

Abstract
Aims
To estimate how much information conveyed by self-reported family history of heart disease (FHHD) is already explained by clinical and genetic risk factors.
Methods and results
Cross-sectional analysis of UK Biobank participants without pre-existing coronary artery disease using a multivariable model with self-reported FHHD as the outcome. Clinical (diabetes, hypertension, smoking, apolipoprotein B-to-apolipoprotein AI ratio, waist-to-hip ratio, high sensitivity C-reactive protein, lipoprotein(a), triglycerides) and genetic risk factors (polygenic risk score for coronary artery disease [PRSCAD], heterozygous familial hypercholesterolemia [HeFH]) were exposures. Models were adjusted for age, sex, and cholesterol-lowering medication use. Multiple logistic regression models were fitted to associate FHHD with risk factors, with continuous variables treated as quintiles. Population attributable risks (PAR) were subsequently calculated from the resultant odds ratios. Among 166 714 individuals, 72 052 (43.2%) participants reported an FHHD. In a multivariable model, genetic risk factors PRSCAD (OR 1.30, CI 1.27–1.33) and HeFH (OR 1.31, 1.11–1.54) were most strongly associated with FHHD. Clinical risk factors followed: hypertension (OR 1.18, CI 1.15–1.21), lipoprotein(a) (OR 1.17, CI 1.14–1.20), apolipoprotein B-to-apolipoprotein AI ratio (OR 1.13, 95% CI 1.10–1.16), and triglycerides (OR 1.07, CI 1.04–1.10). For the PAR analyses: 21.9% (CI 18.19–25.63) of the risk of reporting an FHHD is attributed to clinical factors, 22.2% (CI% 20.44–23.88) is attributed to genetic factors, and 36.0% (CI 33.31–38.68) is attributed to genetic and clinical factors combined.
Conclusions
A combined model of clinical and genetic risk factors explains only 36% of the likelihood of FHHD, implying additional value in the family history.
Keywords: Family history of heart disease, Risk factor, Genetics, Polygenic risk score, Prevention, Cardiovascular disease, UK Biobank
Structured Graphical Abstract
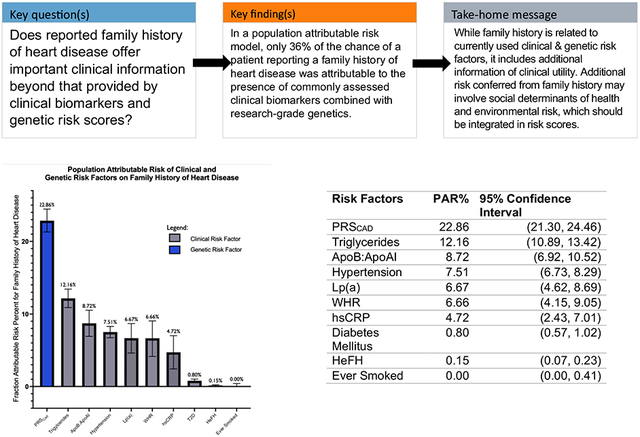
Population attributable risk percent for family history of heart disease by individual risk factors. Here, we present the population attributable risk for each risk factor, individually modeled for reported family history of heart disease, controlling for age and sex. For all continuous variables, the top four quintiles were compared to the bottom quintile, as indicated by [2nd–5th quintiles vs. 1st quintile (reference)]. A lower bound of 0 was created for PAR.
Lay Summary
With advances in genetics, it is tempting to assume that the ‘family history’ of a patient is an imperfect proxy for information we can already glean from genetics and laboratory tests. However, this study shows that much of the information contained in the self-reported family history of heart disease is not captured by currently available genetic and clinical biomarkers and highlights an important knowledge gap.
Clinically used biomarkers explained only 21.9% of the likelihood of a patient reporting a family history of heart disease, while genetics explained 22.2%, and a combined model explained 36% of this likelihood
The majority of the risk of reporting a family history went unexplained, implying that family history still has major relevance in clinical practice.
Introduction
With greater availability of polygenic risk scores (PRS), the related interpretation of a self-reported family history of heart disease (FHHD) will be increasingly important. FHHD has been long recognized as a cardiovascular disease (CVD) risk factor, with a graded increase in CVD risk associated with the number of first-degree relatives with CVD and relatives’ age of CVD onset. Clinicians will be increasingly faced with the challenge of interpreting whether the information contained in family history is completely captured in genetics and to what extent non-genetic modifiable risk factors play a role.1-4
The use of family history in CVD risk prediction is variable. Some clinical risk calculators such as the pooled cohort equations (PCE)5 do not integrate family history, while others such as the Reynolds risk score do.6 Additionally, the Dutch lipid clinical network score (DLCNS) for heterozygous familial hypercholesterolemia (HeFH) uses family history to potentially enhance the yield of genetic testing.7 In this study, we estimate the extent to which FHHD can be explained by common clinical biomarkers alongside PRS and HeFH genotypes.
Here, we leverage the UK Biobank to characterize the extent to which genetic and clinical factors are responsible for a self-reported FHHD. Clinical risk factors in this paper include medical history (diabetes, hypertension), physical measurements (waist-to-hip ratio [WHR]), and laboratory values (apolipoprotein B, apolipoprotein AI, lipoprotein(a) [Lp(a)], triglycerides, and high sensitivity C-reactive protein [hsCRP]).8-12 Included genetic risk factors are HeFH status and polygenic risk score for coronary artery disease (PRSCAD.)13,14 In this paper, we assess the extent to which the risk conveyed by a self-reported FHHD can be explained by clinical risk factors, genetic risk factors, or the two in combination. We further evaluate to what extent this risk remains unexplained to attempt to quantify the utility of FHHD at the population level.
