependencies of the AML CRC therefore represents a promising approach to target the cell-intrinsic mechanisms sustaining this malignancy.
Excitingly, the recent development of selective small-molecule inhibitors of SWI/SNF chromatin remodeling complexes (11) has provided new opportunities to target AML based on its dependency on SWI/SNF activity (12). In AML cell lines, genetic knockdown of SWI/SNF ATPases (13) or treatment with BRM014 (14), a selective dual inhibitor of SWI/SNF ATPase subunits SMARCA4 (BRG1) and SMARCA2 (BRM), reduces MYC expression and induces leukemic differentiation. Loss of MYC is caused by disruption of the hematopoietic-specific blood enhancer cluster (BENC) (15), which is regulated by SWI/SNF and occupied by the hematopoietic TF PU.1 and other members of the AML CRC.
However, SWI/SNF and PU.1 are both essential for many aspects of normal hematopoiesis. SWI/SNF complexes play several roles in hematopoietic stem cell (HSC) maintenance (16,17), erythropoiesis (18,19), myeloid (20,21) and lymphoid (22–24) development, and interact with many hematopoietic lineage TFs, including PU.1 (25). PU.1 is a pioneer-like transcription factor (25–27) that is essential for myeloid and B cell differentiation (28–31), and interacts with a large number of other TFs that regulate hematopoiesis (32,33). Given these critical functions in normal hematopoiesis, important questions regarding the use of SWI/SNF inhibitors against AML have remained.
Here we evaluated the mechanisms and outcomes associated with SWI/SNF inhibition in clinically relevant models of AML and normal hematopoiesis. We establish that SWI/SNF blockade in AML cells disrupts PU.1-directed enhancer programs utilized by CRC members, and drives a genome-wide shift of PU.1 from enhancers to the promoters of many genes, including those involved in leukemic differentiation. We find that SWI/SNF inhibition is well tolerated by mice but has specific off-tumor effects on PU.1-dependent hematopoietic lineages. We also demonstrate potent sensitivity to SWI/SNF inhibition in an immunocompetent mouse model and in primary human AML samples. Our results identify altered PU.1-directed activity at enhancers as a critical on- and off-tumor effect of SWI/SNF inhibition, and establish SWI/SNF blockade as a promising anti-leukemic therapy.
Materials and Methods
Human cell lines.
THP-1 (RRID: CVCL_0006), MV-4–11 (RRID: CVCL_0064), MOLM-13 (RRID: CVCL_2119), OCI-AML3 (RRID: CVCL_1844), and HL-60 cells (RRID: CVCL_0002) were acquired from ATCC or Leibniz Institute DSMZ and maintained in RPMI supplemented with 10% heat-inactivated FBS, 10 mM HEPES, GlutaMAX, nonessential amino acids, 1 mM sodium pyruvate, 2-mercaptoethanol, and pen/strep. Cells were cultured in a humidified incubator maintained at 37 °C and 5% CO2. Common laboratory procedures for these cells are described in Supplementary Methods. After thawing, cells were passaged a minimum of 3 times before use in experiments and were regularly tested for Mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza).
Flow cytometry.
Measurements were made on a Sony SH800 (RRID: SCR_018066), BD FACSCanto (RRID: SCR_018055), BD LSR II (RRID: SCR_002159), or BD FACSAria II (RRID: SCR_018934). Single-stained cells or compensation beads (Invitrogen, 01–3333-41) were used as compensation controls. Antibodies and filter sets for each instrument are listed in Supplementary Methods. All data were analyzed using FlowJo v10.4.1 (RRID: SCR_008520).
Colony forming assays.
Cells were counted and resuspended in IMDM 10% heat-inactivated FBS 1% pen/strep (human) or DMEM 2% heat-inactivated FBS 1% pen/strep at a 1:50 dilution (murine). Cells were plated in triplicate at 10,000 (human AML), 500 (human CD34+), or 1,000 (murine MLL-AF9) viable cells/plate using human or mouse MethoCult methylcellulose media (StemCell Technologies, 04435 or 03434) with indicated concentrations of BRM014 or DMSO. After 10–14 days, plates were scored for colony counts and cells analyzed by flow cytometry where indicated.
Genome-wide analyses
ATAC library preparation and sequencing.
Cells treated with 1 uM BRM014, 3 uM DB2313, or DMSO for indicated durations were prepared using the Omni-ATAC protocol (34) described in Supplementary Methods. Paired-end sequencing was performed on an Illumina NextSeq 500 (RRID: SCR_014983) high-output flow cell.
ChIP-seq library preparation and sequencing.
Cells were treated with 1 uM BRM014, 3 uM DB2313, or DMSO for 72 h. ChIP libraries were prepared from a single-cell suspension fixed for 10 min in 1% formaldehyde (TFs) or dual-fixed for 30 min in 2 mM disuccinimidyl glutarate followed by 10 min in 1% formaldehyde (SMARCA4/BRG1). Excess formaldehyde was quenched by the addition of glycine to 125 mM. Fixed cells were washed, pelleted, and snap-frozen using liquid nitrogen. Chromatin immunoprecipitation was performed as described previously (35). The following antibodies were used for ChIP: PU.1 (Cell Signaling, 2266s, RRID: AB_10692379), RUNX1 (Abcam, ab23980, RRID: AB_2184205), MEIS1 (Abcam, ab19867, RRID: AB_776272), LMO2 (R&D Systems, AF2726, RRID: AB_2249968), and SMARCA4/BRG1 (Proteintech, 21634–1-AP, RRID: AB_10858784).
ATAC-seq and ChIP-seq analysis.
As previously described (35,36), reads were mapped to the hg38 human reference genome using Bowtie 2.4.1 (RRID: SCR_005476) (37). Duplicate fragments and reads with mapping quality <10 were discarded. Peak calling was performed by MACS 2.1.1 (RRID: SCR_013291) (38). DESeq2 (RRID: SCR_000154) was used for differential peak calling, with size factors determined using the top quartile of accessible sites. Differential peak calls were made by requiring fold changes of >1.5-fold in either direction and FDR-adjusted P-values <0.10. TF motif enrichment was measured using HOMER (v4.11, RRID: SCR_010881) (31) and ChromVAR (39). Overlap of peaks was assessed using bedtools (2.28.0) (RRID: SCR_006646). Calculation of mean densities and preparation of genome-wide heat maps was performed using bwtool (RRID: SCR_003035) (40).
Single-cell library preparation and sequencing.
Bone marrow and peripheral blood were pooled by condition (N=3 per condition). Peripheral red blood cells were lysed via osmotic shock and vortexing, and live non-granulocyte PBMCs and bone marrow cells were isolated via FACS based on FSC/SSC profile and propidium iodide viability staining for multiome. Samples were processed on the 10x Chromium platforms for single-cell 5’ RNA-seq (v2) or single-nuclei multiome (ATAC + Gene expression) kits according to the manufacturer’s instructions. The resulting libraries were sequenced on an Illumina NovaSeq 6000 flow cell (RRID: SCR_016387).
5’ single-cell RNA-seq and single-nuclei multiome analysis.
scRNA-seq transcripts within each cell were counted using the 10x Cell Ranger 5.0.1 pipeline (RRID: SCR_017344). Multiome features within each cell were counted and linked using the Cell Ranger ARC 2.0.1 pipeline. All mapping was to the mm10 mouse genome build. Multiplets were removed using DoubletFinder 2.0.3 (RRID: SCR_018771) (41) and AMULET 1.1 (42). To identify immune cell populations, gene expression data were visualized using UMAP embedding in Loupe Browser 6.1.0 (RRID: SCR_018555). For multiome, cell types incompatible with sn-multiome, including granulocytes and clusters with uniformly low ATAC reads were not considered for analysis. Cell type-specific transcripts and heat maps were generated using Seurat 3.2.3 (RRID: SCR_007322) (43).
Human primary leukemias and CD34+ HSPCs
Culturing primary AML samples.
Viably frozen de-identified primary AML samples obtained via leukapheresis were procured from the Texas Children’s Cancer Center tissue repository under IRB–approved protocols. Written informed consent and assent where appropriate were obtained from each participant and/or his or her guardian in accordance with the declaration of Helsinki prior to any study procedures. Cells were thawed at 37 °C and transferred to fresh RPMI with 10% heat-inactivated FBS and 1% pen/strep at 1:50 dilution. After a 2-h rest, cells were counted by trypan blue and resuspended in IMDM with 10% heat-inactivated FBS and 1% pen/strep. For liquid cultures, cells were seeded at 2e5 cells/mL and maintained in IMDM with 10% heat-inactivated FBS and 1 uM BRM014 or DMSO for 48 h.
Isolation of CD34+ cord blood stem/progenitor cells.
Freshly isolated umbilical cord blood units from 3 anonymous healthy donors were obtained from MD Anderson Cancer Center on their IRB-approved protocol and diluted 1:3 with PBS. Mononuclear cells were isolated using density centrifugation. CD34+ cells were isolated by MACS cell separation using the human CD34 MicroBead Kit UltraPure (Miltenyi Biotec, 130–100-453) and resuspended in fresh DMEM with 10% heat-inactivated FBS and 1% pen/strep.
Mice.
Female 6-to-8-week-old C57BL/6J mice were purchased from the BCM Animal Core Facility. All animal experiments performed in this study were approved by the BCM IACUC and BCM Veterinary Office.
Hematopoiesis mouse model
Dosing and sample collection.
The treatment group (N=5) received once daily oral gavage of 20 mg/kg of BRM014 in 10% DMSO and 1% methylcellulose (Sigma, M0430–100G), and the control group (N=6) received vehicle alone for 14 days. ~18 h after the last treatment, peripheral blood was collected. A random subset of mice (N=3 per condition) were euthanized, and bone marrow was collected via crushing of bilateral femurs, tibias and fibulas and homogenized using 40-μm sterile cell strainers (Fisher Scientific, 22–363-547).
Competitive HSC transplant.
