

Abstract
Severe acute respiratory syndrome coronavirus (SARS-CoV) poses a considerable threat to human health. Activation of the viral spike (S)-protein by host cell proteases is essential for viral infectivity. However, the cleavage sites in SARS-S and the protease(s) activating SARS-S are incompletely defined. We found that R667 was dispensable for SARS-S-driven virus-cell fusion and for SARS-S-activation by trypsin and cathepsin L in a virus-virus fusion assay. Mutation T760R, which optimizes the minimal furin consensus motif 758-RXXR-762, and furin overexpression augmented SARS-S-activity, but did not result in detectable SARS-S cleavage. Finally, SARS-S-driven cell-cell fusion was independent of cathepsin L, a protease essential for virus-cell fusion. Instead, a so far unknown leupeptin-sensitive host cell protease activated cellular SARS-S for fusion with target cells expressing high levels of ACE2. Thus, different host cell proteases activate SARS-S for virus-cell and cell-cell fusion and SARS-S cleavage at R667 and 758-RXXR-762 can be dispensable for SARS-S activation.
Keywords: SARS coronavirus, spike protein, proteolytic cleavage, cathepsin L, furin
Introduction
A novel coronavirus (CoV) has been identified as the causative agent of severe acute respiratory syndrome (SARS), which claimed almost 800 lives in 2002–03 (Drosten et al., 2003; Ksiazek et al., 2003; Peiris et al., 2004). Coronaviruses, including SARS-CoV, harbour three envelope proteins, spike (S), membrane (M), and envelope (E), which are required for virion assembly, release and infectious entry into target cells (Masters, 2006). The SARS-CoV-S-protein (SARS-S) mediates infectious cellular entry (Hofmann et al., 2004b; Simmons et al., 2003; Yang et al., 2004) and constitutes the major target of the neutralizing antibody response (Hofmann and Pöhlmann, 2004; Nie et al., 2004b; Nie et al., 2004a). The carboxypeptidase angiotensin-converting enzyme 2 (ACE2) is used by SARS-CoV as receptor for cell entry (Li et al., 2003; Wang et al., 2004) and is expressed on type II pneumocytes, the major viral target cells (Hamming et al., 2004; Mossel et al., 2008; To et al., 2004). Several cellular C-type lectins augment or facilitate SARS-S-driven entry (Gramberg et al., 2005; Jeffers et al., 2004; Marzi et al., 2004; Yang et al., 2004). However, ACE2 but not C-type lectin expression correlates with susceptibility to SARS-S-driven infection (Hofmann et al., 2004a; Nie et al., 2004b) and is essential for SARS-CoV spread in experimentally infected mice (Kuba et al., 2005), indicating that ACE2 is a major and likely the only receptor used by SARS-CoV in the infected host. Collectively, SARS-S interacts with host cell factors to mediate the first essential step in the viral life cycle, virus entry into target cells, and constitutes an attractive target for preventive and therapeutic approaches.
The SARS-S-protein is synthesized in the constitutive secretory pathway of infected cells. Amino acid motifs in its cytoplasmic tail slow down transit through the Golgi compartment (McBride et al., 2007) where interactions with the M-protein facilitate virion incorporation (McBride and Machamer, 2010; Voss et al., 2009). The structural organization of SARS-S is similar to that of several other viral envelope proteins, termed class I fusion proteins: The extracellular S1 domain facilitates binding to the receptor, ACE2, while the membrane-anchored S2 domain harbours the functional elements required for fusion of the viral with a target cell membrane (Hofmann and Pöhlmann, 2004). Viral class I fusion proteins are usually synthesized in an inactive form, and require activation by host cell proteases to transit into a fusion-active state (Eckert and Kim, 2001; Harrison, 2008). However, viral strategies to accomplish proteolytic activation can vary. For instance, the majority of strains of the murine coronavirus mouse hepatitis virus (MHV) contain S-proteins that are cleaved by furin in infected cells, and these viruses are believed to enter target cells by receptor-dependent, pH-independent fusion with the plasma membrane (de Haan et al., 2004; Nash and Buchmeier, 1997; Qiu et al., 2006), although some of these findings are controversial (Eifart et al., 2007; Simmons et al., 2005). In contrast, the S-protein of the MHV type 2 strain is not cleaved by furin and the spike protein on incoming virions is activated in target cell vesicles by endosomal proteases of the cathepsin family (Qiu et al., 2006).
Similar to MHV-2, proteolytic activation of SARS-S is mediated by cathepsins in target cells, most importantly by cathepsin L (Simmons et al., 2005). Cleavage-activation by cathepsin L is thought to require previous binding of SARS-S to ACE2, which is believed to induce a conformational change in the S-protein (Simmons et al., 2005), and seems to involve at least two consecutive proteolytic processing steps (Belouzard et al., 2009; Simmons et al., 2005). However, the cleavage sites in SARS-S have been incompletely defined. Arginine 667 was shown to be required for the robust augmentation of SARS-S-driven cell-cell (Belouzard et al., 2009; Follis et al., 2006) and virus-cell fusion (Kam et al., 2009) by trypsin and for the trypsin-dependent circumvention of the entry blockade imposed by lysosomotropic agents (Belouzard et al., 2009). In spite of these results, it is at present unknown if R667 is required for SARS-S activation by cathepsin L. Recent evidence suggests that R797 is a component of a second cleavage site (Belouzard et al., 2009; Watanabe et al., 2008). Thus, similar as for R667, residue R797 was demonstrated to be required for trypsin-dependent augmentation of SARS-S-dependent membrane fusion and for trypsin-dependent resistance to lysosomotropic agents (Belouzard et al., 2009). Nevertheless, efficient proteolytic processing of wild type SARS-S at this site, or at any other site, has so far not been demonstrated in cells (Hofmann et al., 2004b; Simmons et al., 2004; Xiao et al., 2003; Yang et al., 2004; Yao et al., 2004), with one exception (Wu et al., 2004). Notably, the SARS-S-protein contains a minimal furin cleavage site at position 758 – 761 (RNTR), and a peptide comprising this sequence is efficiently cleaved by furin (Bergeron et al., 2005). However, the contribution of the RNTR motif to proteolytic activation of SARS-S is unknown.
In order to explore the role of the minimal furin cleavage site at position 758–761 and to further investigate the importance of the protease sensitive site at position R667, we analyzed the SARS-S mutants R667A and T760R. Our analysis revealed the R667 was required for responsiveness to trypsin-treatment in some experimental systems but had no effect on SARS-S activation by trypsin and cathepsin L in a virus-virus fusion assay, which adequately mimics, in isolation, the conditions required for SARS-S-driven membrane fusion, suggesting that R667 might not play a major role in SARS-S activation in target cells. Mutation T760R, which optimized an existing minimal furin cleavage motif, increased SARS-S activity but no evidence for cleavage of mutant T760R was obtained. Finally, our results demonstrate the cathepsin L activates SARS-S for virus-cell but not cell-cell fusion, which was dependent on the activity of a so far uncharacterized serine protease (for fusion with targets expressing high amounts of ACE2) or addition of exogenous trypsin (for fusion with targets expressing low amounts of ACE2).
Results
Residue R667 but not K672 determines sensitivity of SARS-S to inactivation by trypsin
Based on alignments with other coronaviruses, it has been suggested that residues R667 and K672 define a potential cleavage site for host cell proteases (Bergeron et al., 2005; Follis et al., 2006). To determine the importance of these residues for SARS-S sensitivity to proteolysis, we introduced mutations R667A or K672L into wt SARS-S (Fig. 1). Analysis of the SARS-S mutants by lentiviral pseudotyping, an experimental approach which adequately models SARS-S-driven entry into target cells (Hofmann et al., 2004b; Moore et al., 2004; Simmons et al., 2004), showed that the mutations R667A and K672L were both compatible with robust SARS-S-driven virion incorporation and entry (Fig. 2A,B), albeit the entry efficiency of viruses harbouring the mutant S-proteins was somewhat reduced compared to pseudotypes bearing wt SARS-S (Fig. 2A). Similar results were seen with a R667A and K672L double mutant (wt SARS-S: 44,390 ± 3286 counts per second (c.p.s.), R667A/K672DL: 39,953 ± 1329 c.p.s.), demonstrating that these particular basic residues are not required for infectious entry in tissue culture.