Methods
Participants
The UK Biobank (UKBB) is a population-based cohort of adults followed prospectively, with an age of adults at recruitment ranging between 40 and 69 years (N = 502 629).15 Recruitment occurred between 2006 and 2010, and participants gave written consent. At the baseline study visit, participants provided demographic information, medical history, family history, medication use, and lifestyle information (such as smoking). We restricted the analysis to individuals with available genotype array data and whole exome sequences to enable ascertainment of PRSCAD and HeFH variant. 281 341 individuals were excluded due to either missing genotyping or whole exome sequencing (WES) necessary for HeFH and PRSCAD calculation. 36 169 individuals were excluded due to withdrawn consent, relatedness, or missing coronary artery disease (CAD) status. To focus on primary prevention, 6445 individuals were excluded given CAD present at enrollment, defined as the history of myocardial infarction, coronary artery bypass graft (CABG) or coronary angioplasty; ICD-10 code for acute myocardial infarction; hospitalization for OPCS-4 coded procedure: CABG; and hospitalization for OPSC-4 coded procedure: coronary angioplasty with or without stenting. Finally, 11 860 individuals were excluded for missing Lp(a) laboratory values. The final sample included 166 714 individuals (Figure 1). The secondary use of data for this study was approved by the Massachusetts General Hospital Institutional Review Board (protocol 2021P002228) facilitated through UKB application 7089.
Figure 1.

Study population. The UK Biobank is a population-based cohort of adults who follow prospectively, with ages ranging between 40 and 70 years. Abbreviations: CAD, coronary artery disease; FHHD, family history of heart disease; WES, whole exome sequencing; PAR, population attributable risk; PRSCAD, polygenic risk score for coronary artery disease; Lp(a), Lipoprotein(a) nmol/L.
Baseline characteristics, clinical exposures, and outcome
Baseline characteristics included sex (field code 31) and age at recruitment (field code 21022). Exposures encompassed both clinical and genetic factors. Clinical factors were defined as laboratory data, medical diagnoses, anthropometric measures, and smoking. Laboratory data were linked to the UK Biobank via electronic health records (EHR) and included hsCRP, apolipoprotein B, apolipoprotein AI, Lp(a), total cholesterol, triglycerides, high-density lipoprotein (HDL) cholesterol, and low-density lipoprotein (LDL) cholesterol (field codes 30710, 30640, 30630, 30790, 30690, 30870, 30760, and 30780). ApoB:ApoAI was used in lieu of LDL or total cholesterol levels. Continuous variables ApoB:ApoAI, triglycerides, hsCRP, and Lp(a) were divided into quintiles. Medical diagnoses of diabetes and hypertension were linked to the UKBB via EHR with ICD10 diagnostic codes (I10-I15). Anthropometric measures such as BMI, waist circumference, and hip circumference (to calculate WHR) were ascertained at study intake (field codes 21001, 48, and 49). WHR was also divided into quintiles. Smoking habits were assessed using baseline survey data. Self-reported FHHD (field codes 20111 for a sibling, 20110 for mother, and 20107 for father) in at least one first-degree relative was used as the primary outcome. At study intake and on follow-up visits, participants were asked to indicate if there was a positive FHHD in their mother, father, or sibling(s). Specifically, using a touchscreen-based questionnaire, individuals were instructed to select from a list of common diseases the illnesses from which diseases their father, mother, and siblings suffered. Heart disease was listed as one of these diseases. Participants were not asked to indicate how many siblings have a history of heart disease. Full assay information for laboratory tests is available in Supplementary material online, Supplementary Methods. All field codes for baseline variables are available in Supplementary material online, Table S1.
Genotypic data: polygenic risk score, HeFH status
Individual-level 2.99 million single nucleotide polymorphisms (SNPs) PRSCAD were constructed on unrelated British participants of the UK Biobank as described in a previous report using the AnnoPred framework.16,17 AnnoPred is a Bayesian framework leveraging diverse types of genomic and epigenomic functional annotations to improve risk prediction accuracy. It uses an empirical prior of SNP effect size based on functional annotations of the SNPs and signal enrichment in different annotation classes estimated from GWAS summary statistics.16,17 After the calculation of the PRSCAD, risk scores were binned by quintile.
To ascertain HeFH status, whole exome sequencing (WES) data was used, and a history of premature CAD was not used. The UKBB released WES data for 200 644 individuals in October 2020. Single nucleotide variants (SNVs) that had a minor allele frequency <0.1% in the LDLR, APOB, PCSK9, LDLRAP1, ABCG5, or ABCG8 and were manually curated for association with familial hypercholesteremia (FH). SNVs in LDLR that predicted loss-of-function variants (nonsense, frameshift, and essential splice site) or were classified as likely pathogenic or pathogenic by most analysis sites were considered FH variants. SNVs in APOB and PCSK9 that were annotated in ClinVar as likely pathogenic or pathogenic by most analysis sites were considered FH variants. There were no cases of FH associated with autosomal recessive inheritance (LDLRAP1, ABCG5, or ABCG8). Additional details are available in Supplementary material online, Supplementary Methods and a list of all variants used to define HeFH are available in Supplementary material online, Table S2.
