

Abstract
Activity-dependent modifications of excitatory synapses contribute to synaptic maturation and plasticity, and are critical for learning and memory. Consequently, impairments in synapse formation or synaptic transmission are thought to be responsible for several types of mental disability. BRAG1 is a guanine nucleotide exchange factor (GEF) for the small GTP-binding protein Arf6 that localizes to the postsynaptic density of excitatory synapses. Mutations in BRAG1 have been identified in families with X-linked intellectual disability (XLID). These mutations mapped to either the catalytic domain or an IQ-like motif, however the pathophysiological basis of these mutations remains unknown. Here, we show that the BRAG1 IQ motif binds apo-calmodulin (CaM), and that calcium-induced CaM release triggers a reversible conformational change in human BRAG1. We demonstrate that BRAG1 activity, stimulated by activation of NMDA-sensitive glutamate receptors (-Rs), depresses AMPA-R-mediated transmission via JNK-mediated synaptic removal of GluA1-containing AMPA-Rs in rat hippocampal neurons. Importantly, a BRAG1 mutant that fails to activate Arf6 also fails to depress AMPA-R signaling, indicating that Arf6 activity is necessary for this process. Conversely, a mutation in the BRAG1 IQ-like motif that impairs CaM binding results in hyperactivation of Arf6 signaling and constitutive depression of AMPA transmission. Our findings reveal a role for BRAG1 in response to neuronal activity with possible clinical relevance to nonsyndromic X-linked intellectual disability.
Introduction
The majority of fast excitatory synaptic transmission in the central nervous system is mediated by AMPA- and NMDA-type ionotropic glutamate receptors (Traynelis et al., 2010). A key factor underlying the strength of individual excitatory synapses is the number of AMPA receptors (-Rs) at synapses, which is tightly regulated by AMPA-R trafficking. This regulated trafficking, largely mediated by NMDA-R signaling, plays a key role in both synaptic transmission and plasticity (Kerchner and Nicoll, 2008; Kessels and Malinow, 2009; Anggono and Huganir, 2012). Both hypo- and hyper-regulation of synaptic AMPA-R trafficking reduce the capacity of synaptic plasticity (McCormack et al., 2006), and are thought to underlie numerous cognitive disorders, including mental retardation (Costa and Silva, 2003; Thomas and Huganir, 2004; Stornetta and Zhu, 2011).
The ADP-ribosylation factor (Arf) proteins are a family of six small, ubiquitously expressed GTP-binding proteins (Donaldson and Jackson, 2011). Of these, Arf6 localizes primarily to the plasma membrane/endosomal system, and is best known as a regulator of endocytic trafficking and actin cytoskeleton dynamics (D’Souza-Schorey and Chavrier, 2006; Myers and Casanova, 2008). In hippocampal neurons, Arf6 has been shown to regulate dendritic arborization (Hernandez-Deviez et al., 2002), axonal outgrowth (Hernandez-Deviez et al., 2004), dendritic spine formation (Miyazaki et al., 2005; Choi et al., 2006), and the assembly of clathrin/AP2 complexes at synaptic membranes (Krauss et al., 2003). The human genome contains 15 Arf-GEFs, which catalyze the exchange of GDP for GTP via the evolutionarily conserved catalytic Sec7 domain (Casanova, 2007). The Brefeldin A-Resistant Arf-GEFs (BRAGs) comprise a subfamily of three proteins that are abundantly expressed within the postsynaptic density (Jordan et al., 2004; Peng et al., 2004; Dosemeci et al., 2007).
