

Abstract
The prototypical aminoalkylindole cannabinoid WIN 55,212-2 (WIN-2) has been shown to produce antihyperalgesia through a peripheral mechanism of action. However, it is not known whether WIN-2 exerts this action directly via cannabinoid receptors located on primary afferents or if other, perhaps indirect or noncannabinoid, mechanisms are involved. To address this question, we have examined the specific actions of WIN-2 on trigeminal ganglion (TG) neurons in vitro by quantifying its ability to modulate the evoked secretion of the proinflammatory neuropeptide CGRP as well as the inflammatory mediator-induced generation of cAMP.
WIN-2 evoked CGRP release from TG neurons in vitro (EC50=26 μM) in a concentration- and calcium-dependent manner, which was mimicked by the cannabinoid receptor-inactive enantiomer WIN 55,212-3 (WIN-3). Moreover, WIN-2-evoked CGRP release was attenuated by the nonselective cation channel blocker ruthenium red but not by the vanilloid receptor type 1 (TRPV1) antagonist capsazepine, suggesting that, unlike certain endogenous and synthetic cannabinoids, WIN-2 is not a TRPV1 agonist but rather acts at an as yet unidentified cation channel.
The inhibitory effects of WIN-2 on TG neurons were also examined. WIN-2 neither inhibited capsaicin-evoked CGRP release nor did it inhibit forskolin-, isoproteranol- or prostaglandin E2- stimulated cAMP accumulation.
On the other hand, WIN-2 significantly inhibited (EC50=1.7 μM) 50mM K+-evoked CGRP release by approximately 70%. WIN-2 inhibition of 50mM K+-evoked CGRP release was not reversed by antagonists of cannabinoid type 1 (CB1) receptor, but was mimicked in magnitude and potency (EC50=2.7 μM) by its cannabinoid-inactive enantiomer WIN-3.
These findings indicate that WIN-2 exerts both excitatory and inhibitory effects on TG neurons, neither of which appear to be mediated by CB1, CB2 or TRPV1 receptors, but by a novel calciumdependent mechanism. The ramifications of these results are discussed in relation to our current understanding of cannabinoid/vanilloid interactions with primary sensory neurons.
Keywords: Pain, cannabinoid, vanilloid, sensory neurons, trigeminal ganglion, calcitonin gene-related peptide
Introduction
Cannabinoids exert their pharmacological actions through multiple membrane spanning receptors of both the G-proteincoupled receptor superfamily, including cannabinoid type 1 (CB1) (Matsuda et al., 1990) and cannabinoid type 2 (CB2) (Munro et al., 1993), and the transient receptor potential (TRP) ion channel family, including transient receptor potential vanilloid subfamily type 1 (TRPV1) (Zygmunt et al., 1999; Huang et al., 2002), ANKTM1 (Jordt et al., 2004) and TRPV4, via the production of epoxyeicosatrienoic acids (Watanabe et al., 2003). Cannabinoid pharmacology comprises a diverse family of ligands that can be classified into roughly six categories based on the chemical structure of the compounds (Khanolkar et al., 2000; Palmer et al., 2002). The prototypical aminoalkylindole WIN55,212-2 (WIN-2) is an agonist of both CB1 and CB2 receptors. Prominent among the various behavioral effects produced by WIN-2 in vivo is antinociception, a trait shared by members of other chemical classes of cannabinoid agonists that hold substantial promise for the development of analgesic pharmacotherapeutics in humans.
A growing number of chemically diverse cannabinoid compounds stimulate primary sensory neuron activity through the TRP family of ligand-gated ion channels. Most notably, the endogenous cannabinoid anandamide (AEA) has been demonstrated to be an agonist of TRPV1 (Zygmunt et al., 1999; Smart et al., 2000; Jennings et al., 2003). In conjunction with this, a number of endogenous and exogenous compounds possessing dual vanilloid–cannabinoid pharmacology (i.e. agonize both CB1/CB2 receptors and TRPV1) have been described (Huang et al., 2002; Chu et al., 2003). In addition to these TRPV1-mediated actions of certain cannabinoids, TRPV1-independent stimulation of sensory neurons by the prototypical cannabinoid Δ9-THC as well as the physiologically less active cannabinol have been demonstrated (Zygmunt et al., 2002), and this may involve the recently identified TRP channel ANKTM1 (Jordt et al., 2004). Thus, there are multiple pathways through which cannabinoids may stimulate sensory neurons. However, to date, such an excitatory action of WIN-2 has not been demonstrated.
Apart from this emerging story that cannabinoids exert excitatory actions on the peripheral sensorium, cannabinoids are well known for their analgesic action, a portion of which has been attributed to peripheral sites of action. WIN-2- mediated antinociception involves both CNS and peripheral sites of action. In the CNS, WIN-2 infusion into any of several brain areas, including the amygdala and thalamus, produces antinociception (Martin et al., 1999a), and WIN-2 has been demonstrated to alleviate both nerve injury- (Herzberg et al., 1997; Fox et al., 2001) and inflammation- (Martin et al., 1999b) enhanced nociception through brain and spinal mechanisms. WIN-2 also diminishes nociceptive responses through a peripherally mediated mechanism. In carrageenantreated animals, WIN-2 inhibits hyperalgesia and spinal Fos expression through peripheral CB1 receptors (Nackley et al., 2003). Furthermore, in the sciatic nerve ligation model of neuropathic pain (Fox et al., 2001) and in capsaicin (CAP)- induced thermal hyperalgesia (Johanek et al., 2001), WIN-2 acts through peripheral CB1 receptors to produce antihyperalgesia. In addition, in the trigeminal system, topically applied WIN-2, in an SR141716A-sensitive manner, markedly reduces mustard oil-induced increases in Fos expression in the Vi/Vc, but to a lesser extent in Vc/C1 brainstem regions (Bereiter et al., 2002).
While WIN-2 clearly influences nociceptive processing through a peripheral site, the exact location of the CB1 receptors involved in this response remains elusive. Conflicting evidence exists on the sensory neuronal distribution of CB1 receptors. While one study has shown that CB1 receptors are localized to TRPV1-positive neurons in dorsal root ganglion (DRG) cultures (Ahluwalia et al., 2000), it has recently been demonstrated that CB1 receptors show a much different profile in native DRG, where they are nearly exclusively found in N52-positive neurons (Bridges et al., 2003). Also, we have demonstrated in trigeminal ganglion (TG) that CB1 receptor mRNA is found in large diameter, myelinated neurons and rarely colocalizes with TRPV1 or CGRP (Price et al., 2003). Consistent with these latter two findings is the demonstration that WIN-2 inhibits K+-evoked calcium influx mostly in medium and large diameter DRG neurons in culture, but not in small diameter neurons that are thought to subserve acute nociceptive sensation (Khasabova et al., 2002).
The present studies sought to assess the effects of the aminoalkylindole cannabinoid WIN-2 on TG sensory neurons. We have utilized in vitro primary culture, which is highly enriched for neurons, to clarify the actions of WIN-2 that are specific to sensory neurons. Our findings suggest that WIN-2 acts through a novel mechanism to excite TG neurons. On the other hand, we observed no cannabinoid receptor-dependent inhibitory effects of WIN-2 on TG nociceptors. These findings indicate that the major action of WIN-2 on TG nociceptors in vitro is excitatory, suggesting that the peripherally mediated antihyperalgesic effects of WIN-2 are likely mediated by cannabinoid receptors found in neuronal populations that do not subserve normal nociception and/or in non-neuronal cell types.