Statistical analyses
The primary statistical analysis in this study is the population attributable risk (PAR) model wherein the percent chance of an outcome attributable to a given risk factor in a population is calculated based on that risk factor’s prevalence in the target population. This is done by first constructing regression models to estimate the odds ratio, and then weighting the odds ratio based upon the relative prevalence of the risk factor. Thus, first, multiple logistic regression models were constructed to assess the effect of CVD risk factors on the odds of FHHD being reported. Individual clinical and genetic risk factors were tested using minimal adjustment—controlling only for age, sex, and use of cholesterol-lowering medication. Next, all clinical and genetic risk factors were simultaneously tested in a fully adjusted model. Issues of collinearity were considered as the PRSCAD contains HeFH variants and the LPA locus (determining Lp(a) level). Removal of the LPA locus from a CAD PRS does not substantively alter performance for CAD risk prediction.18 Additionally, in fully adjusted models inclusion of PRSCAD. HeFH, and Lp(a) should have little effect or bias towards the null. As was comparably done in the INTERHEART study,19 continuous variables were treated as polychotomous variables in quintiles, with quintile 1 equating to the lowest risk and quintile five equating to the highest risk. In subsequent regression models, the 1st quintile was considered the reference compared to the 2nd–5th quintiles. P-value of <0.05 was the threshold used for significance.
Then, for models to be utilized in the population attributable risk (PAR) framework, an optimal reference risk level needed to be established. For binary or categorical variables, the absence of the risk factor was used (e.g. absence of diabetes, absence of hypertension, or never smoking). For polychotomous exposures, the PAR is the cumulative effect of bringing all levels to the target or reference value. As was comparably done in the INTERHEART study,19 continuous variables were treated as polychotomous variables in quintiles, with quintile 1 equating to the lowest risk. In subsequent PAR models, any increase in risk above the 1st quintile was treated as the presence of a risk factor. The 1st quintile was considered as the reference compared to the 2nd–5th quintiles. P-value of <0.05 was the threshold used for significance.
Population attributable risk (PAR) was determined from odds ratios derived from multiple logistic regression models and was used to assess the extent to which clinical and/or genetic factors explain a self-reported FHHD. The PAR calculations were conducted in the R package ‘attribrisk.’20 The PAR analyses were calculated with (1) genetic risk factors only, (2) clinical risk factors only, and (3) combined genetic and clinical risk factors. Age, sex, and cholesterol-lowering medication use were controlled for in these analyses. Additionally, individual PAR analyses were performed for each risk factor separately to compare the effect of each factor on the risk of self-reported FHHD, controlling for age, and sex.
95% confidence intervals were computed from a group jackknife estimate of variance based on k = 20 groups.
All analyses were completed using R 4.1.2.
Results
Baseline characteristics
Among the 166 714 participants, 55.7% (92 942) reported female sex at birth and the mean age was 56.2 years (standard deviation [SD] 8.05) and 57.1 (SD 7.5) years in those with positive FHHD. 73 007 (43.8%) reported ever smoking (43.7% among those with positive FHHD), 10 319 (6.2%) had a history of diabetes (6.7% among those with positive FHHD), and median WHR was 0.87 (inter quartile range [IQR] = 0.48–2.97) (0.87 among those with positive FHHD). 93.7% (156 152) identified as White, 2.1% (3556) as South Asian, 1.7% (2755) as Black, 1.0% (1684) as other, 0.7% (1135) as mixed, and 0.3% (574) as Chinese. Baseline characteristics are summarized in Table 1.
Table 1.
Baseline characteristics
| FHHD absent (N = 94 662) |
FHHD present (N = 72 052) |
Overall (N = 166 714) |
P-value | |
|---|---|---|---|---|
| Age | ||||
| Mean (SD) | 55.6 (8.35) | 57.1 (7.54) | 56.2 (8.05) | <0.001 |
| Sex | ||||
| Female | 50 250 (53.1%) | 42 692 (59.3%) | 92 942 (55.7%) | <0.001 |
| Male | 44 412 (46.9%) | 29 360 (40.7%) | 73 772 (44.3%) | |
| WHR | ||||
| Median [Min, Max] | 0.872 [0.506, 2.97] | 0.866 [0.484, 1.65] | 0.870 [0.484, 2.97] | <0.001 |
| Ever smoked status | ||||
| Absent | 53 113 (56.1%) | 40 594 (56.3%) | 93 707 (56.2%) | 0.347 |
| Present | 41 549 (43.9%) | 31 458 (43.7%) | 73 007 (43.8%) | |
| Hypertension | ||||