Fig. 1.

Domain organization of the SARS-CoV spike-protein, including potential cleavage sites. The mutations introduced into the SARS-S are depicted. The box indicates a potential furin cleavage motif. HR, heptad repeat; SP, signal peptide; T, transmembrane domain; CT, cytoplasmic domain.
Fig. 2.

Role of R667 and K672 in SARS-CoV entry and inactivation. (A) Infectivity of pseudovirions bearing wild-type SARS-S and SARS-S variants R667A or K672L or no envelope protein (control) for VeroE6 cells. Results are presented as means and standard deviations of triplicate wells. Similar results were seen in 293T and 293T-ACE2 cells. (B) Expression of SARS-S and mutant SARS-S-proteins on pseudovirions. Equal volumes of virus stocks were concentrated by ultracentrifugation through a sucrose gradient, treated with PBS or trypsin and spike-proteins were detected using a monoclonal antibody directed against the N-terminal portion of SARS-S. (C) Trypsin inactivation of pseudotypes bearing SARS-S or mutant SARS-S proteins. Ultracentrifuge-concentrated virus was pretreated with varying concentrations of TPCK-trypsin for 10 min at 25°C, before trypsin inactivation and spin infection of 293T-ACE2 cells. Results are presented relative to infectivity measured for untreated virions and are means and standard deviations of triplicate wells. (D) Inactivation of SARS-S and SARS-S mutant R667A by various proteases prior to infection of 293T-ACE2 cells. Results are presented relative to the infectivity measured for untreated viruses and are means and standard deviations of triplicate wells. Similar results were seen in two additional experiments.
We next assessed if the exchanges R667A and K672L altered sensitivity of SARS-S-bearing pseudotypes to trypsin treatment, which was previously shown to inactivate cell-free virions (Simmons et al., 2004; Simmons et al., 2005). Indeed, pre-treatment of pseudotypes bearing wt SARS-S and SARS-S mutant K672L reduced viral infectivity in a dose-dependent manner (Fig. 2C). In contrast, SARS-S mutant R667A was resistant to inactivation by trypsin (Fig. 2B,C), indicating that R667 defines a trypsin-sensitive site and that cleavage at this site abrogates infectivity of free virions. To further characterize the importance of R667 for SARS-S sensitivity to proteolysis, we compared pseudotypes bearing wt SARS-S and SARS-S mutant R667A for their sensitivities to inactivation by a panel of proteases. Treatment with trypsin, plasmin factor Xa and, to a lesser extent, thrombin reduced infectivity of viruses harbouring wt SARS-S but not SARS-S mutant R667A (Fig. 2D), further underlining that R667 defines a protease sensitive site. Thermolysin and chymotrypsin treatment also diminished viral infectivity, but wt SARS-S and SARS-S mutant R667A were equally sensitive to inactivation by these proteases (Fig. 2D), indicating that they cleave SARS-S at a site distinct from R667. Collectively, R667 but not K672 defines a protease-sensitive site and proteolysis of cell-free virions at this site abrogates viral infectivity.
Residue R667 is indispensable for trypsin-induced infection of target cells treated with ammonium chloride
Lysosomotropic agents, such as ammonium chloride, interfere with endosomal acidification and block infectious entry of SARS-CoV (Hofmann et al., 2004b; Simmons et al., 2004; Yang et al., 2004), presumably by inhibiting cathepsins, which require low pH for optimal activity. Treatment of cell-bound virus with trypsin was shown to allow infectious SARS-S-driven entry into ammonium chloride-treated cells (Simmons et al., 2005), indicating that trypsin can functionally replace cathepsin L as a SARS-S-activating protease under these conditions (“trypsin bypass”). We asked if R667 is required for a trypsin bypass of endosomal acidification. Treatment of target cells with ammonium chloride markedly inhibited infectious entry driven by wt SARS-S and SARS-S mutant R667A, and infectivity of wt SARS-S bearing viruses could be fully restored by treatment of cell-bound virions with trypsin (Fig. 3). In contrast, infectivity of R667A bearing virions was not rescued by trypsin treatment (Fig. 3), indicating that R667 is required for activation of cell-bound virions by trypsin.
Fig. 3.

Trypsin bypass of wt SARS-S and SARS-S R667A infection of ammonium chloride treated VeroE6 cells. Virus was allowed to bind, but not internalize into VeroE6 cells pretreated with 20 mM ammonium chloride. Subsequently, the cells were treated with TPCK-trypsin (10 μg/ml) to activate SARS-S for membrane fusion. Results are presented as a percentage of no trypsin, no ammonium chloride control and are means and standard deviations of triplicate wells. Similar results were seen on 293T-ACE2 cells.
Residue R667 is dispensable for activation of SARS-S by trypsin and cathepsin L in a virus-virus fusion assay
We next assessed the impact of R667A on SARS-S proteolytic activation in a virus-virus membrane-fusion assay. In this assay, SARS-S bearing viruses (which also contain the Avian Sarcoma Leukosis Virus-A envelope protein, EnvA, in their membrane) are allowed to fuse with ACE2 harbouring viruses. The fusion efficiency is then quantified by addition of virions to leupeptin-treated (to exclude an impact of host cell proteases on SARS-S activation) HeLa cells, which express the EnvA receptor TvA, and which are not susceptible to SARS-S-driven infection (Simmons et al., 2005). Thus, the virus-virus fusion assay is a reductionistic model system, which allows the proteolytic activation of SARS-S to proceed under cell free conditions. Efficient fusion of virions bearing wt SARS-S with virions bearing ACE2 was only observed upon treatment of particles with trypsin and recombinant activated cathepsin L (Fig. 4), in agreement with previously reported results (Simmons et al., 2005). Strikingly, trypsin and cathepsin L treatment activated virions bearing wt SARS-S and SARS-S mutant R667A with similar efficiency (Fig. 4), demonstrating that R667 was dispensable for SARS-S activation under these conditions. In summary, R667 defines a trypsin cleavage site, which is responsible for trypsin-dependent activation of cell-bound and inactivation of cell-free virions, respectively. However, R667 is dispensable in tissue culture infection and for SARS-S activation by trypsin and cathepsin L in a virion-virion fusion assay, and might thus be dispensable for proteolytic activation of SARS-CoV in target cells.
Fig. 4.