Materials
Reagents
AEA, WIN-2, capsazepine (CPZ), iodo-resinferatoxin (IRTX), AM251, suramin, AM630 and SB366791 were from Tocris (Ellisville, MO, U.S.A.). Capsaicin (CAP), WIN55,212- 3 (WIN-3), isoproteranol rolipram and bradykinin (BK) were from Sigma Aldrich/Fluka (St Louis, MO, U.S.A.). The CB1 antagonist SR141716A was from the NIMH chemical synthesis and drug supply program. Prostaglandin E2 (PGE2) was from Cayman Chemical (Ann Arbor, MI, U.S.A.). Nerve growth factor (NGF; 7.0S) was from Harlan (Indianapolis, IN, U.S.A.). [125I]cAMP tracer was from Perkin-Elmer Life Science Products (Boston, MA, U.S.A.) and the anti-cAMP antibody was from ICN Biomedicals (Costa Mesa, CA, U.S.A.). AEA was purchased predissolved in Tocrisolve™; WIN-2, WIN-3, AM251, AM630, SB366791 and SR141716A were dissolved in DMSO to stock solutions of 10–50mM; IRTX, CPZ and PGE2 were dissolved in EtOH to stock solutions of 10mM; and BK was dissolved in H2O to a stock solution of 10mM.
TG culture
Adult, male, Sprague–Dawley rats weighing 250–300 g were used in this study. All procedures utilizing animals were approved by the Institutional Animal Care and Use Committee of The University of Texas Health Science Center at San Antonio and were conducted in accordance with policies for the ethical treatment of animals established by the National Institutes of Health. Animals were euthanized by decapitation and their trigeminal ganglia were rapidly dissected (within ~30 s) and placed in ice-cold Ca2+- and Mg2+-free Hank’s balanced salt solution (HBSS, Gibco, Carlsbad, CA, U.S.A.). Ganglia were enzymatically digested for 30 min with 5.0 mg ml−1 collagenase followed by 25 min with 0.1% Trypsin Type IX supplemented for the last 10 min with 10U of DNase I (Roche, Indianapolis, IN, U.S.A.). TG homogenates were then centrifuged at 2000 r.p.m. for 2 min, triturated briefly by vortexing and then recentrifuged. They were then resuspended in the culture media containing high glucose Dulbeco’s modified Eagle’s media (DMEM, Gibco), 1× pen-strep (Gibco), 1× glutamine (Gibco) 3 μgml−1 5-FDU and 7 μgml−1 uridine. TG homogenates were gently triturated with a Pasteur pipette followed by successive triturations through 19- and 23-gauge needles. TG homogenates were then transferred to a separate container, wherein the volume was adjusted to yield an initial plating density of 5000 neurons well−1.
Evoked CGRP release assays
Experiments were performed in 48-well, poly-D-lysine precoated plates (Becton Dickinson, Franklin Lakes, NJ, U.S.A.). In all experiments, the TG from three animals were used per 48-well plate. In order to standardize for plating density, the entire TG homogenate prepared for each experiment was pooled together and then divided into equal aliquots before plating, at which time 100 ng ml−1 NGF was added. The culture media were changed at 24 and 72 h, including fresh growth factor supplementation, and all CGRP assays were performed on day 5. On the 5th day, TG cultures were washed free of the culture media by two successive washes with release buffer (HBSS; Gibco) supplemented with 10.9mM HEPES, 4.2mM sodium bicarbonate, 10mM dextrose and 0.1% bovine serum albumin (BSA; pH 7.4). NGF was not included in the release buffer. Following washing, TG cultures were exposed to the indicated concentrations of pretreatment compound (e.g. agonists or antagonists) for 10 min, excepting CPZ, IRTX or ruthenium red, which were applied for 5 min, followed by the stimulus drug (in the same buffer) for 10 min, after which the CGRP containing release buffer was removed and transferred to glass culture tubes. All drugs were diluted from their stock solutions (described in Reagents) into 10× concentrations for each condition, in siliconized glass culture tubes, and drugs were added to the culture plate wells with siliconized pipette tips. For K+-evoked CGRP release experiments, neurons were first incubated for 10 min with agonist or agonist + antagonist for 10 min in release buffer, which was then removed and neurons were subsequently challenged with 50mM K+buffer (containing 2.5mM CaCl2, 50mM KCl, 1.2mM MgCl2, 90mM NaCl, 25mM NaHCO3, 1mM NaH2− PO4, 10mM dextrose, 15mM HEPES, 16 μM thiorphan and 0.1% BSA, pH 7.4), including appropriate drugs or vehicle in the media for 10 min. Data for these experiments are presented as the sum of the pretreatment release+K+-evoked release to account for agonist-evoked release (WIN-2 or WIN-3) in the pretreatment condition. CGRP was measured by radioimmunoassay.
CGRP radioimmunoassay
Following culture release assays, individual aliquots of the superfusate (0.5 ml) were incubated with a C-terminally directed anti-CGRP antiserum, kindly donated by Dr Michael Iadarola (NIDCR, NIH, Bethesda, MD, U.S.A.). After 24 h, 100 μl of [125I]CGRP28–37 (approximately 20 000–25 000 c.p.m.) and 50 μl of goat anti-rabbit antibody conjugated to ferric beads were added. Following another 24 h, bound peptide was separated from free peptide via immunomagnetic separation (PerSeptive Biosystems, Framingham, MA, U.S.A.). All incubations were carried out at 4°C. The minimum detection limit for this assay is approximately 1–2 fmol tube−1, with 50% displacement occurring at 20–40 fmol tube−1. To account for the possibility of any nonspecific effects on the RIA, all drugs used in the release experiments were included in separate standard curves for the purposes of data analysis. None of the compounds used in this study significantly modified the standard curve.
cAMP accumulation
TG neuronal cultures were prepared as described above. On day 5, neurons were washed and incubated in release buffer containing 10 μM rolipram for 10 min, after which they were exposed to drug or vehicle for a subsequent 15 min. Cellular cAMP was extracted by the immediate addition of 500 μl of ice-cold ethanol, allowed to incubate overnight at −20°C, evaporated the following day in RIA tubes and resuspended in RIA buffer. cAMP levels were then measured by radioimmunoassay, as described previously (Berg et al., 1994).
Statistics
All data are presented as mean±s.e.m., unless otherwise noted. Significant differences between groups were assessed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparison post-test, unless otherwise stated. Concentration– response curves were analyzed by variable slope nonlinear regression. All data were analyzed with GraphPad Prism 4.0 for Mac OS X (GraphPad, San Diego, CA, U.S.A.)