Donor bone marrow was harvested from the femurs, tibias and fibulas of CD45.2 C57BL/6 mice after treatment with daily BRM014 or vehicle control for 14 days and pooled from N=3 mice per condition. Competitor bone marrow cells were harvested from age-matched, untreated CD45.1 C57BL/6 mice. Recipient C57BL/6 CD45.1 mice were conditioned with a split dose of 10.5 Gy of irradiation then transplanted with donor and competitor whole bone marrow by retro-orbital injection. Each recipient mouse was transplanted with 250 CD45.2+ donor HSCs (defined as Lin−, cKit+, Sca-1+, CD48−, CD150+) and 250 CD45.1 competitor HSCs, with N=4 (vehicle) and N=6 (BRM014). Fifteen weeks after transplantation, recipient mice were humanely euthanized, and lineage distributions of CD45.1 and CD45.2 peripheral blood cells were compared by flow cytometry.
Syngeneic KMT2A-MLLT3 (MLL-AF9) mouse model
Tumor engraftment and dosing.
Mice were sub-lethally irradiated and transplanted with 20,000 GFP+ murine MLL-AF9 cells. Leukemia development was monitored by detection of GFP+ leukemic cells in peripheral blood. Two weeks after engraftment, mice were assigned to treatment or control groups (N=11 vehicle, N=10 BRM014) with essentially equivalent distributions of leukemic burden. For downstream analyses, all sample sizes in treatment and control conditions were chosen in advance but vary in some cases due to humane endpoints in control animals. The treatment group received once daily oral gavage of 20 mg/kg of BRM014 in 10% DMSO and 1% methylcellulose (Sigma, M0430–100G), and the control group received vehicle alone for 14 days. During treatment, mice were randomly assigned to two equivalent groups (N=5 per condition), which were monitored at staggered intervals to minimize the frequency of bleeds.
Cell processing.
For peripheral blood, RBCs were lysed via osmotic shock and vortexing. ~18 h after the last treatment, a random subset (N=3 per condition) of mice was euthanized, and peripheral blood, bone marrow, livers, and spleens were collected. Cell suspensions were obtained by manual trituration and homogenized using 40-μm sterile cell strainers (Fisher Scientific, 22–363-547), and leukemic burden was assessed by flow cytometry.
Complete blood count (CBC) analysis.
For AML studies, peripheral blood was collected three days after the last treatment from MLL-AF9 mice and age-/sex-matched non-leukemic C57BL/6 controls. For all CBC analyses, 15 μl of blood was analyzed using the Heska HT5 Hemocounter. One specimen for hematopoiesis studies was identified as a significant outlier by the Grubb’s test based on anomalously high total WBC count and was excluded from analysis.
Statistics
Statistical tests were performed in R (3.6.1) as two-sided tests (RRID: SCR_001905). For multiple-comparison tests, p-values were adjusted using the Benjamini-Hochberg false discovery rate (FDR) correction procedure. P-values below the 64-bit double precision machine epsilon (2−52=2.22e-16) are reported as p<2.2e-16. Independent biological replicates were chosen in advance and are indicated in figure legends. All N values indicate independent biological replicates, genome-wide studies used N=2 per condition in agreement with recommendations of the ENCODE Consortium, flow cytometry data are presented as representative analyses from N≥3 replicates, and single-cell experiments used N=3 pooled animals per condition.
Data availability
High-throughput sequencing data generated for this project have been deposited in the Gene Expression Omnibus (GEO, RRID: SCR_005012) database with SuperSeries accession numbers GSE205572 and GSE190723. Public H3K4me3 ChIP-seq data were analyzed from GEO accession GSM3681460.
Results
PU.1-directed recruitment of SWI/SNF enables binding of the core tumor circuitry
Consistent with previous reports, treatment of a panel of AML cell lines with BRM014 resulted in leukemic differentiation (p<2.2e-16 in all cases, Supplementary Fig. S1A–I, full data in Supplementary Dataset 1–2). To dissect the underlying chromatin mechanisms, we performed ATAC-seq on AML cells treated with BRM014 or DMSO, which revealed loss of accessibility at SWI/SNF sites (represented by SMARCA4 ChIP-seq peaks) across the genome (Supplementary Fig. S2A). Motif profiling identified the most outstanding losses to be at sites bearing the binding motif of PU.1 (Fig. 1A and Supplementary Fig. S2B, full data in Supplementary Dataset 3), a hematopoietic lineage-specific TF and ETS family member.
Figure 1. SWI/SNF-mediated chromatin remodeling is required for PU.1-directed binding of AML CRC members.
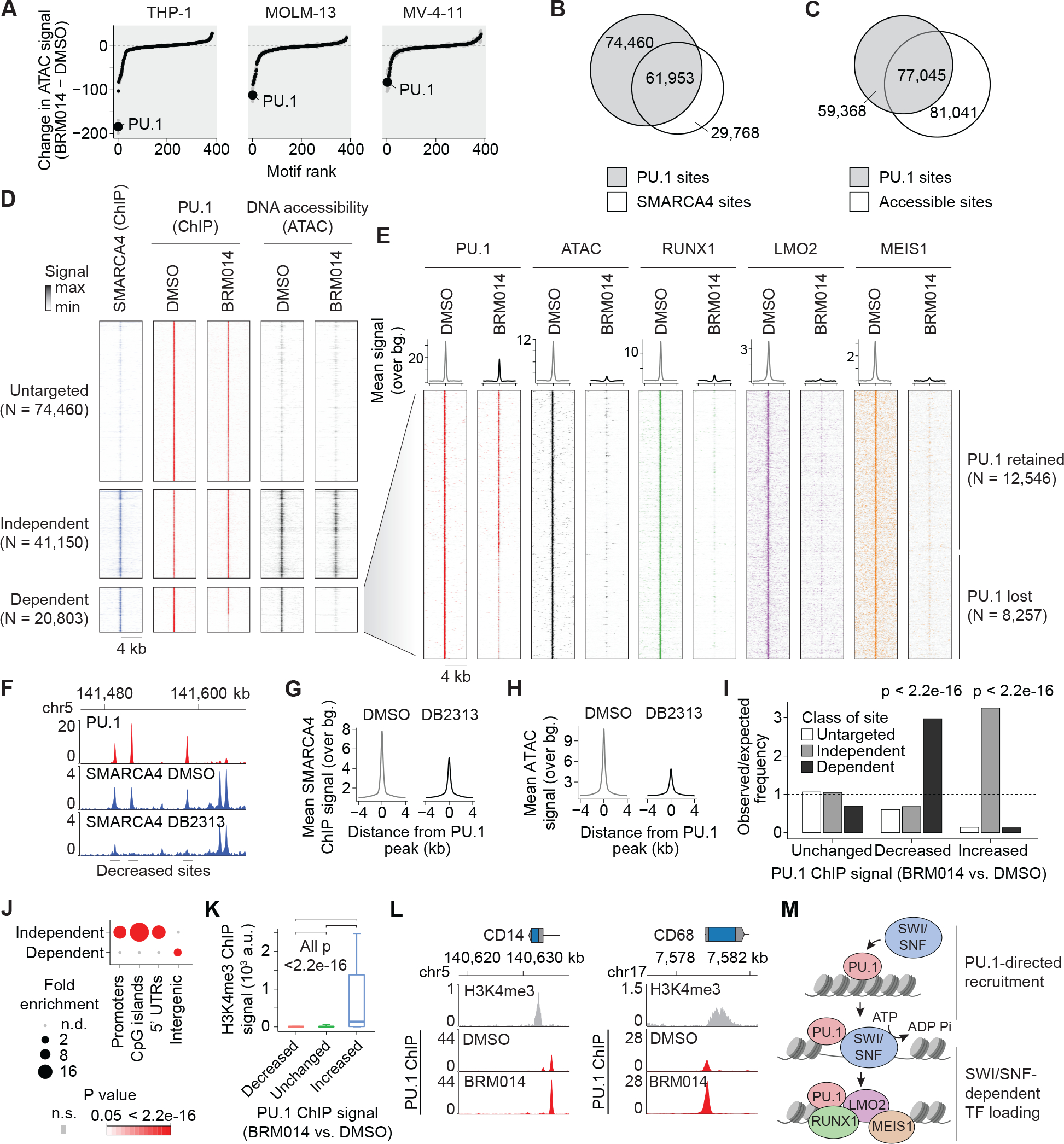
(A) Ranking of differential TF motif accessibility in AML cell lines treated with 1 uM BRM014 versus DMSO control. (B) Overlap of PU.1 and SMARCA4 binding in THP-1 cells. (C) Overlap of PU.1 binding and DNA accessibility in THP-1 cells. (D) SMARCA4 occupancy and DNA accessibility at PU.1 sites in THP-1 cells treated with BRM014 or DMSO control. (E) Binding of AML central regulatory circuit factors RUNX1, LMO2, and MEIS1 at SWI/SNF-dependent PU.1 sites. (F) Representative browser track of SMARCA4 binding at PU.1 sites upon DB2313 treatment. (G) SMARCA4 binding and (H) chromatin accessibility at PU.1 sites in THP-1 cells treated with DB2313. (I) Enrichment of SWI/SNF independent, dependent, and untargeted sites at each class of PU.1 site based on altered PU.1 binding upon treatment with BRM014. Increased, decreased, and unchanged peaks in panels I and K correspond to PU.1 ChIP-seq changes regardless of any other feature. (J) Enrichment of genomic features among SWI/SNF-dependent and -independent sites. (K) H3K4me3 signal at sites with decreased, increased, or unchanged PU.1 occupancy upon BRM014 treatment. Box plot error bars indicate 10%ile and 90%ile range. (L) PU.1 occupancy at the promoters of differentiation-related genes in BRM014- and DMSO-treated cells. (M) Model of PU.1-directed recruitment of SWI/SNF to enhancer sites.