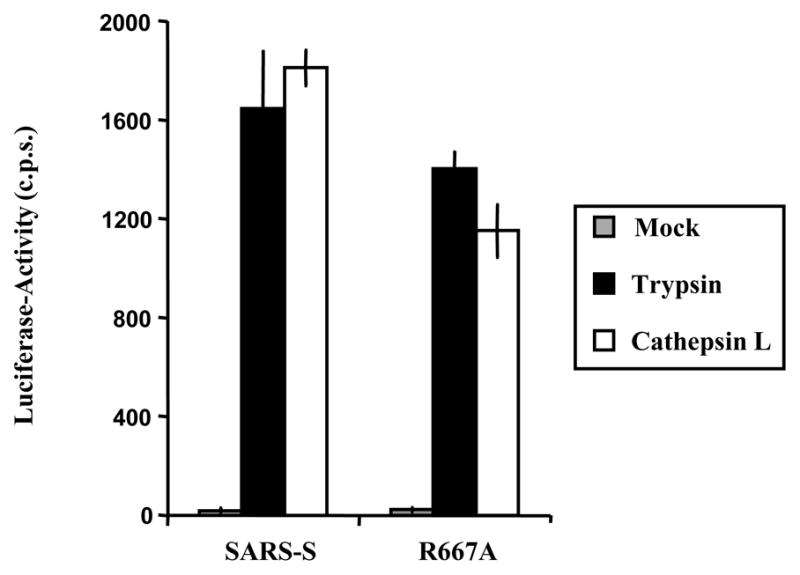
Protease activation of virus-virus fusion mediated by wt SARS-S and mutant R667A. Pseudovirions bearing SARS-S or ACE2 were mixed and virion-fusion activated by trypsin (10 μg/ml) or cathepsin L (2 μg/ml). Mock indicates the mock addition of protease to mixed particles. In addition to this control, further controls of Spike-EnvA or ACE-2 pseudotypes alone treated or untreated with protease are consistently below the level of detection, as previously described (Simmons et al., 2005). Infection by luciferase-encoding particles was determined using luciferase substrate as described by the manufacturer (Promega). Results are presented as means and standard deviations of replicates of four wells. Similar results were seen in 2 additional experiments.
Substitution T760R augments SARS-S-driven infection and cell-cell fusion
We hypothesized that the minimal furin cleavage site (RXXR) at position 758 – 761 might contribute to SARS-S-dependent membrane fusion by allowing SARS-S cleavage, albeit with low efficiency. To investigate the role of this motif in SARS-S-driven entry, we assessed if optimization of the minimal furin cleavage site by exchange of T760R (resulting in the sequence RXRR, Fig. 1) affects SARS-S function. Fluorescence-activated cell sorting analysis revealed that SARS-S mutant T760R and wt SARS-S were expressed to comparable levels at the surface of transfected 293T cells, and could thus be directly compared in functional studies (supplementary figure 1). We first determined if substitution T760R affected S-protein-driven infectious entry. In order to assess infectious entry facilitated by the S-protein variant, pseudotypes were used for infection of control or ACE2-transfected 293T cells (Fig. 5, left panel). The 293T cell line expresses endogenous ACE2 and is therefore susceptible to SARS-S-driven infection (Hofmann et al., 2004b; Hofmann et al., 2004a; Li et al., 2003; Simmons et al., 2004; Yang et al., 2004). Accordingly, infectious entry of SARS-S-bearing pseudovirions into control transfected cells was detectable (Fig. 5, left panel). Notably, entry driven by the T760R variant was about 2-fold more efficient than infection driven by SARS-S wt, and a similar observation (4-fold increase) was made when infection of ACE2-transfected cells was examined (Fig. 5, left panel), suggesting that T760R augments S-protein activity. Indeed, when SARS-S-driven membrane fusion was assessed in a previously reported cell-cell fusion assay (Hofmann et al., 2006), which measures fusion of effector 293T cells expressing S-proteins with 293T target cells expressing ACE2 or pcDNA3, again the S-protein mutant was more active than SARS-S wt (Fig. 5, right panel). Thus, the introduction of an arginine residue at position 760 increases the membrane fusion activity of SARS-S.
Fig. 5.
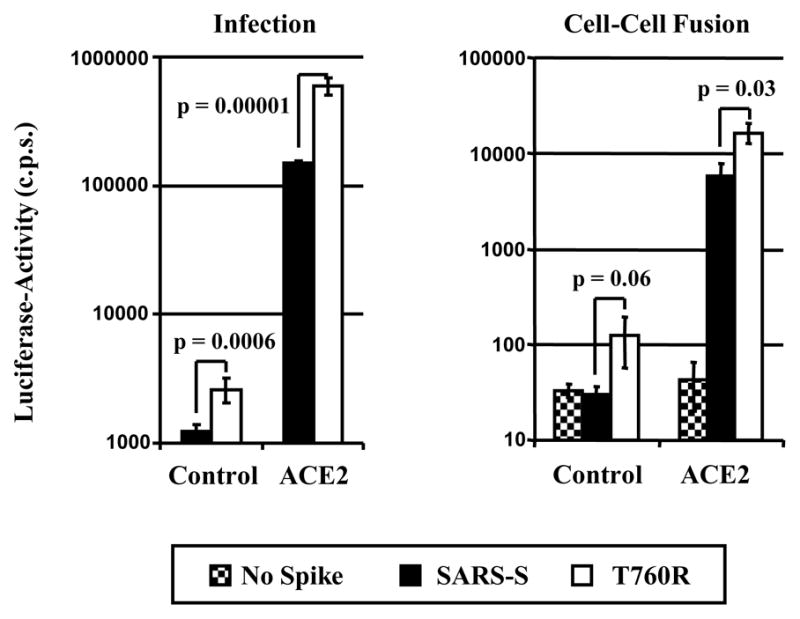
Activity of wt SARS-S and variant T760R in virus-cell (left panel) and cell-cell fusion (right panel) experiments. Left panel: Pseudoparticles harboring the indicated SARS-S proteins or no S-protein as a control were normalized for equal capsid content and used for infection of 293T cells expressing either pcDNA3 or ACE2 in quadruplicates. Luciferase-activities were determined after 72 h. A representative experiment is shown, comparable results were obtained in two independent experiments. Error bars indicate standard deviation (SD). Right panel: 293T effector cells cotransfected with expression vectors encoding the indicated S-proteins (or transfected with empty vector) in combination with a GAL-VP16 expression plasmid were co-cultivated with target cells transfected with either pcDNA3 or ACE2 plasmid together with a plasmid encoding a GAL-VP16-responsive luciferase reporter gene. Luciferase-activities were measured two days after cocultivation. The results of a representative experiment are shown and were confirmed in two separate experiments. Error bars indicate SD.
Overexpression of furin enhances cell-cell and virus-cell fusion driven by SARS-S wt and mutant T760R
Exchange T760R optimizes a minimal furin cleavage site in SARS-S and might increase SARS-S activity by facilitating cleavage by furin or related proteases. We investigated whether furin can augment SARS-S activity by determining the infectivity of p24-normalized SARS-S pseudotypes produced in the absence and presence of overexpressed furin (Fig. 6, left panel). Overexpression of furin moderately increased infectivity of pseudotypes bearing wt SARS-S or mutant T760R. The most notable effect was observed for SARS-S wt pseudotypes, which showed a ~7-fold augmented infectivity (Fig. 6, left panel), albeit this increase was not statistically significant. Comparable observations were made when S-driven cell-cell fusion was examined, and the increase in activity of wt SARS-S and mutant T760R observed upon furin overexpression was statistically significant (Fig. 6, right panel). However, fusion driven by SARS-S wt and T760R were augmented to similar degrees (about 2–3 fold) by furin overexpression. Thus, high levels of furin enhance SARS-S-protein driven cell-cell and virus-cell fusion.
Fig. 6.

Impact of furin expression on virus-cell (left panel) and cell-cell fusion (right panel) driven by wt SARS-S and SARS-S variant T760R. Left panel: Pseudotypes harboring the indicated S-proteins were prepared in the presence (white bars) or absence (black bars) of co-expressed furin. After normalization for p24-content, the particles were used for infection of 293T cells in triplicates. Luciferase-activities in cell lysates were determined after 72 h and values measured for SARS-S pseudotypes prepared in the absence of overexpressed furin were set as 100%. The average of four independent experiments is shown, error bars indicate standard error of the mean (SEM). Right panel: 293T effector cells cotransfected with expression plasmids for the indicated S-variants (or transfected with empty vector as control), GAL-VP16 and furin (or transfected with empty vector as control), were co-cultivated with 293T target cells transfected with plasmids encoding a GAL-VP16-responsive luciferase gene and hACE2 (or transfected with empty vector as control) in triplicates. Luciferase-activities in cell lysates were measured two days after co-cultivation. The experiment shown is representative of three independent experiments, error bars indicate SD.