BRAG2/IQSec1 has recently been shown to interact directly with the cytoplasmic domain of the AMPA-R subunit GluA2, and to regulate its synaptic activity-dependent endocytosis (Scholz et al., 2010). In contrast, BRAG1/IQSec2 is reported to interact with NMDA-Rs, but not AMPA-Rs, through an indirect mechanism involving the synaptic scaffolding protein PSD-95 (Sakagami et al., 2008). Recently, Shoubridge et al. identified four non-synonymous single nucleotide polymorphisms (SNPs) in BRAG1 from families with nonsyndromic X-linked intellectual disabilility (XLID) (Shoubridge et al., 2010). Three of these SNPs led to nonconserved amino acid substitutions within the catalytic Sec7 domain, while the fourth was a nonconserved substitution within an IQ motif (Shoubridge et al., 2010).
Here we report that BRAG1 has an integral role in synaptic transmission. We show that expression of exogenous BRAG1 in CA1 hippocampal neurons results in depression of AMPA-R-mediated synaptic transmission, in a manner dependent upon upstream NMDA-R activation. This depression is also dependent upon BRAG1 catalytic activity, indicating that it requires Arf6 activation. We show that BRAG1 binds calmodulin, and that a mutation in the IQ motif that prevents CaM binding results in constitutive (NMDA-R-independent) depression of AMPA-R-mediated transmission. Furthermore, BRAG1 appears to selectively control the trafficking of GluA1-containing AMPA-Rs by stimulating JNK signaling. Together, these results indicate that BRAG1 acts as a calmodulin-responsive switch to control AMPA-R signaling downstream of NMDA-R activation.
Materials and Methods
Reagents and Antibodies
The reagents used in this study include ionomycin (Invitrogen I-24222), NMDA (Sigma M3262), APV (Sigma A5282), Bapta-AM (Invitrogen B-1205), and calmodulin-sepharose 4B (GE Lifesciences). Primary antibodies used were 9E10 α-Myc, 16B12 α-HA (Covance), α-GFP (Molecular Probes), and α-PSD-95 (Thermo Scientific). BRAG1 rabbit antiserum was raised against a peptide, corresponding to amino acids 258-275 (CAVDSPGSQPPYRLSQLP), coupled to keyhole limpet hemocyanin (KLH) as antigen.
DNA Constructs
Human BRAG1 (KIAA0522) cDNA was obtained from the Kasuza DNA Research Institute. The coding sequence of BRAG1 was subcloned into pCMV3A-Myc using HindIII/XhoI. The BRAG1-E849K and BRAG1-IQ mutants were made by site-directed mutagenesis. The BRAG1-ΔN mutant was made by digesting BRAG1-WT with EcoRV/NruI which creates an in-frame deletion of the N-terminal 213 amino acids. To create Cherry-tagged versions, BRAG1 was digested out of pCMV3A-Myc using HindIII/XhoI, and ligated into mCherry-C2 (Clontech) using HindIII/SalI. The BRAG1-mCherry fusions were digested out of the mCherry-C2 plasmid using NheI/XbaI and ligated into pSinRep5 using XbaI to make Sindbis virus constructs.
Cell Culture
Hela cells were cultured and transfected as described previously (Santy and Casanova, 2001). Dissociated hippocampal neuron cultures were prepared and transfected as described in (Zhang et al., 2003). For ionomycin stimulation, HeLa cells were switched 4–6 hours post-transfection into serum-free DMEM for 16–20 additional hours. Cells were then moved into fresh phenol red-free, serum-free DMEM (plus 25 mM HEPES for timelapse imaging) containing either DMSO vehicle or 5 μM ionomycin for 3 minutes.
Pulldown Assays
Arf6-GTP levels were measured using a GST-GGA3 pulldown assay as described previously (Santy and Casanova, 2001). Results are reported as mean ± s.e.m. and statistical differences were determined using the Wilcoxon matched pairs test. For CaM binding, HeLa cells transiently expressing myc-tagged BRAG1 constructs were lysed on ice in buffer A (150 mM NaCl, 1% Triton X-100, 50 mM Tris, protease inhibitors; pH 7.4) containing either 1 mM CaCl2 or 1 mM EGTA, and incubated with CaM-sepharose for 2 hours. Densitometry was performed using Image J Software (NIH) to quantify protein expression levels.