ChIP-seq revealed 136,513 PU.1 sites across the genome (Fig. 1B). Despite substantial overlap, the majority of PU.1 sites did not overlap with SWI/SNF peaks (55%) and many lacked DNA accessibility peaks (44%, Fig. 1C and Supplementary Fig. S2C), consistent with its pioneer-like activity (44–46). Following SWI/SNF inhibition with BRM014, PU.1 binding was largely preserved at 74,460 sites not bound by SMARCA4 (here referred to as “untargeted sites”, Fig. 1D), as well as at 41,150 sites that were bound by SMARCA4 but did not show reduced accessibility upon BRM014 treatment (“independent sites”). PU.1 was furthermore retained at ~60% of the 20,803 sites that showed reduced DNA accessibility following BRM014 treatment (“dependent sites”), reflecting a high capacity to bind its target sites in a SWI/SNF-independent manner. In contrast to PU.1, TFs and other factors that frequently co-bind with PU.1, including the AML CRC members RUNX1, LMO2, and MEIS1, were highly dependent on SWI/SNF activity and were uniformly reduced to near-background levels at dependent sites upon treatment with BRM014 (Fig. 1E). These losses occurred despite their continued expression (Supplementary Dataset 1) and the preservation of binding at sites not targeted by SWI/SNF (Supplementary Fig. S2D).
These findings led us to hypothesize that PU.1 recruits SWI/SNF to promote accessibility needed for other CRC TFs at PU.1 sites. To test this, we treated THP-1 cells with DB2313, an established small-molecule inhibitor that selectively blocks PU.1 binding to its target sites and reduces the expression of PU.1 target genes (Supplementary Fig. S2E–F) (47–49). ChIP-seq of SMARCA4 following treatment with DB2313 but not DMSO selectively disrupted SMARCA4 recruitment to individual PU.1 sites (Fig. 1F), which reduced SMARCA4 binding (Fig. 1G) and DNA accessibility (Fig. 1H) at these sites genome-wide. PU.1 motifs were the highest enriched motifs at sites with decreased SMARCA4 binding or accessibility upon DB2313 treatment, demonstrating a high degree of selectivity (Supplementary Fig. S2G–H), and confirming PU.1-dependent recruitment of SWI/SNF.
Examination of altered PU.1 targeting following SWI/SNF inhibition revealed that the ~11% of genome-wide sites with decreased PU.1 occupancy were enriched at SWI/SNF-dependent sites, while the ~3% of sites with increased PU.1 occupancy were enriched at SWI/SNF-independent sites (all p<2.2e-16, Fig. 1I). While SWI/SNF-dependent sites were differentially enriched at intergenic regions, SWI/SNF-independent sites were enriched at promoters (Fig. 1J). In agreement, PU.1 increases were particularly associated with sites containing high levels of the promoter mark H3K4me3 (Fig. 1K). Enhanced PU.1 occupancy, including at myeloid differentiation-related genes, such CD14 and CD68 (Fig. 1L), corresponded with increased expression genome-wide (Supplementary Fig. S2I–K). Our data show that PU.1 recruits SWI/SNF to generate accessibility needed for CRC factor binding at distal sites (Fig. 1M). SWI/SNF inhibition reduces DNA accessibility required for CRC factors at these sites, and results in re-localization of PU.1 from a minority of its sites to promoters.
PU.1 and SWI/SNF activity converge on the blood enhancer cluster (BENC)
In hematopoietic cells, MYC expression is regulated by the BENC module (13,15). In AML cells, BRM014 treatment induced loss of DNA accessibility and CRC factor binding at BENC compared to DMSO (Fig. 2A and Supplementary Fig. S3A–B). While PU.1 was retained at some BENC sites, binding partners such as RUNX1 showed strong reductions at SWI/SNF-bound sites across the region (Fig. 2A). Inhibition of PU.1 resulted in reduced SMARCA4 recruitment to PU.1 sites within BENC (Supplementary Fig. S3C), consistent with the PU.1-directed SWI/SNF recruitment described above.
Figure 2. Collaborative regulation of the BENC module underlies convergent differentiation induced by SWI/SNF or PU.1 inhibition in AML.
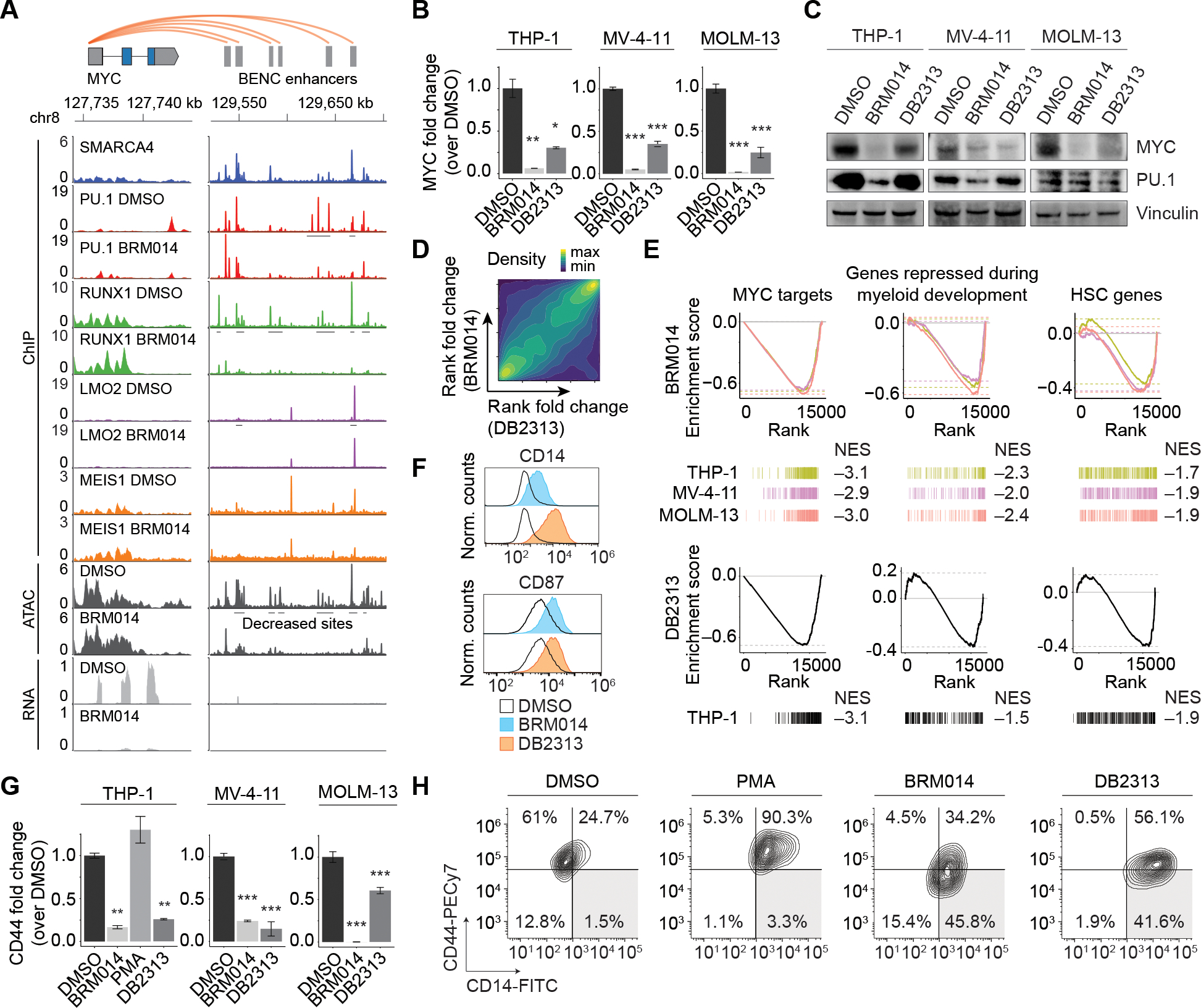
(A) Overlay of SMARCA4, PU.1, RUNX1, LMO2, and MEIS1 occupancy with ATAC-seq and RNA-seq data at MYC and BENC in THP-1 cells. Other cell lines presented in Figure S3A–B. Expression of MYC and PU.1 in AML cells lines treated with BRM014 or DB2313 by (B) RNA-seq (THP-1) or RT-qPCR (MV-4–11 and MOLM-13) at 72 h, N=3, and (C) Western blot at 24 h of treatment. (D) Genome-wide correlation of BRM014- and DB2313-induced transcriptional changes in THP-1 cells based on RNA-seq. (E) Gene sets downregulated upon BRM014 and DB2313 treatment. Normalized enrichment scores (NES) are indicated. (F) Cell surface expression of myeloid differentiation markers CD14 and CD87 in cells treated with DMSO, BRM014, or DB2313. (G) Expression of CD44 in cells treated with DMSO, BRM014, DB2313, or PMA measured by RNA-seq (THP-1) or RT-qPCR (MV-4–11 and MOLM-13) at 72 h, N = 3. (H) Expression of CD14 and CD44 in cells treated with DMSO, BRM014, DB2313, or PMA measured by flow cytometry. Error bars: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
Treatment with either BRM014 or DB2313 caused loss of MYC RNA expression in AML cell lines (Fig. 2B). MYC protein levels were similarly reduced in MV-4–11 and MOLM-13 cells within 24 h, but required 72 h for complete reduction in THP-1 cells treated with DB2313 (Fig. 2C and Supplementary Fig. S3D). Despite modest reduction of PU.1 levels in some cell lines consistent with its autoregulatory function (50), PU.1 remained expressed and was retained at its chromatin binding sites. However, loss of accessibility at PU.1 motifs (Fig. 1A) arose even in cells lacking strong PU.1 expression changes (Fig. 2C).
Transcriptional changes induced by BRM014 and DB2313 treatment were strongly correlated genome wide (Spearman R=0.38 p<2.2e-16; Fig. 2D), and included downregulation of MYC targets, genes repressed during myeloid development, and HSC-related genes (Fig. 2E). Both inhibitors induced leukemic differentiation, as confirmed by increased expression of the cell surface markers CD14 and CD87 (Fig. 2F). Interestingly, SWI/SNF and PU.1 inhibition also induced loss of the stemness marker (51) CD44, which is not a feature of phorbol 12-myristate 13-acetate (PMA)-induced differentiation of AML cells (Fig. 2G–H and Supplementary Fig. S3E), reflecting a unique signature of differentiation via PU.1 deregulation. BRM014 and DB2313 also exhibited largely additive effects (Fig. S3F), consistent with convergence on the same pathway. Together, our findings reveal that PU.1 and SWI/SNF collaboratively regulate MYC expression in AML and that inhibition of either factor converges on myeloid differentiation.