Results
As a number of cannabinoid agonists have also been shown to be TRPV1 agonists, we explored the ability of WIN-2 to evoke CGRP release from TG neurons, a common property of TRPV1 agonists. WIN-2 evoked CGRP release from TG neurons with an EC50 of 26 μM (Figure 1a). The calcium dependence of WIN-2-evoked CGRP release was tested by excluding extracellular calcium from the release buffer. 50 μM WIN-2-evoked CGRP release was blocked by the exclusion of extracellular calcium (Figure 1b). In addition, we utilized WIN-3, the cannabinoid-inactive enantiomer of WIN-2, to test the stereospecificity of the compound for evoking CGRP release from TG neurons. WIN-3 (25 or 50 μM) also evoked CGRP release, although less effectively than WIN-2 at the same concentration (Figure 1c). To identify the receptor(s) mediating the secretogogue effect of WIN-2 on TG neurons, we evaluated the potential for a number of antagonists to block this effect. The TRPV1 antagonists, I-RTX (200 nM, Figure 2a), CPZ (10 μM, Figure 2a) and SB 366791 (10 μM, data not shown) each did not inhibit 50 μM WIN-2-evoked CGRP release, but, in simultaneously performed experiments, completely blocked 100 nM CAP- and 30 μM AEA-evoked CGRP release (Price et al., 2004). Furthermore, neither of the CB1 receptor antagonists SR141716A (Figure 2b) nor AM251 (Figure 2c) was capable of reducing 50 μM WIN-2-evoked CGRP release. EDG receptors have high homology to cannabinoid receptors (Yamaguchi et al., 1996; Molderings et al., 2002), and EDG3 receptors are expressed in rat TG (T.J. Price, A.W. Akopian and C.M. Flores, unpublished observations); hence, we hypothesized that EDG receptors might be involved in WIN-2-evoked CGRP release. On the contrary, the EDG-3 (Ancellin & HLA, 1999; Himmel et al., 2000) and P2Y receptor antagonist suramin did not inhibit WIN-2-evoked CGRP release (Figure 2d). On the other hand, the nonspecific cation channel blocker ruthenium red (1 μM) reversed WIN-2-evoked CGRP release by approximately 75% (Figure 2e). We next tested the ability of CAP pretreatment (10–600 nM) to desensitize the subsequent WIN-2-evoked CGRP release response. WIN-2-evoked CGRP release (50 μM) was not altered significantly by CAP pretreatment (10–300 nM) except at the highest concentration of CAP utilized (600 nM), wherein a small decrease in WIN-2-evoked CGRP release was observed (Figure 2f, 5.32±0.42-fold increase over baseline without CAP pretreatment vs 3.61±0.19-fold increase over baseline following 600 nM CAP pretreatment).
Figure 1.
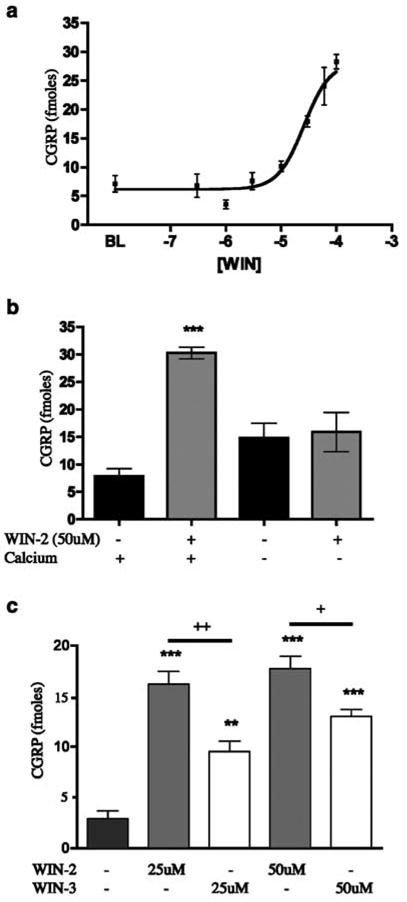
WIN-2 evokes CGRP release from TG neurons: (a) Concentration–response curve for WIN-2-evoked CGRP release from TG neurons following 10 min exposure (n=6, all concentrations). (b) The calcium-dependence of WIN-2-evoked CGRP release was tested by the exclusion of calcium from the extracellular solution (n=6, ***P<0.001 vs baseline with calcium). (c) Both WIN-2 and the enantiomer, WIN-3, evoke CGRP release from TG neurons (n=6, **P<0.01, ***P<0.001 vs no drug control; +P<0.05, ++P<0.01 WIN-2 vs WIN-3 comparisons).
Figure 2.
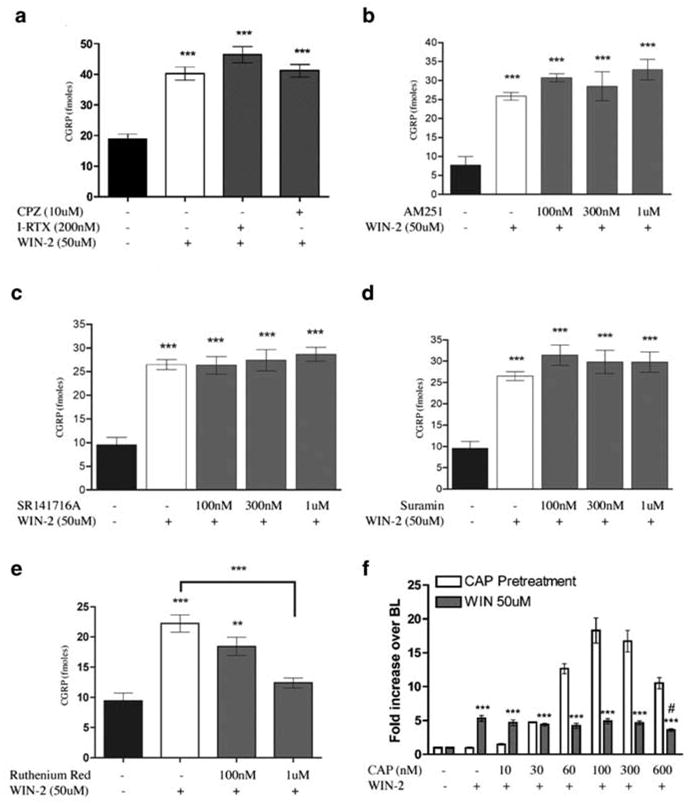
Effect of antagonists and CAP pretreatment on WIN-2-evoked CGRP release: the dependence of WIN-2-evoked CGRP release on TRPV1 was assessed using CPZ or I-RTX (a). In addition, the potential role of CB1 receptors was evaluated using AM251 (b) or SR141716A (c), while that of EDG or P2Y receptors was evaluated using suramin (d). The dependence of WIN-2- evoked CGRP release on non-TRPV1 cationic channels was evaluated using the nonspecific calcium channel blocker ruthenium red (e). (f) To examine the potential for CAP to desensitize the WIN-2 response, TG neurons were pretreated with the indicated concentrations of CAP or vehicle for 10 min after which the release buffer was removed and reserved for CGRP analysis and then replaced with fresh buffer. Following 5min, vehicle or WIN-2 (final concentration of 50 μM) in release buffer was added for 10 min. Both the CAP pretreatment and WIN-2 treatments were assayed for CGRP release. Clear bars indicate CAP-evoked CGRP release and gray bars illustrate the subsequent WIN-2-evoked CGRP response (n=6, ***P<0.001 vs no drug control, #P<0.5 vs WIN-2 50 μM without CAP pretreatment; no comparisons were made for CAP pretreatment only).
Figure 5.
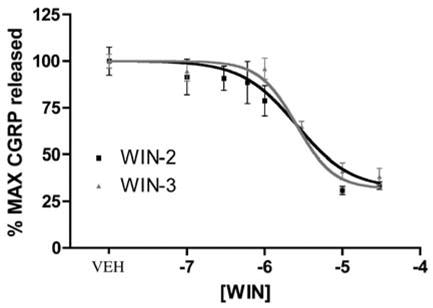
WIN-2 and WIN-3 inhibit 50mM K+-evoked CGRP release: concentration–response curves for WIN-2- (black squares) or WIN-3- (gray triangles) mediated inhibition of 50mM K+-evoked CGRP release (n=6). In order to account for the fact that WIN-2 and WIN-3 evoke CGRP release on their own, data are presented as the sum of the WIN-2 or WIN-3 pretreatment + K+-evoked CGRP release in the continued presence of WIN-2 or WIN-3.