Furin overexpression does not facilitate detectable cleavage of wt SARS-S and mutant T760R
Our functional data showed that mutation of T760R and overexpression of furin augmented SARS-S activity, presumably by facilitating proteolytic processing of the S-protein. To assess SARS-S cleavage, we conducted Western blot analyses of lysates of S-protein transfected 293T cells, using a serum specific for the S2-portion of SARS-S. Expression of β-actin served as loading control. Our results revealed a prominent band of approximately 160 kDa for both wt SARS-S and mutant T760R (Fig. 7), which is expected for uncleaved SARS-S (Hofmann et al., 2004b). Treatment of cells with trypsin before lysis reduced the signal obtained for uncleaved SARS-S, and a band of approximately 90 kDa appeared, in accordance with the previously reported size of the S2-fragment of SARS-S (Bergeron et al., 2005; Follis et al., 2006). Coexpression of furin with SARS-S wt or the S-protein variant T760R did not result in the appearance of a S2-band, indicating that furin-mediated cleavage was inefficient or absent.
Fig. 7.
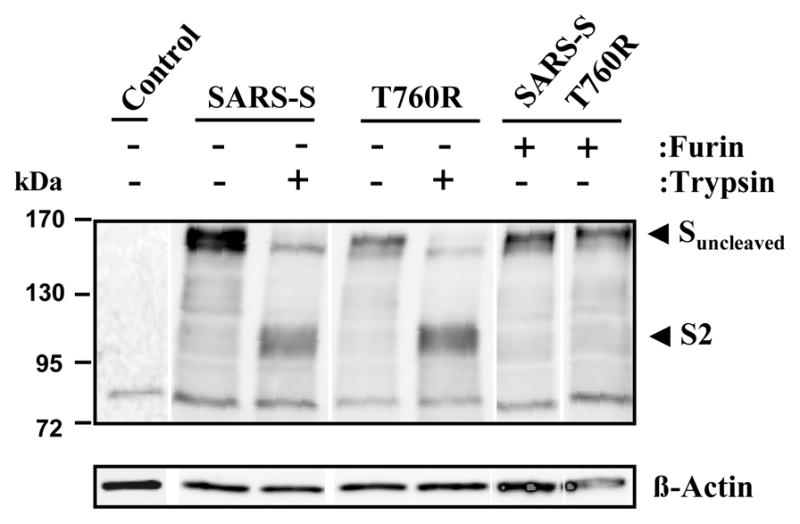
Proteolytic processing of wt SARS-S and SARS-S mutant T760R in the presence and absence of furin overexpression. The indicated S-proteins were transiently expressed in 293T cells in the absence and presence of furin (expression plasmids for SARS-S and furin were transfected at a 1:1 ratio), the cells treated with PBS or trypsin as indicated and cell lysates analyzed with S2-specific antiserum. As control, β-actin expression in cell lysates was assessed. The results of a single gel are shown from which irrelevant lanes were removed.
Virus-cell fusion but not cell-cell fusion by SARS wt and T760R depends on cathepsin activity
We next investigated whether exchange T760R altered the sensitivity of SARS-S-dependent membrane fusion to inhibition by the cathepsin L and B inhibitor MDL 28170. In agreement with published data (Simmons et al., 2005), MDL 28170 efficiently reduced infection by SARS-S-bearing pseudotypes (Fig. 8, left panel). Similar inhibition was observed with the SARS-S mutant T760R, indicating that this change did not modulate cathepsin-dependence of viral entry. In stark contrast, MDL 28170 had no inhibitory effect on wt SARS-S and T760R dependent cell-cell fusion (Fig. 8, right panel), indicating the SARS-S-protein-driven fusion of cellular membranes does not depend on cathepsin activity.
Fig. 8.

Impact of the cathepsin B/L inhibitor MDL 28170 on virus-cell (left panel) and cell-cell fusion (right panel) driven by wt SARS-S and SARS-S mutant T760R. Left panel: 293T target cells were pre-incubated with the indicated concentrations of MDL 28170 for 1 h and thereafter infected in quadruplicates with infectivity-normalized pseudotypes bearing the indicated S-proteins. Luciferase activities in cell lysates were determined at 72 h post infection. The results of a representative experiment carried out in triplicates are shown and were confirmed in two separate experiments. Right panel: 293T target cells transfected with gal5-luc and either pcDNA3 (control) or ACE2 were mixed with effector cells expressing GAL-VP16 in combination with the indicated S-proteins. After mixing, cells were either left untreated or treated with 1 μM MDL 28170. Luciferase-activities were determined two days after mixing. The results of a representative experiment are shown; comparable results were obtained in an independent experiment. Error bars indicate SD.
SARS-S-dependent cell-cell fusion is inhibited by leupeptin
Since SARS-S-driven cell-cell fusion was not dependent on cathepsin-activity, we asked if the activity of other proteases might be required. For this, we first sought to clarify to which extend SARS-S-driven cell-cell fusion depends on the presence of exogenous trypsin, since previous studies reported that efficient SARS-S-driven cell-cell fusion occurred only upon treatment of SARS-S expressing cells with trypsin (Howard et al., 2008; Simmons et al., 2004), while in our experiments trypsin was dispensable for robust cell-cell fusion (Figs. 5, 6 and 8). When we examined fusion of SARS-S or VSV-G transfected 293T cells with control transfected 293T cells (which express low levels of endogenous ACE2 (Hofmann et al., 2004a; Li et al., 2003; Simmons et al., 2004)), we found that trypsin treatment was required for SARS-S- but not VSV-G-driven cell-cell fusion, and that fusion driven by VSV-G but not SARS-S was induced by low pH (Fig. 9A). However, when cell-cell fusion with ACE2 transfected 293T cells was examined, robust SARS-S-driven fusion was already observed in the absence of trypsin treatment, and the fusion activity was further augmented in the presence of trypsin (Fig. 9A). Thus, SARS-S-driven cell-cell fusion depends on trypsin-activation of SARS-S only if receptor levels are limiting. If ACE2 is expressed at high levels, robust fusion occurs in the absence of trypsin and is most likely due to SARS-S activation by a host cell protease other than cathepsins.
Fig. 9.

SARS-S-dependent cell-cell fusion depends on the activity of a cysteine or serine protease. (A) Target 293T cells cotransfected with a plasmid encoding β-galactosidase ω fragment and either pcDNA3 (control) or plasmid encoding ACE2 were mixed with 293T effector cells transiently expressing β-galactosidase α peptide in combination with either the SARS-S or VSV-G proteins. After one hour, cells were pulsed with different pH’s or TPCK-trypsin. After a further five hours, β-galactosidase trans-complementation was assessed and expressed as relative light units. The results of a representative experiment carried out in triplicates are shown. Error bars indicate SD. Similar results were obtained in an independent experiment. (B) Target 293T cells transfected with gal5-luc and ACE2 were mixed with 293T effector cells expressing GAL-VP16 in combination with the SARS-S-protein. Effector cells transfected with empty vector (no spike) served as negative controls. Target cells were pre-incubated with PBS or the indicated concentrations of protease inhibitors before mixing with effector cells. After mixing of cells, which involved detachment and washing of cells, the inhibitors were replenished and cocultures were maintained for 48 h. Thereafter, luciferase-activities in cell lysates were determined. The results of a representative experiment carried out in triplicates are shown, cell-cell fusion measured in the absence of inhibitor was set as 100%. Error bars indicate SD. Similar results were obtained in two independent experiments. (C) 293T-hACE2 cells were incubated with the indicated inhibitors of 1 h and subsequently infected with infectivity-normalized pseudotypes bearing VSV-G or SARS-S. Luciferase activities in cell lysates were determined at 72 h post infection. The results of a representative experiment performed in triplicates are shown; error bars indicate SD. Similar results were obtained in two independent experiments.