| Absent | 68 354 (72.2%) | 47 334 (65.7%) | 115 688 (69.4%) | <0.001 |
| Present | 26 308 (27.8%) | 24 718 (34.3%) | 51 026 (30.6%) | |
| History of diabetes | ||||
| Absent | 89 170 (94.2%) | 67 225 (93.3%) | 156 395 (93.8%) | <0.001 |
| Present | 5492 (5.8%) | 4827 (6.7%) | 10 319 (6.2%) | |
| Cholesterol medication use | ||||
| Absent | 83 528 (88.2%) | 59 047 (82.0%) | 142 575 (85.5%) | <0.001 |
| Present | 11 134 (11.8%) | 13 005 (18.0%) | 24 139 (14.5%) | |
| HeFH status | ||||
| Absent | 94 380 (99.7%) | 71 731 (99.6%) | 166 111 (99.6%) | <0.001 |
| Present | 282 (0.3%) | 321 (0.4%) | 603 (0.4%) | |
| PRSCAD | ||||
| Mean (SD) | 48.3 (28.8) | 53.3 (28.6) | 50.5 (28.9) | <0.001 |
| Lipoprotein(a) | ||||
| Median [Min, Max] | 18.0 [0.0200, 722] | 21.1 [0.0200, 743] | 19.3 [0.0200, 743] | <0.001 |
| hsCRP | ||||
| Median [Min, Max] | 1.30 [0.0800, 78.4] | 1.37 [0.0800, 79.5] | 1.33 [0.0800, 79.5] | <0.001 |
| Total cholesterol | ||||
| Mean (SD) | 221 (42.7) | 223 (43.5) | 222 (43.1) | <0.001 |
| LDL cholesterol | ||||
| Mean (SD) | 138 (32.5) | 140 (33.1) | 139 (32.8) | <0.001 |
| HDL cholesterol | ||||
| Mean (SD) | 56.5 (14.3) | 56.7 (14.1) | 56.6 (14.2) | 0.00839 |
| Triglycerides | ||||
| Median [Min, Max] | 129 [20.5, 988] | 134 [25.4, 999] | 131 [20.5, 999] | <0.001 |
| ApoA concentration | ||||
| Mean (SD) | 1.54 (0.259) | 1.55 (0.258) | 1.55 (0.258) | <0.001 |
| ApoB concentration | ||||
| Mean (SD) | 1.03 (0.232) | 1.05 (0.234) | 1.04 (0.233) | <0.001 |
| ApoB/ApoA ratio | ||||
| Mean (SD) | 0.687 (0.197) | 0.693 (0.197) | 0.690 (0.197) | <0.001 |
Baseline characteristics for the study population. The mean value is listed for normally distributed values. The median value is shown for non-normally distributed values.
ApoAI, apolipoprotein AI; ApoB, apolipoprotein B; ApoB:ApoAI, apolipoprotein B-to-apolipoprotein AI ratio; HDL, high-density lipoprotein; FHHD, family history of heart disease; HeFH, heterozygous familial hypercholesteremia; hsCRP, high sensitivity C-reactive protein; LDL, low-density lipoprotein; Lp(a), lipoprotein(a); PRSCAD Percentile, polygenic risk score for coronary artery disease; WHR, waist-to-hip ratio.
Family history, polygenic score, and risk of heart disease
The prevalence of self-reported FHHD for first-degree relatives was 43.2% (72 052). Among those with an FHHD, 47 821 (28.7%) reported a father, 30 663 (18.4%) reported a mother, and 13 078 (7.8%) reported at least one sibling with a history of heart disease (HD) (see Supplementary material online, Table S3). The reported frequency of family history of HD, stroke, hypertension, and diabetes in the study cohort is displayed in Supplementary material online, Table S3. Among those with an FHHD, the average age was 57.1 (SD 7.54), 31 458 (43.7%) endorsed ever smoking, 4827 (6.7%) had a history of diabetes and median WHR was 0.87 (IQR = 0.48–1.65). Probands who reported a greater number of first-degree relatives with HD had greater incident CAD risk in a dose-dependent manner (Figure 2A). The incidence of CAD among individuals with 0 family members with HD was 4.7%, the incidence among individuals with one family member with HD was 6.2%, the incidence among individuals with two family members with HD was 8.8%, and the incidence among individuals with three or more family members with HD was 13.2%. Furthermore, probands with more family members with HD had higher PRSCAD (Figure 2B).
Figure 2.

Graded relationship between the number of family members with (A) incident CAD and (B) polygenic risk score for CAD. χ2-test for trend was significant to P < 0.001 for both (A) and (B). Average follow-up time 10.7 years. Abbreviations: CAD, coronary artery disease; PRSCAD, polygenic risk score of coronary artery disease.
Regression models
Results of both minimally adjusted models assessing individual risk factors, and the fully adjusted model assessing all risk factors simultaneously, are reported in Table 2. When examining individual risk factors in univariate models, the largest odds (OR = odds ratio) of FHHD were found among genetic risk factors: OR 1.33 (95% CI 1.30–1.36) for the elevated PRSCAD [2nd–5th quintiles vs. 1st quintile (reference)], and OR 1.33 (95% CI 1.13–1.57) for HeFH. Following genetic risk factors, individual clinical risk factors that increased the odds of FHHD included: Lp(a) concentration [2nd–5th quintiles vs. 1st quintile (reference)] (OR 1.20, 95% CI 1.17–1.23), hypertension (OR 1.18, 95% CI 1.15–1.2), ApoB:ApoAI ratio [2nd–5th quintiles vs. 1st quintile (reference)] (OR 1.17, 95% CI 1.08–1.13), and triglycerides [2nd–5th quintiles vs. 1st quintile (reference)] (OR 1.11, 95% CI 1.08–1.13).
Table 2.