Having established a clear excitatory effect for WIN-2 at mid-μM concentrations, we examined the possible inhibitory effects of WIN-2 at lower concentrations more in line with its established pharmacology at cannabinoid receptors. To investigate peptide secretion from a de facto nociceptive subset of TG neurons, we examined the capacity of WIN-2 to inhibit CAP-evoked CGRP release. WIN-2 (100–1000 nM) did not inhibit CAP-evoked CGRP release produced by either an Emax (100 nM; Figure 3a) or EC50 (50 nM; Figure 3b) concentration of CAP (Figure 3a). To evaluate the hypothesis that WIN-2 might inhibit GPCR-mediated release, we evaluated its ability to block the neurosecretory effects of the inflammatory mediators PGE2 plus BK. However, WIN-2 did not inhibit PGE2- (10 μM) plus BK- (300 nM) evoked CGRP release (Figure 3c). As WIN-2 might mediate an inhibition of sensory neuron activity through pathways other than those involved with neuropeptide secretion, we examined the effect of WIN-2 on cAMP accumulation. As shown in Figure 4a, neither did WIN-2 (10–500nM) by itself affect cAMP accumulation in TG neurons nor did it inhibit that stimulated by forskolin (1 μM), isoproterenol (10 μM) or PGE2 (10 μM). Likewise, 50 μM WIN-2, a concentration that evokes CGRP release, did not stimulate cAMP accumulation over baseline levels, while 30 μM AEA did (Figure 4b).
Figure 3.
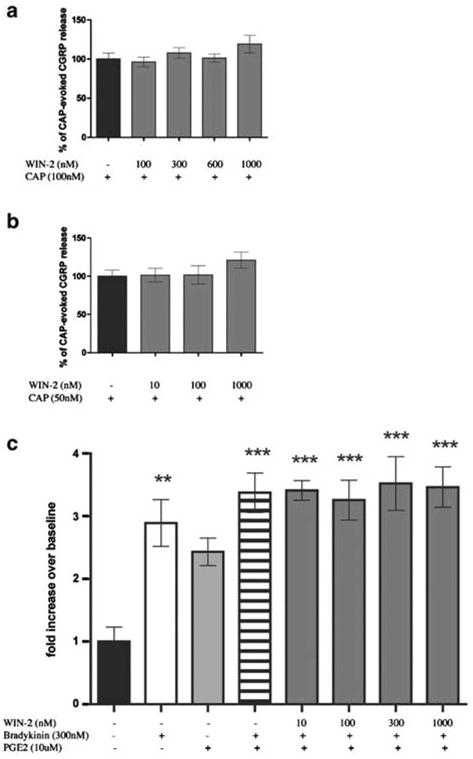
WIN-2 does not inhibit CAP- or BK plus PGE2- evoked CGRP release: the effect of WIN-2 on CAP-evoked CGRP release was tested against both 100 nM (a) and 50 nM (b) CAP (n=6). (c) WIN-2 did not inhibit BK plus PGE2-evoked CGRP release (n=6, **P<0.01, P<0.001 vs no drug control).
Figure 4.
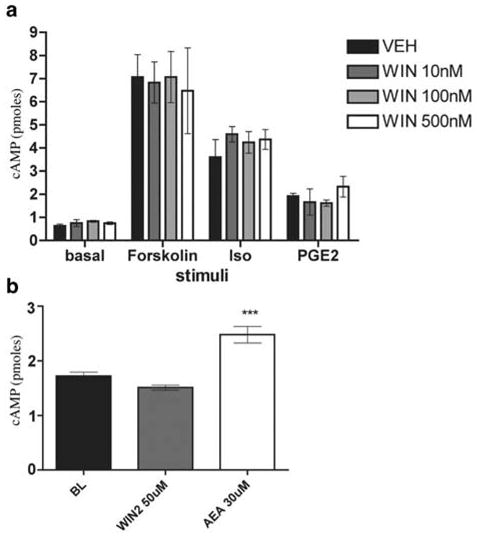
WIN-2 does not inhibit stimulated cAMP accumulation or stimulate cAMP accumulation on its own: (a) Assessment of the effect of 10 min pretreatment with WIN-2 on 15min treatment with 1 μM forskolin-, 10 μM isoproterenol- or 10 μM PGE2-stimulated cAMP accumulation in the continued presence of WIN-2 (n=3, all conditions). (b) WIN-2 and AEA, at concentrations that evoke CGRP release, were exposed to TG neurons for 10 min to determine their effects on cAMP accumulation (n=6, P<0.001 vs baseline).
It has been reported that CAP-evoked CGRP release from sensory neurons in culture is not dependent on voltage-gated calcium channels (Evans et al., 1996). As this might indicate a possible difference between in vivo and in vitro conditions, at least with respect to the utility of CAP, we utilized 50mM K+evoked CGRP release as an alternative model for studying the effects of WIN-2 on neuropeptide secretion. In contrast to the CAP and BK plus PGE2 findings, WIN-2 inhibited 50mM K+-evoked CGRP release from TG neurons in a concentration- dependent manner with an IC50 of 1.7 μM (Figure 5). WIN-3 also concentration dependently inhibited 50mM K+evoked CGRP release with a similar IC50 of 2.7 μM (Figure 5). 10 μM WIN-2-mediated inhibition of 50mM K+-evoked CGRP release was not sensitive to the CB1 receptor antagonists SR141716A (Figure 6a) or AM251 (Figure 6b). Moreover, the CB2 receptor antagonist AM630, at concentrations relevant to CB2 receptor antagonism (i.e. 0.1–1.0 μM), also failed to prevent the inhibitory effect of WIN-2, although higher concentrations (i.e. 10 μM) did (Figure 6c). Finally, 10 μM WIN-2-mediated inhibition of 50mM K+-evoked CGRP release did not appear to be dependent on Gi/oprotein- coupled receptors, as treatment with 500 ng ml−1 pertussis toxin (PTX) for 18 h did not alter this effect (Figure 6d).
Figure 6.
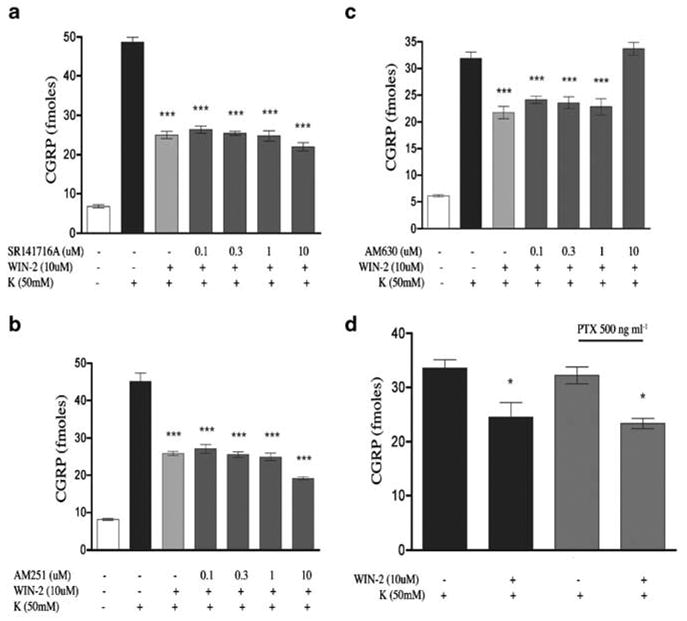
G-protein-coupled receptor dependence of WIN-2 inhibition of 50mM K+-evoked CGRP release: 10 μM WIN-2 inhibition of 50mM K+-evoked CGRP release was assessed in the presence of the CB1 antagonists, SR141716A (a) and AM251 (b), as well as the CB2 antagonist, AM630 (c), n=6, all experiments, P<0.001 vs K+alone). (d) The effect of Gi/o G protein uncoupling was tested by 18 h pretreatment with PTX (n=6, *P<0.05 vs K+alone). All data are presented as the sum of the agonist7antagonist pretreatment + the K+-evoked CGRP release treatment to account for the ability of WIN-2 to evoke CGRP release on its own.