In order to investigate the need for a host cell protease for trypsin-independent SARS-S-driven cell-cell fusion, we inhibited the wt SARS-S-driven cell-cell fusion reaction by leupeptin, an inhibitor of cysteine and serine protease, AEBSF, a serine protease inhibitor, and the cathepsin inhibitors E64c and MDL28170. All inhibitors were used at non-cytotoxic concentrations, as determined by a commercially available cytotoxicity assay (Promega, Madison, USA) and by the lack of inhibition of luciferase expression in cells cotransfected with the reporter plasmids employed to quantify cell-cell fusion (supplementary figure 2). Of all inhibitors tested, only leupeptin inhibited SARS-S-driven cell-cell fusion, and inhibition was dose-dependent (Fig. 9B). Finally, leupeptin, E64c and MDL28170 inhibited SARS-S-driven virus-cell fusion (Fig. 9C), as expected from the results shown in Fig. 8 and from previous work (Simmons et al., 2005). Thus, SARS-S-driven cell-cell fusion depends on the activity of a so far uncharacterized cysteine or serine protease, while virus-cell fusion requires cathepsin activity.
Discussion
The processing of SARS-S by cellular proteases might determine route and efficiency of viral entry into target cells and might have important consequences for development of preventive and therapeutic strategies (Belouzard et al., 2009; Simmons et al., 2005; Watanabe et al., 2008). However, the sites in SARS-S, which are recognized by host cell proteases, are incompletely defined. We show that R667 defines a trypsin sensitive site, which is required for inactivation of cell-free virus by trypsin and for trypsin-dependent infectious entry of cell-bound virus into targets pretreated with ammonium chloride. In contrast, the integrity of R667 was dispensable for infectious entry in cell culture and activation of SARS-S-driven virus-virus fusion by trypsin and cathepsin L. Optimization of an existing minimal furin cleavage site, 758-RNTR-761, by mutation T760R augmented SARS-S-driven cell-cell and virus-cell fusion. However, no evidence for cleavage of SARS-S at this motif was obtained. Finally, differential blockade of SARS-S-driven virus-cell and cell-cell fusion by protease inhibitors showed that these processes depend on different activating proteases, with a so far poorly characterized serine or cysteine protease being responsible for SARS-S-driven cell-cell fusion.
The spike protein of SARS-CoV contains several potentially protease sensitive sites, which have been implicated in proteolytic processing of SARS-S (Belouzard et al., 2009; Bergeron et al., 2005; Follis et al., 2006). The most N-terminal motif, amino acids 657 to 676, shows some similarity to cleavage sites of other coronavirus S-proteins but does not comprise an RXXR motif recognized by proprotein convertases in the context of cleavable coronavirus S-proteins (Follis et al., 2006). Nevertheless, mutation of R667 was reported to block the increase in SARS-S membrane fusion activity observed for wt SARS-S upon furin overexpression, and introduction of a furin consensus sequence at this site was shown to increase SARS-S-driven cell-cell fusion (Belouzard et al., 2009; Follis et al., 2006). Our results demonstrate that this site is important for SARS-S cleavage by trypsin, and that trypsin cleavage depends on the presence of R667 but not K672. Thus, cell-free virions bearing wt SARS-S but not SARS-S mutant R667A were inactivated by trypsin, while cell-bound virions bearing wt SARS-S but not mutant R667A were activated by trypsin for infection of ammonium chloride pretreated target cells (trypsin bypass). These findings are in agreement with published work (Belouzard et al., 2009; Follis et al., 2006), and highlight that R667 defines a protease sensitive site in SARS-S. Unexpectedly, however, mutation of R667 was compatible with robust SARS-S activation by trypsin and cathepsin L in a virion-virion fusion assay. The reasons for the differential requirement for R667 in the trypsin bypass and virion-virion fusion assays are at present unclear, but might relate to different properties of target cell membrane relative to virion membrane, in terms of receptor concentration and presence of cellular proteases. Regardless of the underlying mechanism, the present data suggest that R667 might be dispensable for activation of SARS-CoV by trypsin and by cathepsin L. Such a scenario would be in agreement with results by Bosch and colleagues, who mapped the cathepsin L cleavage site in SARS-S to amino acid T678 (Bosch et al., 2008).
Belouzard and colleagues suggested that proteolytic activation of SARS-S might be a two-step process and might involve cleavage at R667 and at R797 (Belouzard et al., 2009). Evidence for an important role of R797 in proteolytic activation of SARS-S was obtained in cell-cell fusion and trypsin bypass experiments, in which the SARS-S mutant R797N was found to be refractory to activation by trypsin (Belouzard et al., 2009). In addition, the insertion of a furin consensus motif at this site increased SARS-S activity in a cell-cell fusion assay (Belouzard et al., 2009) and, as documented by an independent study, allowed cathepsin-independent infectious entry into target cells (Watanabe et al., 2008). Notably, a minimal furin cleavage site, RXXR, is present in SARS-S at amino acids 758 to 761, and a peptide spanning this motif was previously shown to be cleaved by furin, while peptides spanning R667 and R797 were not recognized by the protease (Bergeron et al., 2005). We found that optimizing the minimal furin site at position 758 (mutant T760R) significantly increased SARS-S activity in cell-cell and virus-cell fusion assays, and we noted that the virus-cell fusion activity of wt SARS-S but not mutant T760R was augmented by overexpression of furin in virus-producer cells, although this effect was not statistically significant. These results suggest that amino acids 758 to 761 might constitute an alternative processing site, which might be recognized by proprotein convertases or related enzymes. However, proteolytic processing of wt SARS-S or variant T760R upon furin overexpression could not be demonstrated, indicating that cleavage was inefficient or absent. Therefore, alternative scenarios for the role of amino acids 758 to 761 in SARS-S activation must be considered. Thus, it is possible that augmentation of SARS-S activity upon furin overexpression and mutation T760R were separate effects, with T760R potentially modulating protease sensitivity of other sites in SARS-S.
Cell to cell fusion assays are commonly used to functionally analyze SARS-S, including the characterization of potential proteolytic processing sites in SARS-S (Belouzard et al., 2009; Follis et al., 2006; Hofmann et al., 2006; Simmons et al., 2003). In most studies, SARS-S-driven cell-cell fusion was examined upon activation of SARS-S by exogenous trypsin. However, it has so far not been determined if SARS-S-driven cell-cell fusion can also be activated by a host cell protease. For instance, it is unknown if the proteases responsible for proteolytic activation of virus-associated SARS-S, cathepsins B and L, can also activate cell-associated SARS-S. We found that trypsin-activation of SARS-S was only required for fusion with cells expressing low amounts of ACE2, while fusion with target cells expressing high levels of ACE2 proceeded efficiently in the absence of trypsin, in agreement with a recent study (Glowacka et al., 2011). Under the latter conditions, cell-cell fusion driven by SARS-S was inhibited by leupeptin but not cathepsin inhibitors, indicating that a serine protease, or a cysteine protease other than cathepsins B and L, can activate SARS-S for cell-cell fusion. The identity of the responsible protease(s) is at present unclear. A role for factor Xa, a serine protease, has been suggested but the results await conformation (Du et al., 2007). The type II transmembrane serine proteases (TTSPs) TMPRSS2 and TMPRSS4 can activate the influenza virus hemagglutinin by cleavage (Bottcher et al., 2006; Chaipan et al., 2009) and these proteases were recently shown to also activate SARS-S for membrane fusion (Glowacka et al., 2011; Matsuyama et al., 2010; Shulla et al., 2011). However, appreciable expression of TMPRSS2 or TMPRSS4 was not detected in 293T cells in a previous study (Bertram et al., 2010), and SARS-S-driven cell-cell fusion was not inhibited by the serine protease inhibitor AEBSF (present study), which was shown to be active against other TTSPs (Beliveau et al., 2009). Therefore, TTSPs and related serine proteases are unlikely to account for SARS-S activation under the conditions tested here, and future studies should focus on the role of cysteine proteases.