Immunofluorescence Microscopy
HeLa cells were grown on glass coverslips, fixed and processed for microscopy as described in (Santy and Casanova, 2001). Fixed images were acquired using a 60x objective on a Nikon Eclipse E800 microscope and a Q-Imaging Retiga CCD camera. Live cells were imaged in extracellular solution (150 mM NaCl, 10 mM HEPES, 5 mM KCl, 30 mM glucose; pH 7.4) with or without 2 mM CaCl2 using a 60x objective on a DeltaVision deconvolution microscope (Applied Precision). For quantitation of BRAG1 condensation, NIS-Elements software (Nikon) was used to set a defined background threshold and automatically count puncta up to 2 μm2.
Hippocampal Slice Cultures
Cultured rat hippocampal slices were prepared from postnatal 6–7 day old rats of either sex, infected with Sindbis virus after 7–14 days in vitro to deliver recombinant proteins into CA1 pyramidal neurons as described previously (Qin et al., 2005; McCormack et al., 2006). Hippocampal extracts were prepared by homogenizing hippocampal CA1 regions isolated from cultured slices (Zhu et al., 2000; Zhu et al., 2002), Viral expression efficacy of recombinant proteins in these experiments was high (>95% of CA1 neurons). Homogenizing solution contained (in mM or percentage): HEPES 10, NaCl 150, EDTA 10, EGTA 4, PMSF 0.2, NaPPi 0.1, NaF 0.5, Na3VO4 1, and Triton 1%. Membranes were blotted with anti-phospho-JNK (1:2000; Cell Signaling Technology, Beverly, MA) and anti-phospho-p38 MAPK antibody (1:2000; Cell Signaling), stripped and reblotted with anti-JNK (1:2000; Cell Signaling) and anti-p38 MAPK antibody (1:1000; Cell Signaling). Western blots were quantified by chemiluminescence and densitometric scanning of the films under linear exposure conditions. The dendritic and spine expression of mCherry-BRAG1 was imaged with a custom-made two-photon laser scanning microscope (Zhu et al., 2000; Kolleker et al., 2003).
Electrophysiology
Simultaneous whole-cell recordings were obtained from nearby infected and non-infected neuron pairs, under visual guidance using fluorescence and transmitted light illumination with two Axopatch-200B amplifiers (Axon Instruments, Foster City, CA) as previously described (Qin et al., 2005; McCormack et al., 2006; Zhu, 2009). Cultured rat organotypic slices exhibit relatively high spontaneous activity comparable to intact brains. Thus, high calcium and magnesium bath solution (29±1.5°C), containing (in mM): NaCl 119, KCl 2.5, CaCl2 4, MgCl2 4, NaHCO3 26, NaH2PO4 1, glucose 11, picrotoxin 0.1, and 2-chloroadenosine 0.002, at pH 7.4, was used to prevent epileptic activity after blocking inhibition. The bath solutions were gassed with 5% CO2/95% O2. Patch recording pipettes (3–6 MΩ) contained (in mM): cesium methanesulfonate 115, CsCl 20, HEPES 10, MgCl2 2.5, Na2ATP 4, Na3GTP 0.4, sodium phosphocreatine 10, EGTA 0.6, and spermine 0.1, at pH 7.25. Synaptic responses were evoked by bipolar electrodes with single voltage pulses (200 μs, up to 20 V) placed in hippocampal s. radiatum ~300 μm away from the recorded hippocampal CA1 pyramidal neurons. To minimize the effect from AMPA responses, the peak NMDA responses at +40 mV were measured after digital subtraction of estimated AMPA responses at +40 mV. Results are reported as mean ± s.e.m. and statistical differences were determined using Wilcoxon test.