Discussion
We have demonstrated that WIN-2 evokes CGRP release from cultured primary sensory neurons in a calcium-dependent manner. Moreover, WIN-2-evoked CGRP release did not appear to be stereospecific, as the cannabinoid receptorinactive enantiomer WIN-3 also stimulated CGRP excocytosis, albeit less effectively at the single concentration tested. Unlike certain effects of other cannabinoids that are also agonists of TRPV1, the stimulatory effects of WIN-2 did not appear to be mediated by TRPV1, as these were not blocked by CPZ, I-RTX or SB 366791 (Gunthorpe et al., 2004). In addition, CB1 and EDG3/P2Y receptor antagonists failed to inhibit WIN-2-evoked CGRP release. Furthermore, the cAMP/PKA pathway does not seem to mediate the stimulatory effect of WIN-2 on TG neurons, as concentrations of WIN-2 that evoked CGRP release failed to promote cAMP accumulation. On the other hand, WIN-2-evoked CGRP release was antagonized by ruthenium red, a nonselective blocker of numerous cation channels. That CPZ, I-RTX and SB 366791 did not antagonize WIN-2-evoked CGRP release, while ruthenium red did, indicates that the action of ruthenium red was likely achieved through antagonism at sites separate from TRPV1, but possibly including other TRP channels. This is further supported by the finding that CAP pretreatment, which is well known to desensitize its own response with repeated applications, did not desensitize the WIN-2 response, except at the highest concentration of CAP (600 nM) at which only a small but significant effect was observed. In this regard, it is interesting that Δ9-THC and cannabinol have been shown to evoke CGRP release from sensory nerves in rat mesenteric arteries in a CPZ-independent, but ruthenium red-dependent manner that persevered in TRPV1-knockout mice (Zygmunt et al., 2002). Moreover, Δ9=THC has recently been identified as an ANKTM1 agonist (Jordt et al., 2004). While specific antagonists of ANKTM1 have yet to be developed, ruthenium red blocks calcium entry through this channel, raising the possibility that WIN-2 might be an ANKTM1 agonist. Lastly, it is possible that the excitatory effects of WIN-2 are indirectly mediated by TRPV4 via the production of epoxyeicosatrienoic acids as has been shown for the endocannabinoid AEA (Watanabe et al., 2003).
While we have demonstrated that WIN-2 has excitatory effects on TG neurons, there are multiple lines of evidence indicating that WIN-2 is capable of inhibiting nociceptive transmission through a peripheral site of action. For example, WIN-2 attenuates carrageenan-evoked hyperalgesia, allodynia and spinal Fos expression through a peripheral mechanism that is blocked by either CB1 or CB2 receptor antagonists (Nackley et al., 2003). Furthermore, WIN-2, acting through peripheral CB1 receptors, attenuates mechanical hyperalgesia in a rat model of neuropathic pain (Fox et al., 2001). The novel CB2 receptor agonist AM1241 has also been shown to reverse tactile and thermal hypersensitivity in the Chung model of neuropathic pain (Ibrahim et al., 2003), raising the interesting possibility that WIN-2, which is approximately 20-fold more potent at CB2 than CB1 receptors, might achieve clinical utility through actions at CB2 receptors at doses insufficient to engage central CB1 receptors and the various side effects they mediate. In any case, these data indicate that WIN-2 is peripherally efficacious in behavioral assays of both inflammatory and neuropathic models of enhanced nociception.
However, in a study to ascertain the actions of WIN-2 in the superficial medullary dorsal horn, no presynaptic activity of WIN-2 on glutamatergic neurotransmission was observed, indicating that the actions of WIN-2 were mediated at sites located remotely from the central terminals of sensory neurons that terminate in the superficial medullary dorsal horn or did not involve glutamatergic neurotransmission (Jennings et al., 2001). Indeed, two recent studies have demonstrated that CB1 receptors are localized nearly exclusively to large diameter, myelinated sensory neurons in both DRG (Bridges et al., 2003) and TG (Price et al., 2003), while CB2 receptor transcripts are not found in TG at all. Consistent with this, we were unable to demonstrate an inhibitory effect of WIN-2 on a de facto nociceptor response (i.e. CAP-evoked CGRP release). In addition, WIN-2 neither inhibited CGRP release evoked by cotreatment with the inflammatory mediators BK plus PGE2 nor diminished forskolin- or PGE2-stimulated cAMP accumulation. Futhermore, WIN-2 itself did not stimulate cAMP accumulation while the endocannabinoid AEA did. That AEA-stimulated cAMP accumulation is antagonized by CPZ and I-RTX (Price et al., 2004) lends further evidence supporting the conclusion that WIN-2 likely does not act through TRPV1 to stimulate TG neurons.
With regard to the physiological mechanisms by which WIN-2 can produce both cannabinoid receptor-dependent effects on peripheral sensory neuronal activity and peripherally mediated antinociception, several findings are worth noting. Aβ-fibers have been implicated in the development and maintenance of hyperalgesia (Campbell et al., 1988; Garcia- Nicas et al., 2001; Kim et al., 2001) and, because many of these fibers likely express CB1 receptors (Bridges et al., 2003), they represent one possible site of action. In an elegant demonstration of the effects of WIN-2 on CAP-evoked hyperalgesia, it was shown that WIN-2 produced a peripherally-mediated, dose-dependent inhibition of CAP-evoked thermal hyperalgesia but not mechanical allodynia (Johanek et al., 2001). Importantly, WIN-2 did not alter the duration of the nocifensive response to CAP. Insofar as the initial, nocifensive response to CAP is due to nociceptor activation, and the corresponding hyperalgesia is mediated, at least in part, by central sensitization (Sang et al., 1996; Magerl et al., 1998; Fang et al., 2002), the effect of WIN-2 to reduce thermal hyperalgesia might be explained by a reduction in the activation of Aβ-fibers through CB1 receptor agonism. A second possible mechanism would involve cannabinoid receptors expressed by non-neuronal peripheral cell types. Both CB1 and CB2 receptors have been localized to a number of immune cells (Berdyshev, 2000; Klein et al., 2001), and especially, CB2 receptors are expressed by mast cells (Facci et al., 1995), macrophages (Carlisle et al., 2002) and lymphocytes (Galiegue et al., 1995). In addition, WIN-2 is capable of inhibiting the release (Berdyshev et al., 1998; Facchinetti et al., 2003) and production (Puffenbarger et al., 2000; Croxford & Miller, 2003) of proinflammatory cytokines from a variety of immune cells in both cannabinoid receptor-dependent and -independent manners. Indeed, the peripheral effects of WIN-2 have been found to be both CB1 and CB2 receptor-dependent in the inflammatory carrageenan model (Nackley et al., 2003), and the involvement of proinflammatory cytokines released from immune cells and epithelia during nociceptor sensitization is an important component of nociceptive processing (Ferreira, 1993; Cunha & Ferreira, 2003; Watkins et al., 2003). In addition, TRPV1 has recently been demonstrated in keratinocytes (Denda et al., 2001; Inoue et al., 2002), which also express cannabinoid receptors (Maccarrone et al., 2003). While the potential role of TRPV1 activation in keratinocytes as regards nociception has yet to be fully realized, TRPV1- mediated release of proinflammatory cytokines from a human keratinocytic cell line has been reported (Southall et al., 2003). Accordingly, the epidermis represents another possible site of action for the extrasensory neuronal effects of WIN-2 in peripherally mediated nociceptive processing.