Materials and Methods
Plasmid construction and in vitro-mutagenesis
Expression plasmids pCAGGS-SARS-S, encoding the spike proteins of SARS-CoV strain Frankfurt (Hofmann et al., 2004b) or Urbani (Simmons et al., 2003), and pcDNA3-hACE2, encoding the human ACE2 receptor, have been described previously (Hofmann et al., 2004b; Hofmann et al., 2004a). Site-directed mutagenesis of the SARS-S-protein was performed by overlap-extension PCR. For generation of mutation T760L the following overlapping primers were used: p5 SARS-S T760R (5-CCGACGTGAAGTGTTCGCTCAAGTC-3) and p3 SARS-S T760R (5-GACTTGAGCGAA CACTTCACGTCGGTTGCGATCCTGTTCAGCAGCAATACC-3). To facilitate generation of PCR fragments bearing the desired mutations, only the 3′ prime portion of the S-sequence was amplified using overlapping PCR. Subsequently, the PCR-amplified fragments were introduced into a pCAGGS variant harbouring the corresponding 5′ portion of the SARS-S sequence. Variants R667A, K672L and KPTKR to EPTED were generated using Quikchange site directed mutagenesis (Stratagene), with SARS-S in pcDNA as template, and then transferred to pCAGGS. All PCR amplified sequences were confirmed by automated sequence analysis.
Cell culture
293T cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin and streptomycin, and grown in a humified atmosphere of 5% CO2. 293T cells stably expressing ACE2 (293T-hACE2) were generated by transfection of plasmid pcDNA3.1zeo-hACE2 (Hofmann et al., 2004a) into 293T cells followed by selection of resistant cells with zeocin (Invitrogen) at 50μg/ml. Surface expression of ACE2 on clonal cells was confirmed by FACS analysis.
Cell-cell fusion assays
For analysis of cell-cell fusion, 293T effector cells seeded in 6-well plates at 3×105/well were CaPO4-cotransfected with plasmid pGAL4-VP16, encoding the Herpes Simplex VP16 transactivator fused to the DNA binding domain of the yeast transcription factor GAL4 (Stamminger et al., 2002), and plasmids encoding SARS-S-variants (or empty plasmid) and furin (or empty plasmid). In parallel, 293T target cells were seeded in 48-well plates at 3×104/well and transfected with pcDNA3 or the hACE2 expression plasmid together with plasmid pGal5-luc, in which luciferase reporter gene expression is controlled by five GAL4 binding sites (Stamminger et al., 2002). The day after transfection, effector cells were diluted in fresh medium and added to the target cells. Cell-cell fusion was quantified by determination of luciferase activities in cell lysates 48 h after cocultivation using a commercially available kit (Promega, Madison, USA). Results generated in the experimental system described above are shown in figures 5, 6, 8 and 9B. Alternatively, cell-cell fusion was assayed using α-complementation of β-galactosidase fragments, as previously described for HIV (Holland et al., 2004). For this, effector 293T cells were co-transfected with plasmids encoding SARS-S or VSV-G and a plasmid encoding an N-terminal fragment of β-galactosidase (amino-acids 1–80; termed α peptide), while target 293T cells were co-transfected with a plasmid encoding ACE2 or with the corresponding empty plasmid and a plasmid encoding the remaining C-terminal portion of β-galactosidase (amino-acids 80–1023; termed ω fragment). The day after transfection, effector cells were diluted in fresh medium and added to the target cells at a ratio of 1:1. After one hour incubation for attachment and binding, cells were washed in serum-free medium and pulsed with medium adjusted to pH5.0, pH7.0, pH8.0 or containing TPCK-trypsin at 15 μg/ml for 10 minutes at 37°C. pH or trypsin were neutralized by the addition of excess medium containing serum and trypsin inhibitor. Cells were then incubated for a further five hours. Upon viral envelope-driven membrane fusion the α peptides and ω fragments of β-galactosidase trans-complement each other to give functional β-galactosidase enzymatic activity, which was detected in cell lysates employing a commercially available kit (Galacton Plus substrate, Applied Biosystems). Results generated in this experimental system are shown in figure 9A.
Production of lentiviral pseudotypes and infection experiments
For generation of lentiviral pseudoparticles, CaPO4 transfections were performed as described (Hofmann et al., 2004b; Simmons et al., 2003). In brief, 293T cells were transiently cotransfected with pNL4-3 E-R- Luc (Connor et al., 1995) and expression plasmids for SARS-S-variants or VSV-G. For some experiments, human furin was co-expressed during production of pseudoparticles. The culture medium was replaced at 16 h and harvested at 48 h post transfection. The supernatants were passed through 0.45 μm filters, aliquotted and stored at −80°C. Capsid contents (p24) in harvested supernatants were determined using a commercially available kit (Murex, Wiesbaden, Germany). For infection, 293T cells or 293T cells transiently transfected with pcDNA3 or hACE2 or 293T cells stably expressing hACE2 were incubated with pseudotypes, normalized for infectivity or p24-capsid protein content, for three days before cells were lysed and luciferase-activities determined using a commercially available kit (Promega, Madison, USA).
Blockade of pseudotype infection by cathepsin inhibitors
293T cells in 96-wells were pre-incubated with the cathepsin L and B inhibitor MDL 28170 (Calbiochem, Darmstadt, Germany) for 30 min. Thereafter, pseudotypes of comparable infectivity were added for 12–16 h, the culture medium was replaced and luciferase activities in cell extracts were determined after 72 h as described above.
Protease inactivation of pseudotyped viruses
Infectivity normalized pseudovirions bearing wild-type or mutant SARS-S protein were incubated with varying amounts of protease for 30 minutes at room temperature. Proteolysis was halted by the addition of an equal volume of medium containing 10%FBS, 100 μg/ml soy bean trypsin inhibitor (STI) and 100 μg/ml aprotinin. Virus was then plated on 293T-ACE2 cells, spin infected for 90 minutes at 2500 rpm and incubated at 37°C. After 4 hours, medium was replaced with fresh medium, and cells were incubated for 48 hours before measurement of luciferase activity. All proteases were obtained from Sigma, and final concentrations used in the assay were determined by preliminary experiments, or the maximal practical level achievable based on the stock solutions of 1mg/ml. Proteases were diluted in PBS. Trypsin and chymotrypsin were used at a final concentration of 25 μg/ml, elastase and factor Xa at 50 μg/ml, thermolysin and thrombin at 125 μg/ml and plasmin at 250 μg/ml.
Trypsin bypass
Trypsin bypass experiments were performed as described (Simmons et al., 2005). Briefly, 293T-ACE2 cells were pretreated for 1 h with cold medium containing ammonium chloride (40 mM). An equal volume of diluted cold pseudovirion mixture (virus was ultracentrifuge-concentrated and resuspended in PBS to remove FBS) was added, and the cells were spin-infected at 4°C to allow virus-binding to cells. The medium was replaced with warm serum-free medium containing ammonium chloride (20 mM) and incubated at 37°C for 15 min. Subsequently, the medium was removed, and fresh medium containing TPCK-trypsin (10 μg/ml) was added for 10 minutes at 25°C. The trypsin-containing medium was then removed and medium supplemented with STI (75 μg/ml) and ammonium chloride (20 mM) was added. After a 12 h incubation period, medium was replaced with fresh medium without ammonium chloride, and cells were incubated for a further 36 hours before luciferase activity was measured.