Odds ratios from fully adjusted and partially adjusted multiple logistic regression models
| Minimally adjusted model (individual risk factors) |
Fully adjusted model (all risk factors) |
|||||
|---|---|---|---|---|---|---|
| Odds ratio | 95% Confidence interval | P-value | Odds ratio | 95% Confidence interval | P-value | |
| Age | — | — | — | 1.02 | (1.02, 1.02) | 1.60e-160 |
| Male sex | — | — | — | 0.74 | (0.73, 0.76) | 4.20e-144 |
| Chol med | — | — | — | 1.51 | (1.46, 1.56) | 7.18e-142 |
| PRSCAD | 1.33 | (1.30, 1.36) | 5.48e-116 | 1.30 | (1.27, 1.33) | 8.98e- 98 |
| HeFH status | 1.33 | (1.13, 1.57) | 5.64e-4 | 1.31 | (1.11, 1.54) | 1.25e-3 |
| Lp(a) | 1.20 | (1.17, 1.23) | 2.49e- 48 | 1.17 | (1.14, 1.20) | 1.44e- 36 |
| Hypertension | 1.18 | (1.15, 1.20) | 9.80e-45 | 1.18 | (1.15. 1.21) | 2.09e- 43 |
| ApoB:ApoAI | 1.17 | (1.15, 1.20) | 3.14e- 36 | 1.13 | (1.10, 1.16) | 9.70e- 20 |
| Triglycerides | 1.11 | (1.08, 1.13) | 2.63e-15 | 1.07 | (1.04, 1.10) | 1.33e-6 |
| hsCRP | 1.02 | (1.00, 1.05) | 0.05 | 0.995 | (0.97, 1.02) | 0.67 |
| WHR | 0.99 | (0.96, 1.02) | 0.55 | 0.961 | (0.93, 0.99) | 6.75e-3 |
| Ever smoked | 0.96 | (0.95, 0.99) | 2.20e-3 | 0.967 | (0.95, 0.99) | 1.08e-3 |
| Diabetes mellitus | 0.92 | (0.88, 0.96) | 8.41e- 5 | 0.889 | (0.85, 0.93) | 1.24e-7 |
The table above presents multiple logistic regression modeling of self-reported FHHD, where continuous variables were divided into quintiles. The 2nd–5th quintiles were compared to the 1st quintile, serving as the reference. In the minimally adjusted model, individual risk factors are modeled as predictors of reported FHHD, controlling for age, sex, and cholesterol-lowering medication use. In the fully adjusted model, analysis is adjusted for all other variables in the model.
ApoB:ApoAI, apolipoprotein B-to-apolipoprotein AI ratio [2nd–5th quintiles vs. 1st quintile (reference)]; Chol Med, cholesterol-lowering medication use; PRSCAD, polygenic risk score for coronary artery disease [2nd–5th quintiles vs. 1st quintile (reference)]; HeFH, heterozygous familial hypercholesteremia; hsCRP, high sensitivity C-reactive protein mg/L [2nd–5th quintiles vs. 1st quintile (reference)]; Lp(a), Lipoprotein(a) nmol/L [2nd–5th quintiles vs. 1st quintile (reference)]; triglycerides, triglycerides mmol/L [2nd–5th quintiles vs. 1st quintile (reference)]; WHR, waist to hip ratio [2nd–5th quintiles vs. 1st quintile (reference)].
When all genetic and clinical risk factors were simultaneously incorporated into a multivariate model, the results were similar with genetic risk factors exhibiting the strongest influence on the risk of FHHD: HeFH (OR 1.31, 95% CI 1.11–1.54) and PRSCAD [2nd–5th quintiles vs. 1st quintile (reference)] (OR 1.30, 95% CI 1.27–1.33). These were followed by clinical risk factors: hypertension (OR 1.18, 95% CI 1.15–1.21), Lp(a) [2nd–5th quintiles vs. 1st quintile (reference)] (OR 1.17, 95% CI 1.14–1.20), ApoBAI ratio [2nd–5th quintiles vs. 1st quintile (reference)] (OR 1.13, 95% CI 1.10–1.16), triglycerides [2nd–5th quintiles vs. 1st quintile (reference)] (OR 1.07, CI 1.04–1.10) (Table 2). These findings were similar in a sensitivity analysis, where all continuous variables were standardized and non-normally distributed variables were logarithmically transformed (see Supplementary material online, Table S4), before being divided into quintiles (see Supplementary material online, Table S5). Reassuringly, the trends in the sensitivity analyses are consistent with the trends found in the primary analyses.
Population attributable risk
PAR was calculated with (1) genetic risk factors only, (2) clinical risk factors only, and (3) combined clinical and genetic risk factors (Table 3). PAR for individual risk factors was determined by adjusting only for age and sex (Table 2; Structured Graphical Abstract). In the genetic risk factor-only model, 22.16% (CI 20.44–23.88) of the risk of reporting FHHD is explained by genetic factors alone. In the clinical risk factor-only model, 21.91% (CI 18.19–25.63) of the risk of reporting FHHD was explained by clinical risk factors alone. In the combined model, 35.99% (CI 33.31–38.68) of the risk of reporting FHHD was explained by a combination of clinical and genetic factors. Structured Graphical Abstract illustrates the attributable risk of specific clinical and genetic risk factors for those who reported a positive FHHD. 22.86% (CI 21.30–24.4) of FHHD is explained by PRSCAD [2nd–5th quintiles vs. 1st quintile (reference)], 12.16% (CI 10.89–13.42) by triglycerides [2nd–5th quintiles vs. 1st quintile (reference)], 8.72% (CI 6.92–10.52) by ApoB:ApoAI [2nd–5th quintiles vs. 1st quintile (reference)], 7.51% (CI 6.73–8.29) by hypertension, 6.67% (CI 4.62–8.69) by Lp(a) [2nd–5th quintiles vs. 1st quintile (reference)], 6.66% (CI 4.15–9.05) by WHR [2nd–5th quintiles vs. 1st quintile (reference)], 4.72% (CI 2.43–7.01) by hsCRP [2nd–5th quintiles vs. 1st quintile (reference)], 0.80% (CI 0.57–1.02) by diabetes, and 0.15% (CI 0.07–0.23) by HeFH. A prior history of smoking appeared not to affect a self-reported FHHD (PAR −0.79%, CI −1.20–0.41). To verify the high PAR for triglycerides, we additionally adjusted the individual PAR for triglycerides for ApoA1 level (in addition to age, sex, and cholesterol medication use) which did not materially alter the result, changing the PAR from 12.16% to 10.42%.