While we were unable to demonstrate WIN-2-mediated, CB1 receptor-dependent inhibition of sensory neuron activation by CAP or inflammatory mediators, WIN-2 did inhibit K+-evoked CGRP release in a cannabinoid receptor-independent manner. Although this effect was blocked by the CB2 receptor antagonist AM630 at 10 μM, the concentration at which this effect was observed was not consistent with CB2 receptor-dependent effects (e.g. 100 nM– 1 μM). While we cannot rule out that these effects are due to a CB2-like receptor-dependent mechanism, a number of additional findings would tend to rule out the involvement of CB2 receptors: (1) WIN-2 inhibition of K+-evoked CGRP release was mimicked by the cannabinoid-inactive enantiomer WIN-3; (2) WIN-2 inhibition of K+-evoked CGRP release was not affected by PTX; and (3) we have previously demonstrated that CB2 receptor mRNA is not found in TG neurons (Price et al., 2003). It has been demonstrated that 1 μM WIN-2, acting in a CB1 receptor-dependent manner, inhibits K+-evoked intracellular calcium accumulation in intermediate-sized (800–1500 μM2) DRG neurons in culture (Khasabova et al., 2002). As CGRP is primarily localized to relatively smaller diameter neurons, many of which are C- or Aδ-fibers (Mccarthy & Lawson, 1990; Lawson et al., 1993) and primarily do not contain CB1 receptors (Bridges et al., 2003; Price et al., 2003), it is likely that the CB1 receptor-independent effects of WIN-2- and WIN-3 to inhibit K+-evoked CGRP release observed here were measured from a different neuronal population than the CB1 receptordependent effects reported by Khasabova et al. (2002). Furthermore, although we also saw a generally similar inhibitory effect with WIN-2, we utilized a 10-fold higher WIN-2 concentration (vis-á-vis that used to produce stimulation), and this inhibitory effect was not blocked by CB1 receptor antagonists.
In hippocampus, Shen and Thayer (1998) have demonstrated that WIN-2 or WIN-3 inhibits glutamate release at μM concentrations, likely through a direct blockade of voltagegated calcium channels. While we have no direct evidence for this, it is possible that the same or a similar mechanism is at play here, as K+-evoked CGRP release is known to depend on voltage-gated calcium channels (Evans et al., 1996). Despite the nonspecificity of this effect, voltage-gated calcium channels play an important role in nociceptive processing in sensory neurons (Matthews & Dickenson, 2001; Todorovic et al., 2001; Murakami et al., 2002), pointing to another possible mechanism of WIN-2-induced effects on sensory neurons, albeit in a cannabinoid receptor-independent manner.
The present studies demonstrate that WIN-2 has both excitatory and inhibitory actions on TG neurons, both of which, interestingly, seem to involve cannabinoid receptorindependent effector mechanisms. As CB1 and CB2 receptors are found in a number of cell types that might contribute to the behavioral or in vivo neurochemical effects of WIN-2, we have utilized an in vitro primary culture model that is substantially enriched for neurons to distinguish between neuronal and nonneuronal mechanisms of action. The findings herein demonstrate that the sensory neuron-mediated effects of WIN-2 cannot be explained by the previously known pharmacology of the compound, suggesting the existence of additional mechanisms. Whether or not these descriptions are idiotypic of WIN-2 or will be generalizable to a broader group of cannabinoid compounds will await further study, as we more fully appreciate the complex diversity of cannabinoid pharmacology. In addition, this work indicates that a broader understanding of the mechanism(s) by which cannabinoid receptors on Aβ-fibers as well as on extraneuronal cell types contribute to cannabinoid receptor-mediated antihyperalgesia would inform efforts to develop a novel class of peripherally active pain therapeutics.
Acknowledgments
We wish to thank Kelly Berg, Bill Clarke and Joe Vela for assistance with cAMP accumulation assays. This work was supported by National Institute of Drug Abuse Grants DA06085 and DA11959.
Abbreviations
- AEA
anandamide
- BK
bradykinin
- CAP
capsaicin
- CB1
cannabinoid type 1
- CB2
cannabinoid type 2
- CPZ
capsazepine
- DRG
dorsal root ganglion
- I-RTX
iodo-resiniferatoxin
- PGE2
prostaglandin E2
- TG
trigeminal ganglion
- TRP
transient receptor potential|TRPV1, transient receptor potential vanilloid subfamily type 1
- WIN-2
WIN55, 212-2
- WIN-3
WIN55,212-3
References
- AHLUWALIA J, URBAN L, CAPOGNA M, BEVAN S, NAGY I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience. 2000;100:685–688. doi: 10.1016/s0306-4522(00)00389-4. [DOI] [PubMed] [Google Scholar]
- ANCELLIN N, HLA T. Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1, EDG-3, and EDG-5. J Biol Chem. 1999;274:18997–19002. doi: 10.1074/jbc.274.27.18997. [DOI] [PubMed] [Google Scholar]
- BERDYSHEV E, BOICHOT E, CORBEL M, GERMAIN N, LAGENTE V. Effects of cannabinoid receptor ligands on LPS-induced pulmonary inflammation in mice. Life Sci. 1998;63:PL125–PL129. doi: 10.1016/s0024-3205(98)00324-5. [DOI] [PubMed] [Google Scholar]
- BERDYSHEV EV. Cannabinoid receptors and the regulation of immune response. Chem Phys Lipids. 2000;108:169–190. doi: 10.1016/s0009-3084(00)00195-x. [DOI] [PubMed] [Google Scholar]
- BEREITER DA, BEREITER DF, HIRATA H. Topical cannabinoid agonist, WIN55,212-2, reduces cornea-evoked trigeminal brainstem activity in the rat. Pain. 2002;99:547–556. doi: 10.1016/S0304-3959(02)00271-3. [DOI] [PubMed] [Google Scholar]
- BERG KA, CLARKE WP, SAILSTAD C, SALTZMAN A, MAAYANI S. Signal transduction differences between 5- hydroxytryptamine type 2A and type 2C receptor systems. Mol Pharmacol. 1994;46:477–484. [PubMed] [Google Scholar]
- BRIDGES D, RICE AS, EGERTOVA M, ELPHICK MR, WINTER J, MICHAEL GJ. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience. 2003;119:803–812. doi: 10.1016/s0306-4522(03)00200-8. [DOI] [PubMed] [Google Scholar]
- CAMPBELL JN, RAJA SN, MEYER RA, MACKINNON SE. Myelinated afferents signal the hyperalgesia associated with nerve injury. Pain. 1988;32:89–94. doi: 10.1016/0304-3959(88)90027-9. [DOI] [PubMed] [Google Scholar]