SWI/SNF inhibition has off-tumor effects on PU.1-dependent hematopoietic cell types
Because SWI/SNF and PU.1 are both critical regulators of hematopoiesis, we sought to examine the effects of SWI/SNF inhibition on non-tumor hematopoietic cells in vivo. We treated healthy C57BL/6 mice with daily oral gavage of 20 mg/kg BRM014 or vehicle control for 14 days (Fig. 3A), which was well tolerated and did not cause significant weight loss (p=0.49, Supplementary Fig. S4A). After the 14-day treatment period, we observed decreased white blood cell (WBC) counts in the peripheral blood of BRM014-treated mice but no change in hemoglobin levels (Fig. 3B).
Figure 3. SWI/SNF inhibition induces reversible leukopenia in PU.1-dependent non-tumor peripheral blood lineages.
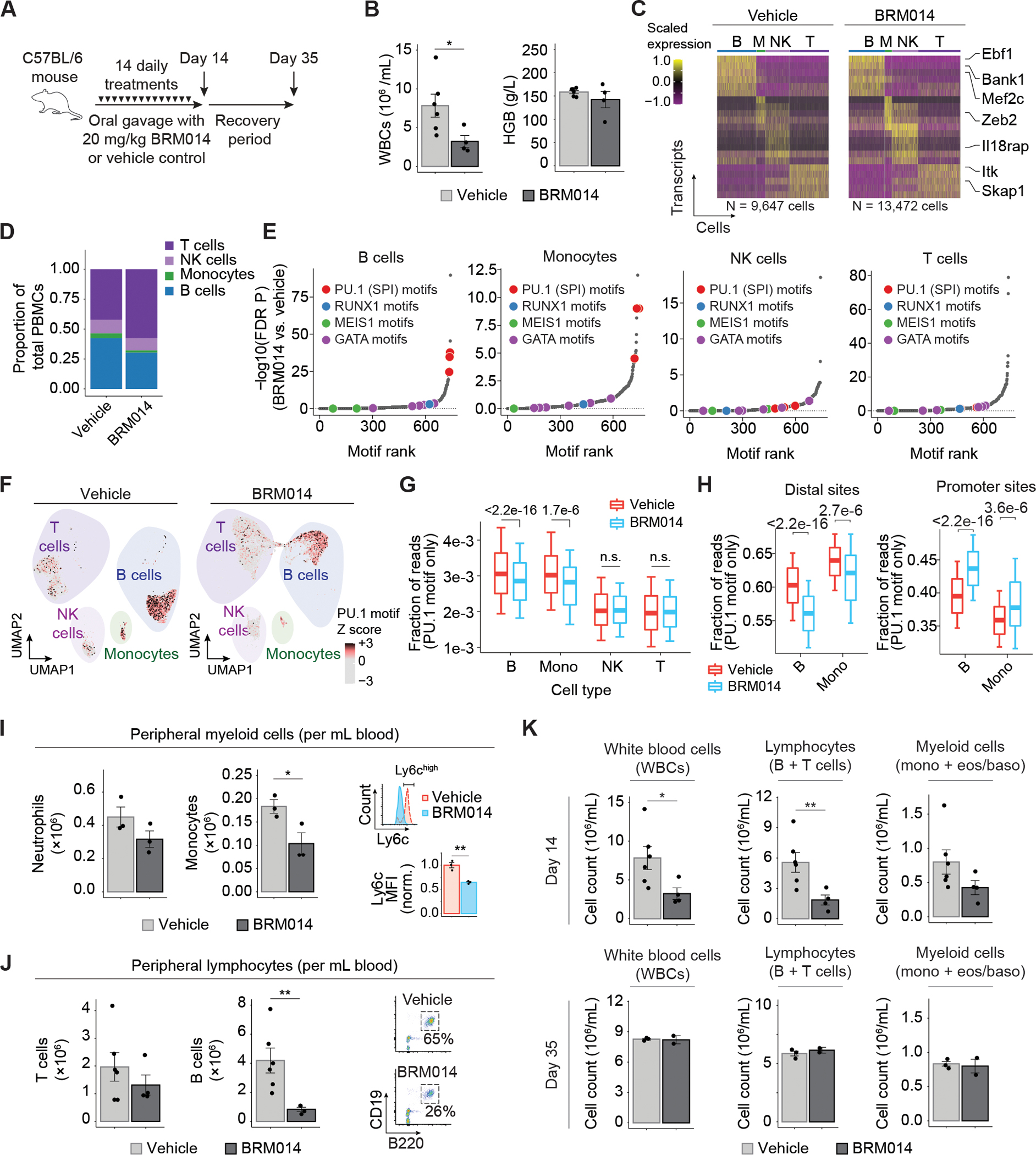
(A) Dosing scheme for in vivo treatment of healthy mice. (B) White blood cell counts (WBC) and hemoglobin (HGB) levels in peripheral blood at day 14, N=6 (vehicle), N=4 (BRM014). (C) Cell-type-specific transcriptional profiles are unchanged by treatment, enabling consistent classification. (D) Composition of non-granulocyte immune cell types in the peripheral blood of BRM014- and DMSO-treated mice. (E) Ranking of significantly reduced differential TF motif accessibility by immune population from mice treated with BRM014 versus vehicle control. (F) UMAP embedding of individual cells by snATAC profile with overlayed PU.1 motif accessibility. (G) Quantification of accessible PU.1 motifs across specific cell types. (H) Shifts of accessible DNA sites containing PU.1 motifs in B cells and monocytes upon BRM014 treatment. (I) Total number of peripheral neutrophils and monocytes at day 14, and Ly6c expression in circulating monocytes, N=3 per condition. (J) Total number of T cells and B cells at day 14, with B cell gating strategy, N=6 (vehicle), N=4 (BRM014). (K) Total WBC, lymphocyte, and myeloid cell counts at day 14 and day 35 (3 weeks post-treatment) measured by CBC. Day 14 N=6 (vehicle), N=4 (BRM014), and day 35 N=3 (vehicle), N=2 (BRM014). Error bars: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
Single-nuclei multiome (sn-multiome; integrated snATAC and snRNA-seq) analysis of peripheral blood collected from BRM014- and vehicle-treated mice (N=3 per condition) revealed cells in distinct clusters, which we identified as B cells, T cells, NK cells, and monocytes using established markers (Fig. 3C and Supplementary Fig. S4B). SWI/SNF inhibition caused a notable reduction in the proportion of peripheral B cells and monocytes (Fig. 3D), but gene expression changes were observed in all cell types (Supplementary Fig. S4C). PU.1 motifs were among the most significantly down-regulated TF motifs in B cells and monocytes, but not in NK or T cells (Fig. 3E). Moreover, reduced cell numbers corresponded to PU.1 motif utilization, with B cells and monocytes exhibiting the highest PU.1 motif accessibility, which was furthermore reduced by SWI/SNF inhibition (Fig. 3F–G). Remarkably, more detailed analysis of sites with PU.1 motifs revealed DNA accessibility losses at distal sites but gains at promoters (Fig. 3H), similar to the effects observed in AML cells.
Flow cytometry (Fig. 3I–J) confirmed that compared to vehicle treatment, BRM014 induced no significant effect on neutrophils (p=0.18) but reduced total monocytes 1.8-fold (p=0.05), with specific loss of the Ly6Chigh population, a marker for monocytes recently derived from bone marrow (52,53) (p=3.7e-3, Fig. 3I and Supplementary Fig. S4D). Within the lymphoid compartment, BRM014 treatment had no significant effect on peripheral T cells (p=0.38) but caused a 5-fold depletion of B cells (p=0.01, Fig. 3J and Supplementary Fig. S4E). Our findings reveal important off-tumor effects of SWI/SNF inhibition that arise through the same mechanisms responsible for therapeutic effects against AML.
To evaluate the reversibility of SWI/SNF inhibition’s effects on hematopoiesis, we assessed complete blood counts (CBCs) from mice on the last day of treatment and 3 weeks after cessation of treatment. This comparison revealed that BRM014-induced effects on peripheral blood cells recover within three weeks of drug withdrawal (Fig. 3K), demonstrating that hematological and immune-related effects of transient SWI/SNF inhibition are reversible.
Effects of SWI/SNF inhibition on hematopoiesis arise within bone marrow
To determine at which stage of hematopoietic development SWI/SNF sensitivity arises, we evaluated the effect of BRM014 on bone marrow (BM) harvested from BRM014- and vehicle-treated mice at day 14 (N=3 per condition). BM from BRM014-treated mice contained fewer total cells than vehicle control (p=0.017, Fig. 4A). To assess the chromatin effects of BRM014 treatment, we performed sn-multiome on BM harvested at day 14, which revealed distinct clusters that we classified as B cells, erythroid cells, monocytes, T cells, plasmacytoid dendritic cells (pDCs), and Kit+ hematopoietic stem and progenitor cells (HSPCs) using standard cell-type-specific markers (Supplementary Fig. S5A). This study revealed the same loss of PU.1 motifs in B cells (p=5.9e-17) and monocytes (p=7.4e-3) in BM as observed in the peripheral blood, with no other cell types showing significant reductions (Fig. 4B and Supplementary Fig. S5A–C).
Figure 4. Transient SWI/SNF inhibition depletes committed hematopoietic cells in bone marrow but preserves HSC function.