RESULTS
BRAG1 interaction with calmodulin
IQ motifs are best known as binding domains for calmodulin. Although BRAG1, BRAG2 and BRAG3 each contain an IQ-like motif N-terminal to the catalytic domain (Figure 1A), it has not yet been demonstrated that any of the BRAGs do indeed bind CaM. Inspection of this motif (residues 354-363) indicated that it fits the consensus sequence for calcium-independent CaM binding (Figure 1B) (Rhoads and Friedberg, 1997). To determine if this is the case, lysates of Hela cells expressing Myc-tagged BRAG1 were incubated with CaM-sepharose in either the presence or absence of Ca2+ (2 mM CaCl2 or 2 mM EGTA, respectively). As shown in Fig. 1C, BRAG1 was robustly precipitated by CaM-sepharose, but not sepharose alone. Moreover, this interaction was strengthened in the presence of EGTA, indicating that BRAG1 preferentially binds to Ca2+-free CaM. Substitution of three conserved residues within the consensus IQ motif (I354QTAFR to AATAFA, hereafter referred to as BRAG1-IQ) completely abrogated CaM binding (Figure 1C). However, mutation of a conserved glutamate residue within the Sec7 domain essential for catalytic activity (E849K, hereafter referred to as BRAG1-EK) (Shoubridge et al., 2010), had no effect on the ability of BRAG1 to bind CaM, indicating that catalytic activity does not impact calmodulin binding (Figure 1C). Deletion of an N-terminal coiled coil domain (BRAG1-ΔN) does appear to result in more efficient CaM binding than BRAG1-WT. This may be a result of the enhanced solubility of BRAG1-ΔN (see below), or it could suggest that the coiled-coil motif regulates accessibility of the IQ motif to CaM.
Figure 1. BRAG1 binds Ca2+-free calmodulin.

(A) Domain organization of the three BRAG family members. (B) Consensus sequence for IQ motifs, compared to the IQ-like motif in wild-type BRAG1. The bottom sequence shows mutations introduced to generate BRAG1-IQ. (C) Calmodulin binding to wild-type and mutant BRAG1. Hela cells were transiently transfected with myc-tagged BRAG1-WT, BRAG1-EK, BRAG1-IQ, or BRAG1-ΔN and lysed in the presence of either 2 mM EGTA or 2 mM CaCl2. Cell lysates were then incubated with CaM-sepharose, and BRAG1 binding was analyzed by anti-myc immunoblotting. Binding was quantified by densitometry. The signal for BRAG1-WT was normalized to a value of 1 and relative binding values are shown in red. The blot shown is representative of 3 independent experiments.
BRAG1 localization to the postsynaptic density
Previous studies have revealed the localization of BRAG1 specifically at the postsynaptic membrane of excitatory synapses using both immunofluorescence and electron microscopy (Murphy et al., 2006; Sakagami et al., 2008). To confirm this localization, we stained dissociated rat hippocampal neurons at 21 days in vitro (DIV) with rabbit antiserum raised against a peptide corresponding to amino acids 258-275 of BRAG1. As expected from previous studies, we detected endogenous BRAG1 at discrete clusters along dendrites that clearly co-label with the excitatory postsynaptic marker, PSD-95 (Figure 2A). We next sought to confirm that exogenously expressed mCherry-tagged BRAG1 fusion proteins localized to excitatory synapses, similar to endogenous BRAG1. Therefore, we transfected dissociated rat hippocampal neurons at DIV 6 with wild-type BRAG1 fused to mCherry at its N-terminus. Neurons were fixed at DIV 19 and counterstained for PSD-95. In contrast to soluble mCherry, which is diffusely distributed and fails to localize to any particular compartment (Figure 2B), mCherry-BRAG1 was found in prominent puncta distributed along the length of dendrites, where it clearly colocalized with PSD-95 (Figure 2C).
Figure 2. BRAG1 mutants localize to excitatory synapses in hippocampal neurons.