- CARLISLE SJ, MARCIANO-CABRAL F, STAAB A, LUDWICK C, CABRAL GA. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int Immunopharmacol. 2002;2:69–82. doi: 10.1016/s1567-5769(01)00147-3. [DOI] [PubMed] [Google Scholar]
- CHU CJ, HUANG SM, DEP ETROCELLIS L, BISOGNO T, EWING SA, MILLER JD, ZIPKIN RE, DADDARIO N, APPENDINO G, DI MARZO V, WALKER JM. N-oleoyldopamine, a novel endogenous capsaicin-like lipid that produces hyperalgesia. J Biol Chem. 2003;278:13633–13639. doi: 10.1074/jbc.M211231200. [DOI] [PubMed] [Google Scholar]
- CROXFORD JL, MILLER SD. Immunoregulation of a viral model of multiple sclerosis using the synthetic cannabinoid R+WIN55,212. J Clin Invest. 2003;111:1231–1240. doi: 10.1172/JCI17652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUNHA FQ, FERREIRA SH. Peripheral hyperalgesic cytokines. Adv Exp Med Biol. 2003;521:22–39. [PubMed] [Google Scholar]
- DENDA M, FUZIWARA S, INOUE K, DENDA S, AKAMATSU H, TOMITAKA A, MATSUNAGA K. Immunoreactivity of VR1 on epidermal keratinocyte of human skin. Biochem Biophys Res Commun. 2001;285:1250–1252. doi: 10.1006/bbrc.2001.5299. [DOI] [PubMed] [Google Scholar]
- EVANS AR, NICOL GD, VASKO MR. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Res. 1996;712:265–273. doi: 10.1016/0006-8993(95)01447-0. [DOI] [PubMed] [Google Scholar]
- FACCHINETTI F, DEL GIUDICE E, FUREGATO S, PASSAROTTO M, LEON A. Cannabinoids ablate release of TNFalpha in rat microglial cells stimulated with lipopolysaccharide. Glia. 2003;41:161–168. doi: 10.1002/glia.10177. [DOI] [PubMed] [Google Scholar]
- FACCI L, DAL TOSO R, ROMANELLO S, BURIANI A, SKAPER SD, LEON A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc Natl Acad Sci USA. 1995;92:3376–3380. doi: 10.1073/pnas.92.8.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FANG L, WU J, LIN Q, WILLIS WD. Calcium– calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 2002;22:4196–4204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERREIRA SH. The role of interleukins and nitric oxide in the mediation of inflammatory pain and its control by peripheral analgesics. Drugs. 1993;46 (Suppl 1):1–9. doi: 10.2165/00003495-199300461-00003. [DOI] [PubMed] [Google Scholar]
- FOX A, KESINGLAND A, GENTRY C, MCNAIR K, PATEL S, URBAN L, JAMES I. The role of central and peripheral cannabinoid 1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92:91–100. doi: 10.1016/s0304-3959(00)00474-7. [DOI] [PubMed] [Google Scholar]
- GALIEGUE S, MARY S, MARCHAND J, DUSSOSSOY D, CARRIERE D, CARAYON P, BOUABOULA M, SHIRE D, LE FUR G, CASELLAS P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- GARCIA-NICAS E, LAIRD JM, CERVERO F. Vasodilatation in hyperalgesic rat skin evoked by stimulation of afferent A beta-fibers: further evidence for a role of dorsal root reflexes in allodynia. Pain. 2001;94:283–291. doi: 10.1016/S0304-3959(01)00365-7. [DOI] [PubMed] [Google Scholar]
- GUNTHORPE MJ, RAMI HK, JERMAN JC, SMART D, GILL CH, SOFFIN EM, LUIS HANNAN S, LAPPIN SC, EGERTON J, SMITH GD, WORBY A, HOWETT L, OWEN D, NASIR S, DAVIES CH, THOMPSON M, WYMAN PA, RANDALL AD, DAVIS JB. Identification and characterisation of SB- 366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology. 2004;46:133–149. doi: 10.1016/s0028-3908(03)00305-8. [DOI] [PubMed] [Google Scholar]
- HERZBERG U, ELIAV E, BENNETT GJ, KOPIN IJ. The analgesic effects of R(+)-WIN 55,212-2 mesylate, a high affinity cannabinoid agonist, in a rat model of neuropathic pain. Neurosci Lett. 1997;221:157–160. doi: 10.1016/s0304-3940(96)13308-5. [DOI] [PubMed] [Google Scholar]
- HIMMEL HM, MEYER ZU HERINGDORF D, GRAF E, DOBREV D, KORTNER A, SCHULER S, JAKOBS KH, RAVENS U. Evidence for Edg-3 receptor-mediated activation of I (KACh) by sphingosine-1-phosphate in human atrial cardiomyocytes. Mol Pharmacol. 2000;58:449–454. doi: 10.1124/mol.58.2.449. [DOI] [PubMed] [Google Scholar]
- HUANG SM, BISOGNO T, TREVISANI M, AL-HAYANI A, DE PETROCELLIS L, FEZZA F, TOGNETTO M, PETROS TJ, KREY JF, CHU CJ, MILLER JD, DAVIES SN, GEPPETTI P, WALKER JM, DI MARZO V. An endogenous capsaicinlike substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci USA. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IBRAHIM MM, DENG H, ZVONOK A, COCKAYNE DA, KWAN J, MATA HP, VANDERAH TW, LAI J, PORRECA F, MAKRIYANNIS A, MALAN TP., JR Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci USA. 2003;100:10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INOUE K, KOIZUMI S, FUZIWARA S, DENDA S, DENDA M. Functional vanilloid receptors in cultured normal human epidermal keratinocytes. Biochem Biophys Res Commun. 2002;291:124–129. doi: 10.1006/bbrc.2002.6393. [DOI] [PubMed] [Google Scholar]
- JENNINGS EA, VAUGHAN CW, CHRISTIE MJ. Cannabinoid actions on rat superficial medullary dorsal horn neurons in vitro. J Physiol. 2001;534:805–812. doi: 10.1111/j.1469-7793.2001.00805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENNINGS EA, VAUGHAN CW, ROBERTS LA, CHRISTIE MJ. The actions of anandamide on rat superficial medullary dorsal horn neurons in vitro. J Physiol. 2003;548:121–129. doi: 10.1113/jphysiol.2002.035063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHANEK LM, HEITMILLER DR, TURNER M, NADER N, HODGES J, SIMONE DA. Cannabinoids attenuate capsaicin-evoked hyperalgesia through spinal and peripheral mechanisms. Pain. 2001;93:303–315. doi: 10.1016/S0304-3959(01)00336-0. [DOI] [PubMed] [Google Scholar]
- JORDT SE, BAUTISTA DM, CHUANG HH, MCKEMY DD, ZYGMUNT PM, HOGESTATT ED, MENG ID, JULIUS D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- KHANOLKAR AD, PALMER SL, MAKRIYANNIS A. Molecular probes for the cannabinoid receptors. Chem Phys Lipids. 2000;108:37–52. doi: 10.1016/s0009-3084(00)00186-9. [DOI] [PubMed] [Google Scholar]
- KHASABOVA IA, SIMONE DA, SEYBOLD VS. Cannabinoids attenuate depolarization-dependent Ca2+influx in intermediate- size primary afferent neurons of adult rats. Neuroscience. 2002;115:613–625. doi: 10.1016/s0306-4522(02)00449-9. [DOI] [PubMed] [Google Scholar]
- KIM HT, PARK SK, LEE SE, CHUNG JM, LEE DH. Non-noxious A fiber afferent input enhances capsaicin-induced mechanical hyperalgesia in the rat. Pain. 2001;94:169–175. doi: 10.1016/S0304-3959(01)00351-7. [DOI] [PubMed] [Google Scholar]