Virus-virus fusion assay
Virus to virus fusion was assayed as described (Simmons et al., 2005). Briefly, equal amounts of pseudovirions bearing either ACE2 and encoding luciferase as a reporter (HIV-luc(ACE2)) or both SARS-CoV Spike (or mutants) and Avian Sarcoma Leukosis Virus-A envelope and encoding GFP (HIV-gfp(S+E)) were mixed and incubated for 30 min on ice to allow binding. The temperature was then raised to 37°C for 15 min to allow induction of conformational rearrangements. Particles were then either treated with 10 ug/ml TPCK-trypsin (Sigma) or the pH was lowered to pH 6 by the addition of 0.1M citric acid, and preactivated recombinant cathepsin L was added to a final concentration of 2 ug/ml. Proteolysis was halted after 10 min at 25°C by addition of soybean trypsin inhibitor and leupeptin. Virus mixtures were then diluted and used to infect HeLa cells stably expressing Tva that had been pretreated with 20 μg/ml leupeptin for 1 h.
Analysis of SARS-S expression by Western blot
Cells transiently expressing SARS-S or VLPs harboring SARS-S were lysed in SDS-Laemmli buffer and boiled for 15–30 min at 95°C. Samples were separated via 12,5% SDS-PAGE and transferred onto nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). SARS-S proteins were detected by staining with SARS-S specific rabbit serum (Imgenex, San Diego, USA) at a 1:1000 dilution, followed by detection of bound antibodies by use of a peroxidase-conjugated anti-rabbit IgG (Dianova, Hamburg, Germany) at a dilution of 1:5000. For loading control, the stripped membranes were incubated with an anti-β-actin antibody (Sigma, Deisenhofen, Germany) at a 1:1000 dilution, followed by incubation with peroxidase-conjugated anti-mouse IgG (Dianova, Hamburg, Germany) at a dilution of 1:5000. Chemiluminescence detection was performed employing a commercially available kit, according to the manufacturer’s protocol (ECL Western detection kit; Amersham Pharmacia Biotech Europe, Freiburg, Germany).
Statistics
Statistical significance was calculated employing a two-tailed student’s t-test for dependent samples.
Supplementary Material
Surface expression of SARS-S variant T760R. 293T cells transiently transfected with SARS-S (black line) or T760R (dark grey) were stained with a polyclonal anti-SARS-CoV antiserum and a FITC-labeled secondary antibody. Cells transfected with pcDNA3 served as control (black filled histogram).
Protease inhibitors used for blockade of cell-cell and virus-cell fusion were employed at non-cytotoxic concentrations. (A) The reporter plasmids pGAL4-VP16 and pGal5-luc (which were used for the cell-cell fusion assay shown in FIG. 9B) were transfected into 293T cells, the transfected cells were treated with PBS or the indicated protease inhibitors for 48 h and luciferase-activities in cell lysates were determined. The results of a representative experiment performed in triplicates are shown, error bars indicate SD. (B) 293T cells were treated with PBS, leupeptin, the only protease inhibitor able to block SARS-S-driven cell-cell fusion in FIG. 9B, or E64c and ATP levels in the cultures were determined employing the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). The results of a representative experiment performed in triplicates are shown, error bars indicate SD.
Acknowledgments
We thank B. Fleckenstein, K. von der Mark and T.F. Schulz for constant support and K. Korn for p24-ELISA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beliveau F, Desilets A, Leduc R. Probing the substrate specificities of matriptase, matriptase-2, hepsin and DESC1 with internally quenched fluorescent peptides. FEBS J. 2009;276:2213–2226. doi: 10.1111/j.1742-4658.2009.06950.x. [DOI] [PubMed] [Google Scholar]
- Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A. 2009;106:5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron E, Vincent MJ, Wickham L, Hamelin J, Basak A, Nichol ST, Chretien M, Seidah NG. Implication of proprotein convertases in the processing and spread of severe acute respiratory syndrome coronavirus. Biochem Biophys Res Commun. 2005;326:554–563. doi: 10.1016/j.bbrc.2004.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram S, Glowacka I, Blazejewska P, Soilleux E, Allen P, Danisch S, Steffen I, Choi SY, Park Y, Schneider H, Schughart K, Pöhlmann S. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J Virol. 2010;84:10016–10025. doi: 10.1128/JVI.00239-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch BJ, Bartelink W, Rottier PJ. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J Virol. 2008;82:8887–8890. doi: 10.1128/JVI.00415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol. 2006;80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, Takeda M, Bugge TH, Kim S, Park Y, Marzi A, Pöhlmann S. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol. 2009;83:3200–3211. doi: 10.1128/JVI.02205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- de Haan CA, Stadler K, Godeke GJ, Bosch BJ, Rottier PJ. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J Virol. 2004;78:6048–6054. doi: 10.1128/JVI.78.11.6048-6054.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C, Gunther S, Preiser W, van der WS, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Du L, Kao RY, Zhou Y, He Y, Zhao G, Wong C, Jiang S, Yuen KY, Jin DY, Zheng BJ. Cleavage of spike protein of SARS coronavirus by protease factor Xa is associated with viral infectivity. Biochem Biophys Res Commun. 2007;359:174–179. doi: 10.1016/j.bbrc.2007.05.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- Eifart P, Ludwig K, Bottcher C, de Haan CA, Rottier PJ, Korte T, Herrmann A. Role of endocytosis and low pH in murine hepatitis virus strain A59 cell entry. J Virol. 2007;81:10758–10768. doi: 10.1128/JVI.00725-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follis KE, York J, Nunberg JH. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell-cell fusion but does not affect virion entry. Virology. 2006;350:358–369. doi: 10.1016/j.virol.2006.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K, Niemeyer D, Schneider H, Drosten C, Pöhlmann S. Evidence that TMPRSS2 activates the SARS-coronavirus spike-protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011 doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramberg T, Hofmann H, Moller P, Lalor PF, Marzi A, Geier M, Krumbiegel M, Winkler T, Kirchhoff F, Adams DH, Becker S, Munch J, Pöhlmann S. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology. 2005;340:224–236. doi: 10.1016/j.virol.2005.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H, Geier M, Marzi A, Krumbiegel M, Peipp M, Fey GH, Gramberg T, Pöhlmann S. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem Biophys Res Commun. 2004a;319:1216–1221. doi: 10.1016/j.bbrc.2004.05.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H, Hattermann K, Marzi A, Gramberg T, Geier M, Krumbiegel M, Kuate S, Uberla K, Niedrig M, Pöhlmann S. S protein of severe acute respiratory syndrome-associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. J Virol. 2004b;78:6134–6142. doi: 10.1128/JVI.78.12.6134-6142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H, Pöhlmann S. Cellular entry of the SARS coronavirus. Trends Microbiol. 2004;12:466–472. doi: 10.1016/j.tim.2004.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H, Simmons G, Rennekamp AJ, Chaipan C, Gramberg T, Heck E, Geier M, Wegele A, Marzi A, Bates P, Pöhlmann S. Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. J Virol. 2006;80:8639–8652. doi: 10.1128/JVI.00560-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AU, Munk C, Lucero GR, Nguyen LD, Landau NR. Alpha-complementation assay for HIV envelope glycoprotein-mediated fusion. Virology. 2004;319:343–352. doi: 10.1016/j.virol.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Howard MW, Travanty EA, Jeffers SA, Smith MK, Wennier ST, Thackray LB, Holmes KV. Aromatic amino acids in the juxtamembrane domain of severe acute respiratory syndrome coronavirus spike glycoprotein are important for receptor-dependent virus entry and cell-cell fusion. J Virol. 