Table 3.
Percent of population risk attributable to a clinical model, genetic model, and combined model
| Model | Risk factor | PAR % | 95% confidence interval |
|---|---|---|---|
| Genetic |
|
22.16% | (20.44–23.88) |
| Clinical |
|
21.91% | (18.19–25.63) |
| Combined: clinical and genetic |
|
35.99% | (33.31–38.68) |
Population attributable risk (PAR) models using (1) genetic, (2) clinical, and (3) combined clinical and genetic risk factors were constructed to describe the percent likelihood a FHHD can be explained by each of the above variables. The PAR analytic framework compares exposed to unexposed, and as such, continuous variables were converted to polychotomous variables. The lowest risk level for all variables was compared to any elevation in risk. Thus for polychotomous variables quintiles 2–5 are compared to the lowest quintile (reference). A combined approach using both clinical and genetic risk factors explained a larger proportion of the likelihood of having a FHHD. Each model was controlled for age, sex, and cholesterol medication use.
ApoB:ApoAI, apolipoprotein B-to-apolipoprotein AI ratio [2nd–5th quintiles vs. 1st quintile (reference)]; HeFH, heterozygous familial hypercholesteremia; hsCRP, high sensitivity C-reactive protein mg/L [2nd–5th quintiles vs. 1st quintile (reference) ntile]; Lp(a), Lipoprotein(a) nmol/L [2nd–5th quintiles vs. 1st quintile (reference)]; PRSCAD. polygenic risk score for coronary artery disease [2nd–5th quintiles vs. 1st quintile (reference)]; WHR, waist to hip ratio [2nd–5th quintiles vs. 1st quintile (reference)].
As a sensitivity analysis while the above analyses sought to understand what fraction of FHHD risk was explained by PRSCAD, we also attempted to predict the opposite—whether FHHD could predict PRSCAD. The presence of FHHD did not reliably and linearly predict the PRSCAD percentile as demonstrated in Supplementary material online, Table S6, though there is a relationship above the 90th percentile. By contrast, restricting the PRSCAD to sequentially higher strata of the PRSCAD percentile (top 25%, 10%, 5%, 1%), had a direct relationship to increasing odds of FHHD (Figure 3).
Figure 3.

(A) Distribution of PRSCAD scores, and (B) odds of family history of HD being present after adjustment for age, sex, cholesterol medication use, ever-smoking, hypertension, diabetes, waist-to-hip ratio, triglycerides, and heterozygous familial HeFH, ApoB:ApoA1 ratio, lp(a), and hsCRP level. ApoB:ApoA1, apolipoprotein B-to-apolipoprotein AI ratio; Lp(a), lipoprotein(a): hsCRP, high sensitivity C-reactive protein, PAR, population attributable risk.
Discussion
In this study, using a PAR framework, we sought to estimate how much information within the family history is already explained in genetic and clinical biomarkers. While it is tempting to assume family history is an imperfect proxy for genetic risk, our analyses suggest otherwise. The genetic model including PRSCAD and HeFH explained 22.2% of the PAR of FHHD. The clinical model similarly explained only 21.9% PAR of FHHD. The combined model of clinical and genetic biomarkers explained 36.0% PAR of FHHD. Using multiple logistic regression models, we found that genetic risk factors PRSCAD and HeFH were most strongly associated with a self-reported FHHD, followed by clinical risk factors hypertension and dyslipidemia. These findings suggest that genetic, modifiable clinical, and unrecognized factors contribute to FHHD. Understanding family based risks for CVD offers prospects for tailored family based surveillance and risk mitigation.
Results from PAR analyses for individual risk factors (adjusted only for age, sex, and cholesterol medication use) demonstrated that PRSCAD had the greatest PAR for FHHD, followed by triglycerides, ApoB:ApoA1 ratio, hypertension, and WHR. As expected of rare variants, HeFH showed a strong effect size in regression models, but its PAR was low given its low prevalence. Triglycerides, independently accounting for substantial PAR of FHHD after controlling for ApoB: ApoA1 ratio, likely served to integrate other risk factors associated with a FHHD, including diet, exercise, insulin resistance, and postprandial lipid metabolism, among other unfavorable qualities. Thus, after polygenic risk, the most prevalent and powerful risk factors were traditional clinical risk factors. These findings emphasize that when a patient reports a family history of cardiac disease, while they are more likely to have a high PRS (or less likely, harboring a rare monogenic variant), traditional modifiable risk factors play a roughly comparable role in the population. Families enriched for CVD but low PRS may benefit from thorough investigations of drivers of risk; while traditional risk factors may often explain enrichment, we demonstrate that much of this enrichment remains unexplained through currently recognized risk factors.