- KLEIN TW, NEWTON CA, FRIEDMAN H. Cannabinoids and the immune system. Pain Res Manage. 2001;6:95–101. doi: 10.1155/2001/326867. [DOI] [PubMed] [Google Scholar]
- LAWSON SN, PERRY MJ, PRABHAKAR E, MCCARTHY PW. Primary sensory neurones: neurofilament, neuropeptides, and conduction velocity. Brain Res Bull. 1993;30:239–243. doi: 10.1016/0361-9230(93)90250-f. [DOI] [PubMed] [Google Scholar]
- MACCARRONE M, DI RIENZO M, BATTISTA N, GASPERI V, ROSSI A, FINAZZI-AGRO A. The endocannabinoid system in human keratinocytes. Evidence that anandamide inhibits epidermal differentiation through CB1 receptor-dependent inhibition of protein kinase C, activating protein-1 and transglutaminase. J Biol Chem. 2003;278:33896–33903. doi: 10.1074/jbc.M303994200. [DOI] [PubMed] [Google Scholar]
- MAGERL W, WILK SH, TREEDE RD. Secondary hyperalgesia and perceptual wind-up following intradermal injection of capsaicin in humans. Pain. 1998;74:257–268. doi: 10.1016/s0304-3959(97)00177-2. [DOI] [PubMed] [Google Scholar]
- MARTIN WJ, COFFIN PO, ATTIAS E, BALINSKY M, TSOU K, WALKER JM. Anatomical basis for cannabinoidinduced antinociception as revealed by intracerebral microinjections. Brain Res. 1999a;822:237–242. doi: 10.1016/s0006-8993(98)01368-7. [DOI] [PubMed] [Google Scholar]
- MARTIN WJ, LOO CM, BASBAUM AI. Spinal cannabinoids are anti-allodynic in rats with persistent inflammation. Pain. 1999b;82:199–205. doi: 10.1016/S0304-3959(99)00045-7. [DOI] [PubMed] [Google Scholar]
- MATSUDA LA, LOLAIT SJ, BROWNSTEIN MJ, YOUNG AC, BONNER TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- MATTHEWS EA, DICKENSON AH. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain. 2001;92:235–246. doi: 10.1016/s0304-3959(01)00255-x. [DOI] [PubMed] [Google Scholar]
- MCCARTHY PW, LAWSON SN. Cell type and conduction velocity of rat primary sensory neurons with calcitonin gene-related peptide-like immunoreactivity. Neuroscience. 1990;34:623–632. doi: 10.1016/0306-4522(90)90169-5. [DOI] [PubMed] [Google Scholar]
- MOLDERINGS GJ, BONISCH H, HAMMERMANN R, GOTHERT M, BRUSS M. Noradrenaline release-inhibiting receptors on PC12 cells devoid of alpha(2(−)) and CB(1) receptors: similarities to presynaptic imidazoline and edg receptors. Neurochem Int. 2002;40:157–167. doi: 10.1016/s0197-0186(01)00076-6. [DOI] [PubMed] [Google Scholar]
- MUNRO S, THOMAS KL, ABU-SHAAR M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- MURAKAMI M, FLEISCHMANN B, DEFE LIPE C, FREICHEL M, TROST C, LUDWIG A, WISSENBACH U, SCHWEGLER H, HOFMANN F, HESCHELER J, FLOCKERZI V, CAVALIE A. Pain perception in mice lacking the beta3 subunit of voltage-activated calcium channels. J Biol Chem. 2002;277:40342–40351. doi: 10.1074/jbc.M203425200. [DOI] [PubMed] [Google Scholar]
- NACKLEY AG, SUPLITA RL, II, HOHMANN AG. A peripheral cannabinoid mechanism suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience. 2003;117:659–670. doi: 10.1016/s0306-4522(02)00870-9. [DOI] [PubMed] [Google Scholar]
- PALMER SL, THAKUR GA, MAKRIYANNIS A. Cannabinergic ligands. Chem Phys Lipids. 2002;121:3–19. doi: 10.1016/s0009-3084(02)00143-3. [DOI] [PubMed] [Google Scholar]
- PRICE TJ, HELESIC G, PARGHI D, HARGREAVES KM, FLORES CM. The neuronal distribution of cannabinoid receptor type 1 in the trigeminal ganglion of the rat. Neuroscience. 2003;120:155–162. doi: 10.1016/S0306-4522(03)00333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRICE TJ, PATWARDHAN A, AKOPIAN AN, HARGREAVES KM, FLORES CM. Modulation of trigeminal sensor, neuron activity by the dual cannabinoid-vanilloid agonists anandamide, N-anachidonoyl-dopamine and anachidonyl-2-chloroethylamide. Br J Pharmacol. 2004 doi: 10.1038/sj.bjp.0705711. Mar 8 (Ep ub) [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUFFENBARGER RA, BOOTHE AC, CABRAL GA. Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia. 2000;29:58–69. [PubMed] [Google Scholar]
- SANG CN, GRACELY RH, MAX MB, BENNETT GJ. Capsaicin-evoked mechanical allodynia and hyperalgesia cross nerve territories. Evidence for a central mechanism. Anesthesiology. 1996;85:491–496. doi: 10.1097/00000542-199609000-00007. [DOI] [PubMed] [Google Scholar]
- SHEN M, THAYER SA. The cannabinoid agonist Win 55, 212-2 inhibits calcium channels by receptor-mediated and direct pathways in cultured rat hippocampal neurons. Brain Res. 1998;783:77–84. doi: 10.1016/s0006-8993(97)01195-5. [DOI] [PubMed] [Google Scholar]
- SMART D, GUNTHORPE MJ, JERMAN JC, NASIR S, GRAY J, MUIR AI, CHAMBERS JK, RANDALL AD, DAVIS JB. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) Br J Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOUTHALL MD, LI T, GHARIBOVA LS, PEI Y, NICOL GD, TRAVERS JB. Activation of epidermal vanilloid receptor-1 induces release of proinflammatory mediators in human keratinocytes. J Pharmacol Exp Ther. 2003;304:217–222. doi: 10.1124/jpet.102.040675. [DOI] [PubMed] [Google Scholar]
- TODOROVIC SM, JEVTOVIC-TODOROVIC V, MEYENBURG A, MENNERICK S, PEREZ-REYES E, ROMANO C, OLNEY JW, ZORUMSKI CF. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron. 2001;31:75–85. doi: 10.1016/s0896-6273(01)00338-5. [DOI] [PubMed] [Google Scholar]
- WATANABE H, VRIENS J, PRENEN J, DROOGMANS G, VOETS T, NILIUS B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- WATKINS LR, MILLIGAN ED, MAIER SF. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol. 2003;521:1–21. [PubMed] [Google Scholar]
- YAMAGUCHI F, TOKUDA M, HATASE O, BRENNER S. Molecular cloning of the novel human G protein-coupled receptor (GPCR) gene mapped on chromosome 9. Biochem Biophys Res Commun. 1996;227:608–614. doi: 10.1006/bbrc.1996.1553. [DOI] [PubMed] [Google Scholar]
- ZYGMUNT PM, ANDERSSON DA, HOGESTATT ED. Delta 9-tetrahydrocannabinol and cannabinol activate capsaicin-sensitive sensory nerves via a CB1 and CB2 cannabinoid receptor-independent mechanism. J Neurosci. 2002;22:4720–4727. doi: 10.1523/JNEUROSCI.22-11-04720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT PM, PETERSSON J, ANDERSSON DA, CHUANG H, SORGARD M, DI MARZO V, JULIUS D, HOGESTATT ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]