2008;82:2883–2894. doi: 10.1128/JVI.01805-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers SA, Tusell SM, Gillim-Ross L, Hemmila EM, Achenbach JE, Babcock GJ, Thomas WD, Jr, Thackray LB, Young MD, Mason RJ, Ambrosino DM, Wentworth DE, Demartini JC, Holmes KV. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci U S A. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam YW, Okumura Y, Kido H, Ng LF, Bruzzone R, Altmeyer R. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS One. 2009;4:e7870. doi: 10.1371/journal.pone.0007870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi A, Gramberg T, Simmons G, Moller P, Rennekamp AJ, Krumbiegel M, Geier M, Eisemann J, Turza N, Saunier B, Steinkasserer A, Becker S, Bates P, Hofmann H, Pöhlmann S. DC-SIGN and DC-SIGNR Interact with the Glycoprotein of Marburg Virus and the S Protein of Severe Acute Respiratory Syndrome Coronavirus. J Virol. 2004;78:12090–12095. doi: 10.1128/JVI.78.21.12090-12095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters PS. The molecular biology of coronaviruses. Adv Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol. 2010;84:12658–12664. doi: 10.1128/JVI.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride CE, Li J, Machamer CE. The cytoplasmic tail of the severe acute respiratory syndrome coronavirus spike protein contains a novel endoplasmic reticulum retrieval signal that binds COPI and promotes interaction with membrane protein. J Virol. 2007;81:2418–2428. doi: 10.1128/JVI.02146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride CE, Machamer CE. A single tyrosine in the severe acute respiratory syndrome coronavirus membrane protein cytoplasmic tail is important for efficient interaction with spike protein. J Virol. 2010;84:1891–1901. doi: 10.1128/JVI.02458-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Dorfman T, Li W, Wong SK, Li Y, Kuhn JH, Coderre J, Vasilieva N, Han Z, Greenough TC, Farzan M, Choe H. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin-converting enzyme 2. J Virol. 2004;78:10628–10635. doi: 10.1128/JVI.78.19.10628-10635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossel EC, Wang J, Jeffers S, Edeen KE, Wang S, Cosgrove GP, Funk CJ, Manzer R, Miura TA, Pearson LD, Holmes KV, Mason RJ. SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology. 2008;372:127–135. doi: 10.1016/j.virol.2007.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash TC, Buchmeier MJ. Entry of mouse hepatitis virus into cells by endosomal and nonendosomal pathways. Virology. 1997;233:1–8. doi: 10.1006/viro.1997.8609. [DOI] [PubMed] [Google Scholar]
- Nie Y, Wang G, Shi X, Zhang H, Qiu Y, He Z, Wang W, Lian G, Yin X, Du L, Ren L, Wang J, He X, Li T, Deng H, Ding M. Neutralizing antibodies in patients with severe acute respiratory syndrome-associated coronavirus infection. J Infect Dis. 2004a;190:1119–1126. doi: 10.1086/423286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y, Wang P, Shi X, Wang G, Chen J, Zheng A, Wang W, Wang Z, Qu X, Luo M, Tan L, Song X, Yin X, Chen J, Ding M, Deng H. Highly infectious SARS-CoV pseudotyped virus reveals the cell tropism and its correlation with receptor expression. Biochem Biophys Res Commun. 2004b;321:994–1000. doi: 10.1016/j.bbrc.2004.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris JS, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med. 2004;10:S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Hingley ST, Simmons G, Yu C, Das SJ, Bates P, Weiss SR. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J Virol. 2006;80:5768–5776. doi: 10.1128/JVI.00442-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. 2011;85:873–882. doi: 10.1128/JVI.02062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G, Reeves JD, Grogan CC, Vandenberghe LH, Baribaud F, Whitbeck JC, Burke E, Buchmeier MJ, Soilleux EJ, Riley JL, Doms RW, Bates P, Pöhlmann S. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology. 2003;305:115–123. doi: 10.1006/viro.2002.1730. [DOI] [PubMed] [Google Scholar]
- Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci U S A. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamminger T, Gstaiger M, Weinzierl K, Lorz K, Winkler M, Schaffner W. Open reading frame UL26 of human cytomegalovirus encodes a novel tegument protein that contains a strong transcriptional activation domain. J Virol. 2002;76:4836–4847. doi: 10.1128/JVI.76.10.4836-4847.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To KF, Tong JH, Chan PK, Au FW, Chim SS, Chan KC, Cheung JL, Liu EY, Tse GM, Lo AW, Lo YM, Ng HK. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: an in-situ hybridization study of fatal cases. J Pathol. 2004;202:157–163. doi: 10.1002/path.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss D, Pfefferle S, Drosten C, Stevermann L, Traggiai E, Lanzavecchia A, Becker S. Studies on membrane topology, N-glycosylation and functionality of SARS-CoV membrane protein. Virol J. 2009;6:79. doi: 10.1186/1743-422X-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Chen J, Zheng A, Nie Y, Shi X, Wang W, Wang G, Luo M, Liu H, Tan L, Song X, Wang Z, Yin X, Qu X, Wang X, Qing T, Ding M, Deng H. Expression cloning of functional receptor used by SARS coronavirus. Biochem Biophys Res Commun. 2004;315:439–444. doi: 10.1016/j.bbrc.2004.01.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Matsuyama S, Shirato K, Maejima M, Fukushi S, Morikawa S, Taguchi F. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved s protein as revealed by pseudotype virus bearing cleaved s protein. J Virol. 2008;82:11985–11991. doi: 10.1128/JVI.01412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XD, Shang B, Yang RF, Yu H, Ma ZH, Shen X, Ji YY, Lin Y, Wu YD, Lin GM, Tian L, Gan XQ, Yang S, Jiang WH, Dai EH, Wang XY, Jiang HL, Xie YH, Zhu XL, Pei G, Li L, Wu JR, Sun B. The spike protein of severe acute respiratory syndrome (SARS) is cleaved in virus infected Vero-E6 cells. Cell Res. 2004;14:400–406. doi: 10.1038/sj.cr.7290240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem Biophys Res Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZY, Huang Y, Ganesh L, Leung K, Kong WP, Schwartz O, Subbarao K, Nabel GJ. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J Virol. 2004;78:5642–5650. doi: 10.1128/JVI.78.11.5642-5650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao YX, Ren J, Heinen P, Zambon M, Jones IM. Cleavage and serum reactivity of the severe acute respiratory syndrome coronavirus spike protein. J Infect Dis. 2004;190:91–98. doi: 10.1086/421280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Surface expression of SARS-S variant T760R. 293T cells transiently transfected with SARS-S (black line) or T760R (dark grey) were stained with a polyclonal anti-SARS-CoV antiserum and a FITC-labeled secondary antibody. Cells transfected with pcDNA3 served as control (black filled histogram).
Protease inhibitors used for blockade of cell-cell and virus-cell fusion were employed at non-cytotoxic concentrations. (A) The reporter plasmids pGAL4-VP16 and pGal5-luc (which were used for the cell-cell fusion assay shown in FIG. 9B) were transfected into 293T cells, the transfected cells were treated with PBS or the indicated protease inhibitors for 48 h and luciferase-activities in cell lysates were determined. The results of a representative experiment performed in triplicates are shown, error bars indicate SD. (B) 293T cells were treated with PBS, leupeptin, the only protease inhibitor able to block SARS-S-driven cell-cell fusion in FIG. 9B, or E64c and ATP levels in the cultures were determined employing the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). The results of a representative experiment performed in triplicates are shown, error bars indicate SD.