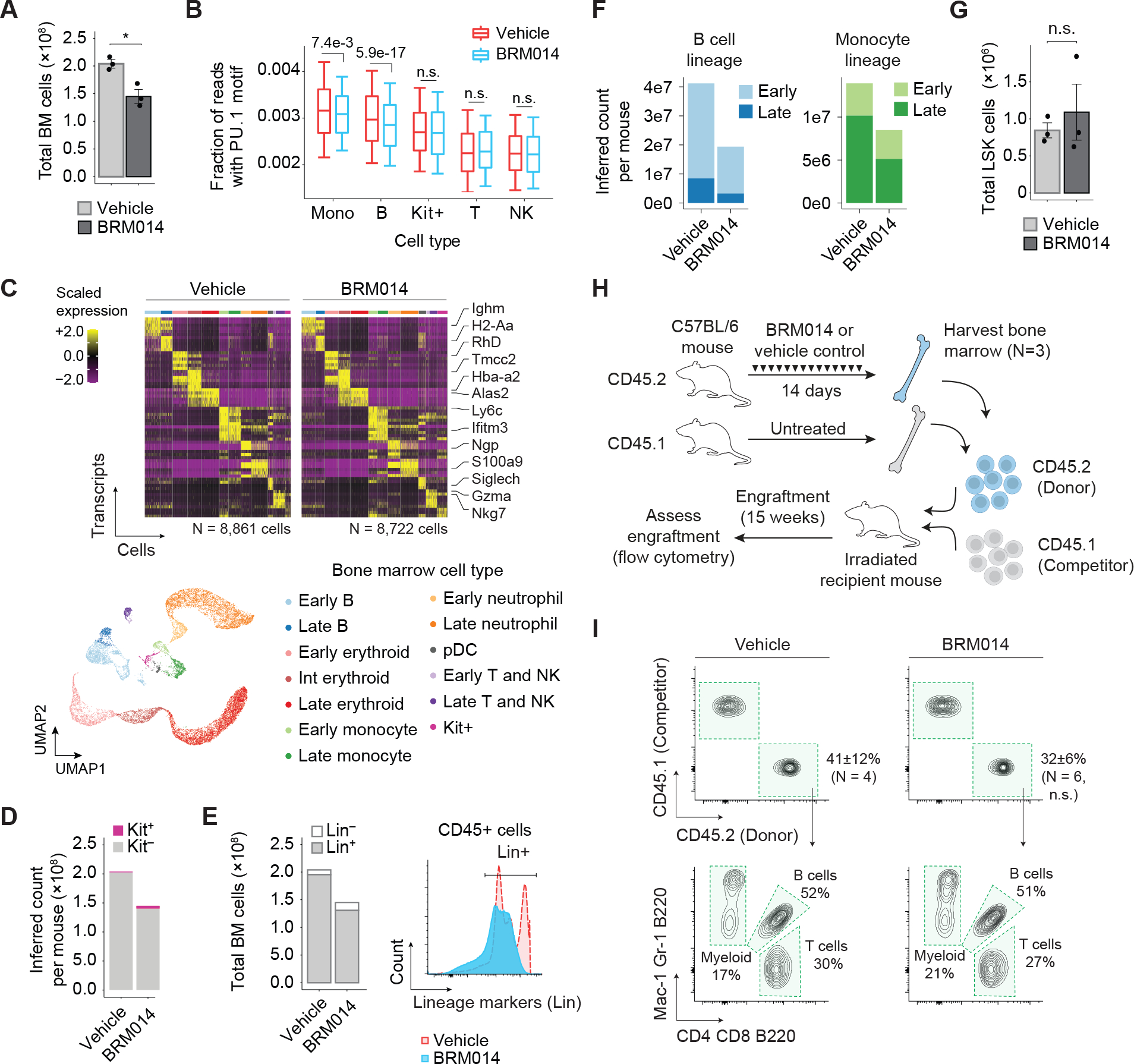
(A) Total number of cells isolated from bone marrow (BM) of BRM014- and vehicle-treated mice at day 14, N=3 per condition. (B) Quantification of accessible PU.1 motifs across specific bone marrow cell types. M = monocytes, B = B cells, Kit+ = Kit+ progenitors, T = T cells, NK = NK cells. (C) Cell-type-specific transcriptional profiles are unchanged by treatment, enabling consistent classification. UMAP embedding of individual cells pooled from all conditions. Resulting clusters are classified by immune cell type based on cell-specific expression profiles. (D) Inferred total number of HSPC Kit+ and lineage committed Kit− cells determined by scRNA-seq. (E) Flow cytometry quantification and histogram of lineage-committed (CD3, CD8, B220, GR1, TER119) and uncommitted CD45+ BM cells. (F) Inferred total number of B cell and monocyte populations in BM determined by scRNA-seq. (G) Total number of LSK (Lin−Sca-1+Kit+) HSPCs in bone marrow by flow cytometry, N=3 per condition. (H) Competitive HSCT workflow and (I) representative flow plots of HSC engraftment and recapitulation of major hematopoietic lineages. N=4 (vehicle), N=6 (BRM014). Error bars: mean ± SEM.
To provide increased resolution of expression changes within BM populations, we performed single-cell RNA-seq (scRNA-seq), which revealed similar clustering of cell identities but with increased granularity (Fig. 4C). Interestingly, scRNA-seq revealed reduced BM cell counts to be caused primarily by a loss of lineage-committed (Kit−) cells (Fig. 4D). This finding was orthogonally confirmed by flow cytometry, which showed that SWI/SNF inhibition differentially affects BM cells that have become committed and lineage-restricted (p=7.0e-3, Fig. 4E and Supplementary Fig. S5D–E). Further, reductions of B cells and monocytes varied between early proliferative cells and late non-proliferative cells across cell types (Fig. 4F), demonstrating that SWI/SNF inhibition has lineage-specific effects and is required at distinct developmental stages for each lineage (Supplementary Fig. S5F–G).
Importantly, BRM014 did not alter the number of LSK (Lin−Sca-1+Kit+) HSPCs in BM (p=0.57, Fig. 4G and Supplementary Fig. S5H). To assess the effects of SWI/SNF inhibition on HSC fitness and function, we performed a competitive HSC transplant of BM harvested from BRM014- and vehicle-treated mice (Fig. 4H). Excitingly, HSCs isolated from BRM014-treated mice retained the ability to engraft and to reconstitute all major hematopoietic lineages (Fig. 4I). We conclude that transient SWI/SNF inhibition does not permanently suppress HSC function.
SWI/SNF inhibition induces regression of AML in an immunocompetent in vivo model
To assess the efficacy of SWI/SNF inhibition against AML in vivo, we employed a syngeneic KMT2A-MLLT3 (“MLL-AF9”) leukemic model in C57BL/6 mice, which is an aggressive and immunocompetent AML model (54) with predictable latency. In vitro, leukemic cells displayed a dose-dependent inhibition of colony forming capacity upon treatment with BRM014 (Supplementary Fig. S6A). We treated mice engrafted with GFP+ MLL-AF9 cells with BRM014 or vehicle control for 14 days (Fig. 5A). Compared to vehicle control, BRM014 strongly prevented leukemic expansion (p<2.2e-16, two-way ANOVA, Fig. 5B–C). Notably, we observed significant regression of disease burden (p=8.3e-3), including one mouse in which the peripheral leukemic burden was reduced from 32% to 7% after 6 days of treatment (Supplementary Fig. S6B).
Figure 5. SWI/SNF inhibition has a significant therapeutic window against AML in an immunocompetent setting.