When clinical biomarkers and genetics explain only a fraction of FHHD, environmental, behavioral, and socioeconomic factors rise to the fore. For example, low parental income during childhood contributes to adverse cardiovascular health for parents and future adult offspring.21 In this study, hypertension and dyslipidemia but not smoking or diabetes are enriched among those with FHHD. The lack of smoking enrichment may reflect secular trends in decreasing smoking prevalence across subsequent generations over the 20th century.22 Lack of enrichment for diabetes among those with FHHD was a surprising meriting further investigation. The reasons for the lack of enrichment are not immediately clear beyond potentially heightened surveillance and risk mitigation in affected families. Additionally, the male sex was a predictor for the absence of an FHHD, but this may be due to differential knowledge of family history compared to females.23
Social determinants of health such as intergenerational wealth, educational, and employment opportunities, health literacy, and environmental exposures are also passed down through families.24 These factors may have a large influence on the heritability of CVD and additional clinical risk factors but are not well-characterized in the UK Biobank.24-27 Additionally, important parameters like sleep and stress have imperfect measures, while parameters such as diet and physical activity reflect imperfect survey data. Nevertheless, when factors like diet, physical activity, ‘neuroticism score’ (for stress), and others were integrated into models they introduced dramatic model instability, likely reflecting the complexity of the biopsychosocial model of disease that is not easily quantified. A limitation of our study is our inability to properly include these risk factors into our analyses. However, it is uncommon for routine clinical assessments to include detailed behavioral phenotyping, socioeconomic assessment, or shared exposures such as air pollution, sound pollution, ambient radiation, structural racism, and poverty.
Our study has several limitations. First, a self-reported FHHD may be limited by recall bias and imprecision. Individuals may be unaware of their family history or have inconsistent definitions for HD (e.g. do they include hypertension, atrial fibrillation, etc., in their definition). Of note, prior analyses in the UK Biobank show that individuals without CAD but with FHHD show similar genetic architecture as individuals with CAD, consistent with PRSCAD being a proxy for genetics related to HD more broadly.28 Nevertheless, more specifically inquiring about a family history of CAD may further improve the estimates provided. Second, the age at diagnosis of family members who had CVD was not available in the UK Biobank. Prior work indicates that early onset CVD in first-degree relatives carries notable risk4,29 We enriched family history-related CVD risk by examining individuals with multiple affected family members in secondary analyses. Third, the combination of healthy participant bias30 in the UKBB and suggestions that a known FHHD affects individuals’ behavior31 in unpredictable ways would have introduced biases into the model for which adjustment may not have been appropriate.24 Fourth, our population is a majority white, relatively healthy population in the United Kingdom, limiting generalizability.15
In summary, this study demonstrates that while commonly obtained clinical biomarkers and research-grade genetic tools contribute approximately equally to self-reported FHHD, family history continues to yields a distinct set of information unexplained by these risk factors, that may function as an integrator of lifelong exposures that we do not routinely assess in clinical or research studies. Future work creating risk scores that integrate family history with clinical and genetic biomarkers will likely be of use.
Supplementary Material
Funding
R.B. is supported by the John S. LaDue Memorial Fellowship in Cardiovascular Research. G.M.P. is supported by National, Heart, Lung, and Blood Institute grants R01HL127564 and R01HL142711. M.C.H. is supported by the American Heart Association (940166, 979465). P.N. is supported by grants from National, Heart, Lung, and Blood Institute (R01HL142711, R01HL127564) and National Human Genome Research Institute (U01HG011719).
Footnotes
Supplementary material
Supplementary material is available at European Journal of Preventive Cardiology.
Conflict of interest: R.B. is a medical advisor to Casana Care Inc, unrelated to present work. A.C.F. holds equity and receives consulting fees from Goodpath, and receives research funding from Abbott Inc., both unrelated to this study. M.C.H. reports consulting fees from CRISPR Therapeutics and serves on the advisory board for Miga Health, both unrelated to this work. P.N. reports investigator-initiated grants from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis. personal fees from Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, Novartis, Roche/Genentech, is a cofounder of TenSixteen Bio, is a scientific advisory board member of Esperion Therapeutics, geneXwell, and TenSixteen Bio, and spousal employment at Vertex, all unrelated to this work.
Data availability
The data underlying this article are available in the UK Biobank at http://www.ukbiobank.ac.uk/using-the-resource/.