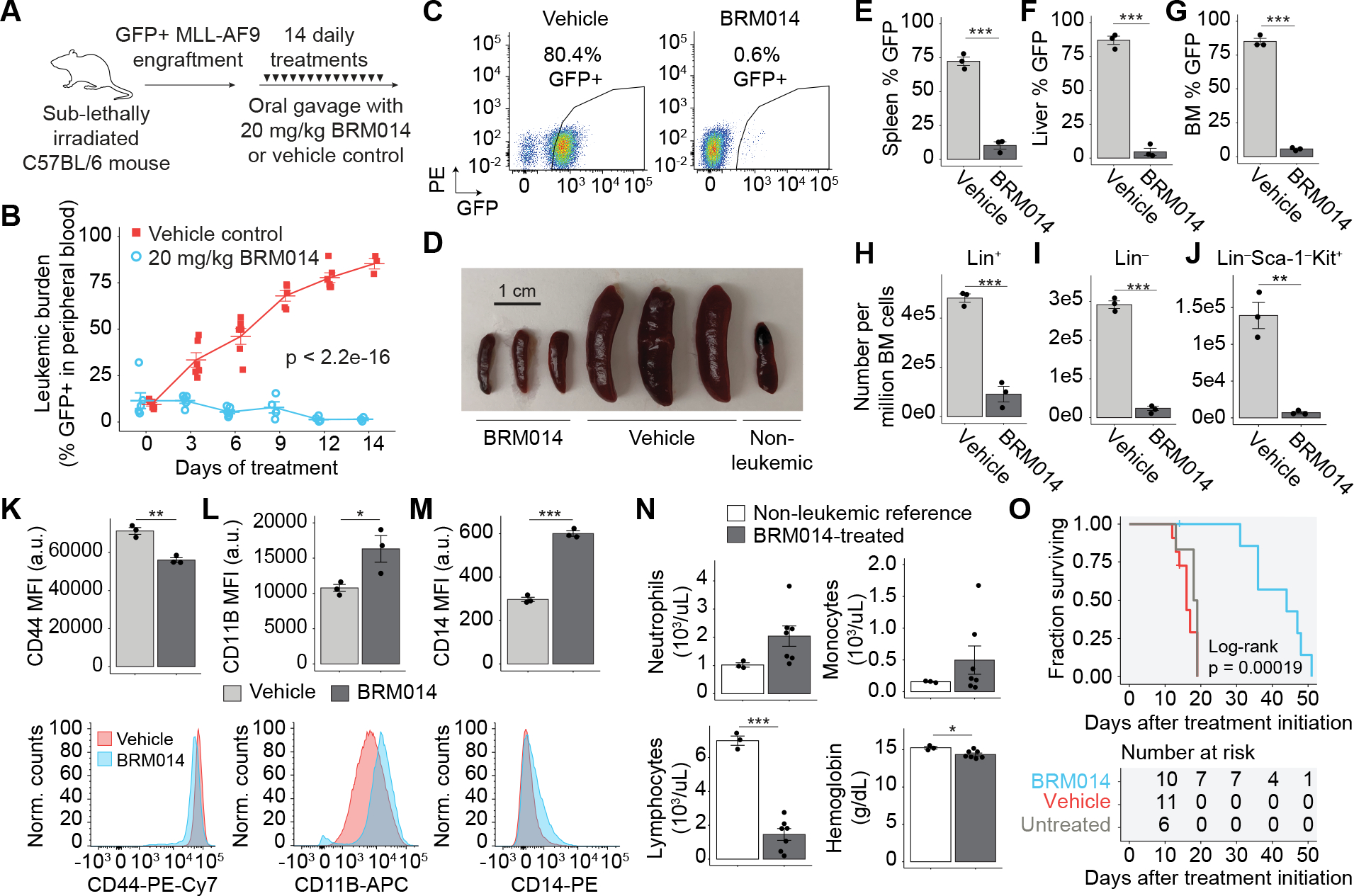
(A) Dosing scheme for in vivo treatment of MLL-AF9 leukemia with BRM014 or vehicle control. (B) Leukemic burden of mice treated with BRM014 or vehicle control over the two-week treatment period and (C) quantification by flow cytometry, N = 11 (vehicle), N=10 (BRM014). (D) Spleens harvested after 14-days of treatment with BRM014 or vehicle control. Leukemic burden in the (E) spleen, (F) liver, and (G) bone marrow of mice treated with BRM014 or vehicle control, N=3 per condition. Effect of BRM014 on the number of (H) committed (Lin+), (I) uncommitted (Lin−), and (J) LSC-enriched Lin−Sca-1−Kit+ stem/progenitor cells within the leukemic population in the bone marrow. Cells in panels (H), (I), and (J) were gated based on GFP+ cells, N=3 per condition. Expression of (K) stemness (CD44) and myeloid differentiation markers (L) CD11B and (M) CD14 on leukemic GFP+ cells in the bone marrow of mice treated with BRM014 or vehicle control, N=3 per condition. (N) CBC neutrophil, monocyte, and lymphocyte counts and hemoglobin levels in BRM014-treated mice versus non-leukemic controls 3 days after completion of treatment course, N=3 (non-leukemic) and N=7 (BRM014). (O) Survival of MLL-AF9 mice treated with BRM014 or vehicle control, N=6 (untreated), N=11 (vehicle), and N=10 (BRM014). Error bars: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
Treatment with BRM014 prevented leukemia-induced splenomegaly (p=2.4e-4) and hepatomegaly (p=5.5e-3, Fig. 5D and Supplementary Fig. S6C–D), with leukemic burden reduced 14.8-fold (p=4.8e-6) in the spleen and 18.8-fold (p=3.3e-5) in the liver (Fig. 5E–F). Importantly, BRM014 also reduced leukemic burden in bone marrow 7-fold compared to vehicle control (p=1.2e-4, Fig. 5G). To understand the leukemic subpopulations affected by SWI/SNF inhibition, we assessed the effects on committed lineage-marker positive (Lin+) and immature (Lin−) leukemic blasts (Fig. 5H–I). BRM014 effectively reduced all leukemic subpopulations, resulting in a 5.3-fold reduction of Lin+ (p=3.9e-4) and a 12-fold reduction of Lin− leukemic BM cells (p=1.9e-5). Notably, Lin−Sca-1−Kit+ leukemic cells, a population enriched for leukemic stem cells (55,56), were reduced 20-fold (p=1.8e-3, Fig. 5J).
To detect leukemic differentiation in vivo, we assessed the expression of CD44 and myeloid differentiation markers CD11B and CD14. Leukemic BM cells from mice treated with BRM014 for 14 days displayed decreased CD44 (p=2e-3) and increased expression of CD11B (p=0.05) and CD14 (p=4.7e-5, Fig. 5K–M). Similar results were observed in the peripheral blood as early as day 9 (Supplementary Fig. S6E–G). Hence our results confirm that leukemic differentiation occurs in both peripheral blood and BM in an immunocompetent context.
BRM014 was well tolerated and caused no significant weight loss (p>0.05, two-way ANOVA, Supplementary Fig. S6H). CBCs 3 days after completion of therapy revealed vehicle-treated mice had high white blood cell (WBC) counts, anemia and thrombocytopenia, reflecting their leukemic state (Supplementary Fig. S6I–K). BRM014-treated mice had reduced WBC (p=2.3e-4) and lymphocyte (p=1.1e-5) counts compared to non-leukemic control mice (Fig. 5N, Supplementary Fig. S6I–K, and Supplementary Dataset 4). Platelets, neutrophils, and monocytes were not significantly different (p>0.05 in all cases). While hemoglobin was mildly reduced in BRM014-treated mice compared to normal controls (p=6.1e-6), BRM014-treated mice were not anemic, with hemoglobin levels within a normal range. The modest inflation of monocytes in BRM014-treated mice was non-significant and caused by the identification of differentiated leukemic blasts as monocytes by CBC. However, flow cytometry of GFP-negative cells revealed significant reduction of non-tumor myeloid cells in BRM014-treated compared to non-leukemic reference mice, confirming similar off-tumor effects in a leukemic setting (Supplementary Fig. S6L–M).
Treatment with BRM014 significantly prolonged survival (p=1.9e-4, log-rank test), with a median survival of 44 days for BRM014-treated mice compared to 16 days for vehicle-treated mice (Fig. 5O). Overall, our results demonstrate that small-molecule inhibition of SWI/SNF ATPase activity significantly reduces disease burden and prolongs survival of AML.
Primary human samples mirror the on- and off-tumor effects of SWI/SNF inhibition in mice
Upon treatment with BRM014, primary human CD34+ HSPCs exhibited dose-dependent reduction of colony forming capacity (Fig. 6A), with an IC50 value of 58 ± 33 nM (mean ± SEM). Notably, we observed selective reduction in the development of myeloid lineage cells, consistent with the PU.1-related effects observed in mice (p=1.5e-3, Supplementary Fig. S7A–C). These results reveal that similar off-tumor effects arise in both mice and in primary human samples.
Figure 6. The relative sensitivity of primary human CD34+ HSPCs and AML specimens defines a therapeutic window for SWI/SNF inhibition.
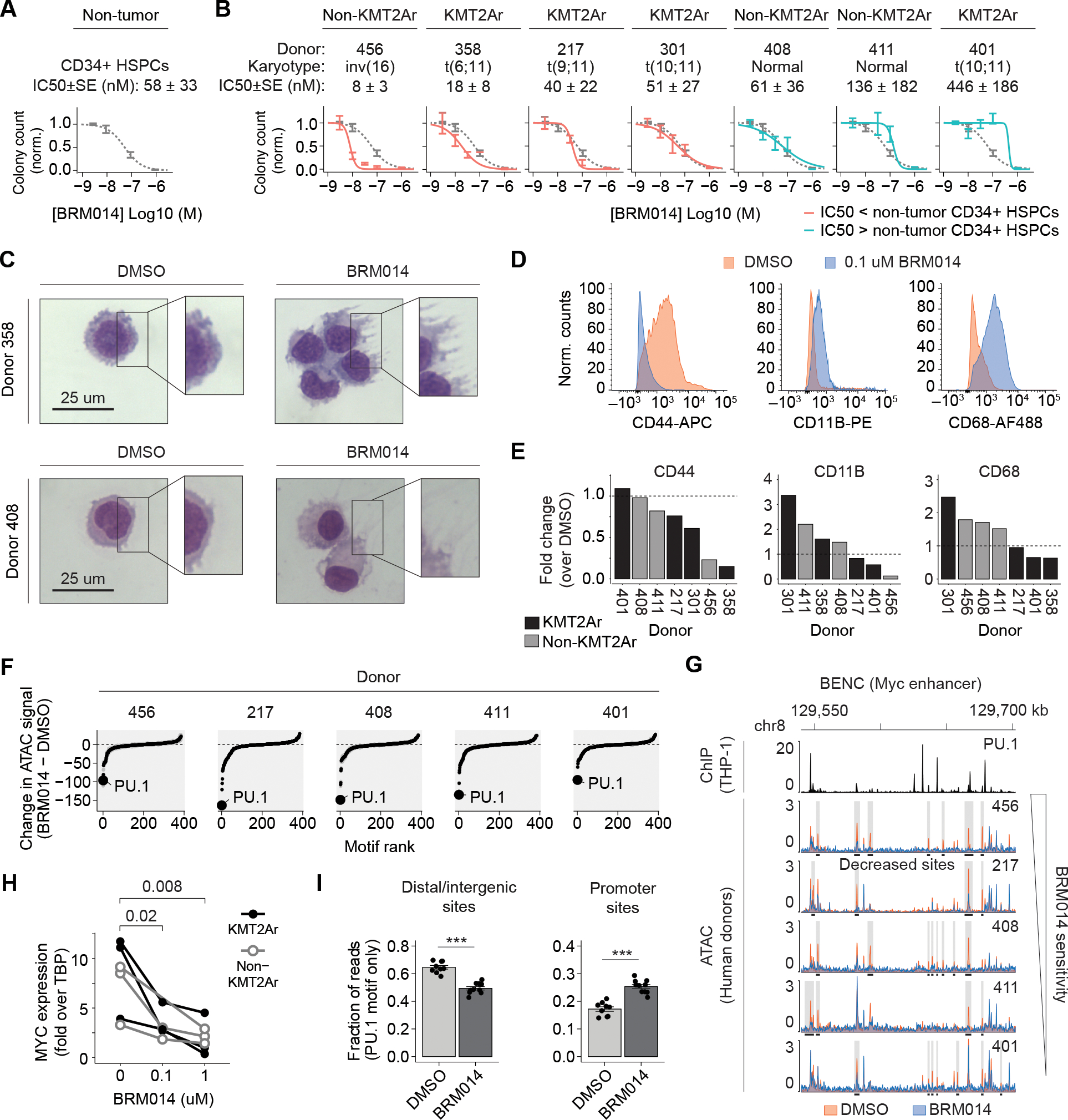
Number of colonies formed by primary human (A) CD34+ HSPCs and (B) AML samples treated with BRM014 or DMSO and IC50 measurements. CD34+ HSPC curves overlayed in (B) for comparison, N=3 per sample. (C) Representative images of May-Grunwald Giemsa stained primary KMT2Ar and non-KMT2Ar leukemia cells from donors treated with BRM014 or DMSO. (D) Representative flow cytometry histograms and (E) quantification of CD44 and myeloid differentiation markers in primary leukemias treated with BRM014 compared to DMSO control. (F) Ranking of differential TF motif accessibility in primary AML specimens treated with 1 uM BRM014 versus DMSO control. (G) Accessibility at BENC module following treatment with DMSO or 1 uM BRM014. (H) MYC expression after treatment with BRM014 or DMSO measured by RT-qPCR. (I) Quantification of accessible PU.1 motifs at intergenic and promoter sites in primary AML specimens treated with 1 uM BRM014 or DMSO, N=5 independent specimens with replicates. Error bars: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
To assess the effect of SWI/SNF inhibition on human AML samples, we obtained leukemic blasts from AML patients and evaluated their sensitivity to BRM014. BRM014 caused dose-dependent reduction of colony formation potential (Fig. 6B and Supplementary Fig. S7D) and altered cell morphology (Fig. 6C). While the majority of AML specimens had a lower IC50 compared to non-tumor CD34+ HSPCs, three of the seven had higher IC50 values. This variation in therapeutic response indicates that the balance between on- and off-tumor effects gives rise to a unique therapeutic window for each patient. Effective concentrations of BRM014 varied between donors, but 1 uM of BRM014 was sufficient to ablate colony formation potential of all leukemias. Remarkably, one patient sample (358) that was highly responsive to BRM014 treatment bore the KMT2A-MLLT4 (MLL-AF6) fusion [t(6;11)], which is the most therapy-refractory KMT2A fusion, associated with event-free survival odds of only 11–17% (57,58). No systematic variation was observed between KMT2Ar and non-KMT2Ar samples, indicating that SWI/SNF inhibition targets mechanisms that contribute to AML maintenance independently of KMT2A fusion status.
Treatment with BRM014 did not result in significant loss of viability of primary leukemia cells within 48 hours, even at concentrations up to 1 μM (Supplementary Fig. S7E). Flow cytometry of human samples confirmed accumulation of myeloid differentiation markers (CD11B, CD68) and loss of the stemness marker CD44, a signature of PU.1 deregulation in AML (Fig. 6D–E). Markers activated upon SWI/SNF inhibition varied by donor, but indicated a general myeloid differentiation phenotype.
Similar to our cell line observations, ATAC-seq revealed PU.1 to be the most decreased motif following BRM014 treatment (Fig. 6F). Moreover, BRM014 caused loss of accessibility at BENC in all specimens (Fig. 6G), with concomitant loss of MYC expression (Fig. 6H). Notably, we observed significant heterogeneity in the specific peaks lost at BENC, and hence variation in chromatin state may underlie crucial aspects of the heterogeneous patient-specific responses to SWI/SNF inhibition. Excitingly, BRM014 caused accessibility losses at intergenic regions with PU.1 motifs (p=2.5e-7) and gains at promoter regions with PU.1 motifs (p=1.4e-6, Fig. 6I), consistent with the mechanisms observed in our cell line studies. Overall, our findings reveal that primary human AMLs are highly sensitive to pharmacological targeting of SWI/SNF and indicate that the balance between on- and off-tumor effects on hematopoietic cells will be an important clinical consideration in humans.
Discussion
Subunits of SWI/SNF complexes act as oncogenic dependencies in multiple cancer types (59–62). In these cancers, pharmacological inhibition of SWI/SNF represents a promising and novel strategy to target tumor-specific chromatin dependencies. Targeting tumor cell-intrinsic dependencies may be particularly valuable for cancers with low mutational burdens, which often have few targetable mutations (63,64) and respond poorly to immunotherapies due to low immunogenicity (65). Hematopoietic malignancies such as leukemia are ideal candidates for this strategy, due to their low mutation burden and demonstrated dependence on SWI/SNF activity.
Acute leukemias rely on SWI/SNF activity to maintain the stemness of leukemic cells and to prevent terminal differentiation. In this setting, SWI/SNF complexes as well as PU.1 and CRC member RUNX1 have been linked to regulation of MYC expression (13,66–68). Our work reveals that PU.1 requires SWI/SNF activity to recruit CRC factors to distal enhancers, including the BENC module that drives MYC. When SWI/SNF is inhibited, PU.1 is retained at many sites, but is also relocalized to H3K4me3-bound promoter sites associated with myeloid differentiation genes.
Variable outcomes regarding the requirement for SWI/SNF subunits for hematopoiesis (59,69,70) have created uncertainty about whether SWI/SNF inhibition would cause adverse hematological effects. Here, we demonstrate that transient SWI/SNF inhibition dysregulates hematopoietic development within bone marrow, with lineage-specific effects similar to PU.1 inhibition, which also impacts committed PU.1-dependent lineages but spares HSCs (47). Nevertheless, long-term PU.1 impairment has been reported to impair HSC function (71), therefore additional studies on the long-term use of SWI/SNF inhibition are warranted. Although our work identifies impaired development in B cells and monocytes, lineage tracing studies can be expected to provide deeper insight into the altered mechanisms within each lineage. Importantly, the immune-related toxicities observed during short-term treatment compare favorably to the dose-limiting and persistent immunosuppression associated with standard-of-care chemotherapy (72,73).
Recent studies showed that ex vivo inhibition of SWI/SNF complexes in T cells enhances memory T cell formation (74) and limits T cell exhaustion (75), which is not assessed in our studies due to the absence of an immune challenge. Importantly, unlike ex vivo treatment, in vivo SWI/SNF inhibition also impacts antigen-presenting cells such as macrophages. Our work shows that full consideration of the distinct effects on diverse cell types is required to understand the impact of SWI/SNF inhibition on immune response in vivo.
The strong leukemic regression achieved by SWI/SNF inhibitors in vivo demonstrates their remarkable potential for use in AML. Still, dosing regimens and treatment duration will require further optimization to achieve optimal outcomes in vivo, for example, by accounting for external factors including drug metabolism and small-molecule pharmacokinetics/pharmacodynamics that limit drug efficacy. Our work highlights that normal hematopoiesis can be affected at doses similar to those needed for therapeutic response in AML specimens, which will be an important consideration for clinical use of SWI/SNF inhibitors against AML and other tumors. Nevertheless, the robust regression of leukemic burden seen over a short two-week treatment period suggests a considerable therapeutic window in immunocompetent settings for some patients, and provides a compelling justification for continued study of SWI/SNF inhibitors in the treatment of AML.
Supplementary Material
Highlights.
PU.1 recruits SWI/SNF to enhancers to generate DNA accessibility for binding of core AML regulators, including RUNX1, LMO2, and MEIS1.
Disruption of PU.1-directed enhancers by SWI/SNF inhibition drives AML differentiation and influences non-tumor B cell and monocyte development without altering HSC function.
Significance:
Disruption of PU.1-directed enhancer programs upon SWI/SNF inhibition causes differentiation of AML cells and induces leukopenia of PU.1-dependent B cells and monocytes, revealing the on- and off-tumor effects of SWI/SNF blockade.
Acknowledgements
We thank Zainab Jagani and Florencia Rago (Novartis) for helpful comments and feedback. This work was supported by grants to H. C. Hodges from Gabrielle’s Angel Foundation, the V Foundation (V2018–003), the Mark Foundation for Cancer Research (20–024-ASP), and the Adrienne Helis Malvin Medical Research Foundation. H. C. Hodges is a CPRIT Scholar supported by grants from the NIH (R35GM137996 and R01CA272769) and the Cancer Prevention and Research Institute of Texas (CPRIT RR170036). C. Chambers is supported by NIH grant F31AI161906. H. D. Lacorazza is supported by NIH grant R01CA207086 and the ASH Bridge Award. This project was also supported by the UTMB Next Generation Sequencing Core, the BCM Cytometry and Cell Sorting Core (CPRIT RP180672, and NIH P30CA125123, S10RR024574), the BCM Single Cell Genomics Core (NIH S10OD018033, S10OD023469, S10OD025240 and P30EY002520), the BCM Genomic and RNA Profiling Core (NIH S10OD023469), the Texas Children’s Cancer Center tissue repository, and the MD Anderson Next-Generation Sequencing Core (CPRIT RP120348).