References
- 1.Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol 2020;76:2982–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chow CK, Islam S, Bautista L, Rumboldt Z, Yusufali A, Changchun X, et al. Parental history and myocardial infarction risk across the world the INTERHEART study. j Am Coll Cardiol 2011;57:619–627. [DOI] [PubMed] [Google Scholar]
- 3.Moonesinghe R, Yang Q, Zhang Z, Khoury MJ. Prevalence and cardiovascular health impact of family history of premature heart disease in the United States: analysis of the national health and nutrition examination survey, 2007–2014. J Am Heart Assoc 2019; 8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd-Jones DM, Nam BH, D’Agostino RB, Levy D, Murabito JM, Wang TJ, et al. Parental cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults a prospective study of parents and offspring. JAMA 2004;291:2204–2211. [DOI] [PubMed] [Google Scholar]
- 5.Lloyd-Jones DM, Braun LT, Ndumele CE, Smith SC, Sperling LS, Salim SC, et al. Use of risk assessment tools to guide decision-making in the primary prevention of atherosclerotic cardiovascular disease: a special report from the American heart association and American college of cardiology. Circulation 2019;139:E1162–E1177. [DOI] [PubMed] [Google Scholar]
- 6.Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women the Reynolds risk score. JAMA 2007;297:611–620. [DOI] [PubMed] [Google Scholar]
- 7.Ray KK, Hovingh GK. Familial hypercholesterolaemia: a common disease. Eur Heart J 2016;37:1395–1397. [DOI] [PubMed] [Google Scholar]
- 8.Sniderman AD, Furberg CD, Keech A, Roeters van Lennep JE, Frohlich J, Jungner I, et al. Apolipoproteins versus lipids as indices of coronary risk and as targets for statin treatment. Lancet 2003;361:777–780. [DOI] [PubMed] [Google Scholar]
- 9.Trinder M, Uddin MM, Finneran P, Aragam KG, Natarajan P. Clinical utility of lipoprotein(a) and LPA genetic risk score in risk prediction of incident atherosclerotic cardiovascular disease. JAMA Cardiol 2021;6:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart j 2010;31: 2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ridker PM. Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation 2003;107:363–369. [DOI] [PubMed] [Google Scholar]
- 12.Aikawa M, Ganz P, Ridker PM, Aday AW. Antiinflammatory therapy in clinical care: the CANTOS trial and beyond. Front Cardiovasc Med 2018;1:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong B, Kruse G, Kutikova L, Ray KK, Mata P, Bruckert E. Cardiovascular disease risk associated with familial hypercholesterolemia: a systematic review of the literature. Clin Ther 2016;38:1696–1709. [DOI] [PubMed] [Google Scholar]
- 14.Hindy G, Aragam KG, Ng K, Chaffin M, Lotta LA, Baras A, et al. Genome-wide polygenic score, clinical risk factors, and long-term trajectories of coronary artery disease. Arterioscler Thromb Vasc Biol 2020;40:2738–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye Y, Chen X, Han J, Jiang W, Natarajan P, Zhao H. Circulation: genomic and precision medicine interactions between enhanced polygenic risk scores and lifestyle for cardiovascular disease, diabetes, and lipid levels. Circ Genom Precis Med 2021;14:3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y, Lu Q, Powles R, Yao X, Yang C, Fang F, et al. Leveraging functional annotations in genetic risk prediction for human complex diseases. PLoS Comput Biol 2017;13:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Damask A, Steg PG, Schwartz GG, Szarek M, Hagström E, Badimon L, et al. Patients with high genome-wide polygenic risk scores for coronary artery disease may receive greater clinical benefit from alirocumab treatment in the ODYSSEY OUTCOMES trial. Circulation 2020;141:624–636. [DOI] [PubMed] [Google Scholar]
- 19.Yusuf PS, Hawken S, Ôunpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004;364:937–952. [DOI] [PubMed] [Google Scholar]
- 20.Schenck L, Atkinson EJ, Crowson CS, Therneau TM. Attributable Risk; 2014. Accessed April 23, 2022. https://rdrr.io/github/bethatkinson/attribrisk/ [Google Scholar]
- 21.Evensen M, Klitkou ST, Tollånes MC, Øverland S, Lyngstad TH, Vollset SE, et al. Parental income gradients in adult health: a national cohort study. BMC Med 2021;19:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics-2022 update: a report from the American heart association. Circulation 2022;145(8):e153–e639. [DOI] [PubMed] [Google Scholar]
- 23.Hull LE, Natarajan P. Self-rated family health history knowledge among all of US program participants. Genet Med 2022;24:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Javed Z, Haisum Maqsood M, Yahya T, Amin Z, Acquah I, Valero-Elizondo J, et al. Race, racism, and cardiovascular health: applying a social determinants of health framework to racial/ethnic disparities in cardiovascular disease. Circ Cardiovasc Qual Outcomes 2022; 15:e007917. [DOI] [PubMed] [Google Scholar]
- 25.Nayak A, Hicks AJ, Morris AA. Understanding the complexity of heart failure risk and treatment in black patients. Circ Heart Fail 2020;13:301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martínez-García M, Salinas-Ortega M, Estrada-Arriaga I, Hernández-Lemus E, García-Herrera R, Vallejo M. A systematic approach to analyze the social determinants of cardiovascular disease. PLoS One 2018;13:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powell-Wiley TM, Baumer Y, Baah FO, Baez AS, Farmer N, Mahlobo CT, et al. Social determinants of cardiovascular disease. Circ Res 2022;130:782–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu JZ, Erlich Y, Pickrell JK. Case-control association mapping by proxy using family history of disease. Nat Genet 2017;49:325–331. [DOI] [PubMed] [Google Scholar]
- 29.Murabito JM, Pencina MJ, Nam BH, D’Agostino RB, Wang TJ, Lloyd-Jones D, et al. Sibling cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults. JAMA ;294:3117–3123. [DOI] [PubMed] [Google Scholar]
- 30.Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen RT, et al. Comparison of sociodemographic and health-related characteristics of UK biobank participants with those of the general population. Am j Epidemiol 2017;186:1026–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mudd-Martin G, Rayens MK, Lennie TA, Chung ML, Gokun Y, Wiggins AT, et al. Fatalism moderates the relationship between family history of cardiovascular disease and engagement in health-promoting behaviors among at-risk rural kentuckians. J Rural Health 2015;31:206–216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the UK Biobank at http://www.ukbiobank.ac.uk/using-the-resource/.