Footnotes
Disclosure of Conflicts of Interest: The authors declare no conflicts of interest
References
- 1.de Kouchkovsky I, Abdul-Hay M. ‘Acute myeloid leukemia: a comprehensive review and 2016 update.’ Blood Cancer J. 2016;6(7):e441–e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lonetti A, Pession A, Masetti R. Targeted Therapies for Pediatric AML: Gaps and Perspective. Front Pediatr. 2019;7:463–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zwaan CM, Kolb EA, Reinhardt D, et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. Journal of Clinical Oncology. 2015;33(27):2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nature Reviews Immunology 2007 7:2. 2007;7(2):105–117. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Xu L, Lin RYT, Müschen M, Koeffler HP. Core transcriptional regulatory circuitries in cancer. Oncogene 2020 39:43. 2020;39(43):6633–6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goyama S, Schibler J, Cunningham L, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123(9):3876–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong P, Iwasaki M, Somervaille TCP, So CWE, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21(21):2762–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diffner E, Beck D, Gudgin E, et al. Activity of a heptad of transcription factors is associated with stem cell programs and clinical outcome in acute myeloid leukemia. Blood. 2013;121(12):2289–2300. [DOI] [PubMed] [Google Scholar]
- 9.Porcher C, Swat W, Rockwell K, et al. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell. 1996;86(1):47–57. [DOI] [PubMed] [Google Scholar]
- 10.Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nature Reviews Cancer 2003 3:2. 2003;3(2):89–101. [DOI] [PubMed] [Google Scholar]
- 11.Papillon JPN, Nakajima K, Adair CD, et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/ SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J Med Chem. 2018;61(22):10155–10172. [DOI] [PubMed] [Google Scholar]
- 12.Shi J, Wang E, Milazzo JP, et al. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33(6):661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi J, Whyte WA, Zepeda-Mendoza CJ, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013;27(24):2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rago F, Rodrigues LU, Bonney M, et al. Exquisite Sensitivity to Dual BRG1/BRM ATPase Inhibitors Reveals Broad SWI/SNF Dependencies in Acute Myeloid Leukemia. Molecular cancer research. 2021;molcanres.MCR-21–0390-A.2021. [DOI] [PubMed] [Google Scholar]
- 15.Bahr C, von Paleske L, Uslu V v., et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018 553:7689. 2018;553(7689):515–520. [DOI] [PubMed] [Google Scholar]
- 16.Krasteva V, Buscarlet M, Diaz-Tellez A, et al. The BAF53a subunit of SWI/SNF-like BAF complexes is essential for hemopoietic stem cell function. Blood. 2012;120(24):4720–4732. [DOI] [PubMed] [Google Scholar]
- 17.Krasteva V, Crabtree GR, Lessard JA. The BAF45a/PHF10 subunit of SWI/SNF-like chromatin remodeling complexes is essential for hematopoietic stem cell maintenance. Exp Hematol. 2017;48:58–71.e15. [DOI] [PubMed] [Google Scholar]
- 18.O’Neill D, Yang J, Erdjument-Bromage H, et al. Tissue-specific and developmental stage-specific DNA binding by a mammalian SWI/SNF complex associated with human fetal-to-adult globin gene switching. Proc Natl Acad Sci U S A. 1999;96(2):349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee CH, Murphy MR, Lee JS, Chung JH. Targeting a SWI/SNF-related chromatin remodeling complex to the β-globin promoter in erythroid cells. Proceedings of the National Academy of Sciences. 1999;96(22):12311–12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kowenz-Leutz E, Leutz A. A C/EBPβ Isoform Recruits the SWI/SNF Complex to Activate Myeloid Genes. Mol Cell. 1999;4(5):735–743. [DOI] [PubMed] [Google Scholar]
- 21.Wu J, Krchma K, Lee J, et al. Requisite Chromatin Remodeling for Myeloid and Erythroid Lineage Differentiation from Erythromyeloid Progenitors. Cell Rep. 2020;33(7):108395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chi TH, Wan M, Lee PP, et al. Sequential roles of Brg, the ATPase subunit of BAF chromatin remodeling complexes, in thymocyte development. Immunity. 2003;19(2):169–182. [DOI] [PubMed] [Google Scholar]
- 23.Gebuhr TC, Kovalev GI, Bultman S, et al. The Role of Brg1, a Catalytic Subunit of Mammalian Chromatin-remodeling Complexes, in T Cell Development. J Exp Med. 2003;198(12):1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loo C-S, Gatchalian J, Liang Y, et al. A Genome-wide CRISPR Screen Reveals a Role for the Non-canonical Nucleosome-Remodeling BAF Complex in Foxp3 Expression and Regulatory T Cell Function. Immunity. 2020;53(1):143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minderjahn J, Schmidt A, Fuchs A, et al. Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat Commun. 2020;11:402-undefined. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwafuchi-Doi M, Zaret KS. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014;28(24):2679–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sunkel BD, Stanton BZ. Pioneer factors in development and cancer. iScience. 2021;24(10):. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science (1979). 1994;265(5178):1573–1577. [DOI] [PubMed] [Google Scholar]
- 29.DeKoter RP, Singh H. Regulation of B Lymphocyte and Macrophage Development by Graded Expression of PU.1. Science (1979). 2000;288(5470):1439–1442. [DOI] [PubMed] [Google Scholar]
- 30.Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene 2007 26:47. 2007;26(47):6816–6828. [DOI] [PubMed] [Google Scholar]
- 31.Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta P, Gurudutta GU, Saluja D, Tripathi RP. PU.1 and partners: regulation of haematopoietic stem cell fate in normal and malignant haematopoiesis. J Cell Mol Med. 2009;13(11–12):4349–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson NK, Foster SD, Wang X, et al. Combinatorial Transcriptional Control In Blood Stem/Progenitor Cells: Genome-wide Analysis of Ten Major Transcriptional Regulators. Cell Stem Cell. 2010;7(4):532–544. [DOI] [PubMed] [Google Scholar]
- 34.Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14(10):959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodges HC, Stanton BZ, Cermakova K, et al. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat Struct Mol Biol. 2018;25:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cermakova K, Demeulemeester J, Lux V, et al. A ubiquitous disordered protein interaction module orchestrates transcription elongation. Science (1979). 2021;374(6571):1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25–R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schep AN, Wu B, Buenrostro JD, Greenleaf WJ. chromVar: inferring transcription-factor- associated accessibility from single-cell epigenomic data. Nat Methods. 2017;14(10):975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pohl A, Beato M. bwtool: a tool for bigWig files. Bioinformatics. 2014;30(11):1618–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8(4):329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thibodeau A, Eroglu A, McGinnis CS, et al. AMULET: a novel read count-based method for effective multiplet detection from single nucleus ATAC-seq data. Genome Biol. 2021;22(1):. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25(21):2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hosokawa H, Ungerbäck J, Wang X, et al. Transcription Factor PU.1 Represses and Activates Gene Expression in Early T Cells by Redirecting Partner Transcription Factor Binding. Immunity. 2018;48(6):1119–1134.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ungerbäck J, Hosokawa H, Wang X, et al. Pioneering, chromatin remodeling, and epigenetic constraint in early T-cell gene regulation by SPI1 (PU.1). Genome Res. 2018;28(10):1508–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antony-Debré I, Paul A, Leite J, et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J Clin Invest. 2017;127(12):4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwasaki H, Somoza C, Shigematsu H, et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood. 2005;106(5):1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Baron RM, Zhu G, et al. PU.1 regulates cathepsin S expression in professional APCs. Journal of immunology. 2006;176(1):275–283. [DOI] [PubMed] [Google Scholar]
- 50.Chen HM, Ray-Gallet D, Zhang P, et al. PU.1 (Spi-1) autoregulates its expression in myeloid cells. Oncogene. 1995;11(8):1549–1560. [PubMed] [Google Scholar]
- 51.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nature Medicine 2006 12:10. 2006;12(10):1167–1174. [DOI] [PubMed] [Google Scholar]
- 52.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. [DOI] [PubMed] [Google Scholar]
- 53.Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kurtz KJ, Conneely SE, O’Keefe M, Wohlan K, Rau RE. Murine Models of Acute Myeloid Leukemia. Front Oncol. 2022;12:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krivtsov A v., Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 2006 442:7104. 2006;442(7104):818–822. [DOI] [PubMed] [Google Scholar]
- 56.Lewis AH, Bridges CS, Moorshead DN, et al. Krüppel-like Factor 4 Supports the Expansion of Leukemia Stem Cells in MLL-AF9-driven Acute Myeloid Leukemia. Stem Cells. 2022;40(8):736–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Balgobind B v., Raimondi SC, Harbott J, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114(12):2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pollard JA, Guest E, Alonzo TA, et al. Gemtuzumab Ozogamicin Improves Event-Free Survival and Reduces Relapse in Pediatric KMT2A-Rearranged AML: Results From the Phase III Children’s Oncology Group Trial AAML0531. J Clin Oncol. 2021;39(28):3149–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buscarlet M, Krasteva V, Ho L, et al. Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood. 2014;123(11):1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jubierre L, Soriano A, Planells-Ferrer L, et al. BRG1/SMARCA4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene. 2016;35(39):5179–5190. [DOI] [PubMed] [Google Scholar]
- 61.Cyrta J, Augspach A, de Filippo MR, et al. Role of specialized composition of SWI/SNF complexes in prostate cancer lineage plasticity. Nat Commun. 2020;11(1):5549–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rago F, Elliott GN, Li A, et al. The Discovery of SWI/SNF Chromatin Remodeling Activity as a Novel and Targetable Dependency in Uveal Melanoma. Mol Cancer Ther. 2020;19(10):2186–2195. [DOI] [PubMed] [Google Scholar]
- 63.TJ L, C M, L D, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daver N, Alotaibi AS, Bücklein V, Subklewe M. T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia. 2021;35(7):1843–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Curtiss BM, VanCampen J, Macaraeg J, et al. PU.1 and MYC transcriptional network defines synergistic drug responses to KIT and LSD1 inhibition in acute myeloid leukemia. Leukemia 2022 36:7. 2022;36(7):1781–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bakshi R, Hassan MQ, Pratap J, et al. The human SWI/SNF complex associates with RUNX1 to control transcription of hematopoietic target genes. J Cell Physiol. 2010;225(2):569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mill CP, Fiskus W, DiNardo CD, et al. RUNX1-targeted therapy for AML expressing somatic or germline mutation in RUNX1. Blood. 2019;134(1):59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willis MS, Homeister JW, Rosson GB, et al. Functional Redundancy of SWI/SNF Catalytic Subunits in Maintaining Vascular Endothelial Cells in the Adult Heart. Circ Res. 2012;111(5):e111–e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roberts CWM, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell. 2002;2(5):415–425. [DOI] [PubMed] [Google Scholar]
- 71.Staber PB, Zhang P, Ye M, et al. Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol Cell. 2013;49(5):934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616–1634. [DOI] [PubMed] [Google Scholar]
- 73.Crawford J, Dale DC, Lyman GH. Chemotherapy-induced neutropenia. Cancer. 2004;100(2):228–237. [DOI] [PubMed] [Google Scholar]
- 74.Guo A, Huang H, Zhu Z, et al. cBAF complex components and MYC cooperate early in CD8+ T cell fate. Nature 2022 607:7917. 2022;607(7917):135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Belk JA, Yao W, Ly N, et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell. 2022;40(7):768–786.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data generated for this project have been deposited in the Gene Expression Omnibus (GEO, RRID: SCR_005012) database with SuperSeries accession numbers GSE205572 and GSE190723. Public H3K4me3 ChIP-seq data were analyzed from GEO accession GSM3681460.