(A) Dissociated rat hippocampal neurons were fixed at DIV 21, and co-immunostained with rabbit polyclonal anti-BRAG1 and mouse anti-PSD-95 antibodies. (B–F) Hippocampal neurons were transfected (DIV 6) with the indicated mCherry-tagged constructs and fixed (DIV 21) for epifluorescence imaging. Cells expressing; mCherry (B), mCherry-BRAG1-WT (C), mCherry-BRAG1-EK (D), mCherry-BRAG1-IQ (E), and mCherry-BRAG1-ΔN (F) were co-stained for PSD-95 antibody to detect excitatory synapses. Scale bars = 10 μm.
BRAG1-EK colocalized with PSD-95 to the same extent as BRAG1-WT, indicating that catalytic activity does not direct or alter BRAG1 localization. We also examined whether the IQ motif of BRAG1 was required for its localization to the PSD. Although the majority of cherry-tagged BRAG1-IQ was localized to the PSD (Figure 2E), we detected the presence of puncta within the shaft of the dendrite that were not observed in cells expressing either BRAG1-WT or BRAG1-EK. The BRAG1-ΔN mutant, which lacks the N-terminal coiled-coil motif, also colocalizes with PSD-95 at synapses. However, we also observed a significant fraction of BRAG1-ΔN diffusely distributed throughout the dendritic shaft (Figure 2F). In summary, these results suggest that neither catalytic activity nor an intact IQ-motif or coiled-coil domain is necessary for the localization of BRAG1 to the PSD.
The IQ motif regulates a calcium-dependent conformational switch in BRAG1
The calcium-dependent release of calmodulin from BRAG1 suggests that changes in intracellular calcium levels may regulate the BRAG1-CaM interaction, and that this might modulate BRAG1 conformation or activity. To test this idea, we examined the effects of calcium influx on mCherry-BRAG1 distribution in live Hela cells stimulated with the calcium ionophore, ionomycin. As shown in Figure 3A, BRAG1 is mostly diffuse at steady state. However, within 30s of ionomycin treatment, we observed the formation of discrete BRAG1 puncta scattered throughout the cell (Figure 3A). These appear to be aggregates of protein, as they do not contain endosomal or other intracellular membranes (data not shown). In contrast, BRAG1-IQ exhibited a punctate distribution even in the absence of ionomycin, and did not undergo a change in its localization upon Ca2+ influx (Figure 3B). These observations suggest that the Ca2+-induced release of CaM triggers a conformational change in BRAG1, manifested in Hela cells as condensation into cytoplasmic puncta.
Figure 3. Ca2+ influx triggers a reversible conformational change in BRAG1.

(A–B) Formation of cytoplasmic puncta in response to calcium influx. Hela cells expressing mCherry-BRAG1-WT (A) or mCherry-BRAG1-IQ (B) were stimulated with 5 μM ionomycin for 3 minutes. Single frames from time-lapse movies beginning at 30 sec before ionomycin treatment (−30) are shown. Note that BRAG1-IQ is punctate in the absence of ionomycin and does not change its distribution after ionomycin addition. (C) Calcium-induced condensation of BRAG1 is reversible. Hela cells were either left untreated (left panel), were treated with 5 μM ionomycin for 3 minutes (center), or were treated with 5 μM ionomycin for 3 minutes followed by incubation with 5mM BAPTA-AM for 10 minutes (right panel). (D) BRAG1-ΔN does not form puncta in response to ionomycin stimulation. Cells expressing mCherry-BRAG1-ΔN were treated with ionomycin for 3 min and imaged as described above. (E) Quantification of calcium-induced puncta formation. Cells expressing BRAG1-WT, BRAG1-IQ or BRAG1-ΔN were treated with either vehicle (DMSO) or ionomycin for 3 min, then fixed and imaged. Puncta were quantified as described in Methods from at least 18 cells from 3 independent experiments. Mean number of BRAG1 puncta per 100 μm2 pre- and post-ionomycin stimulation (WT-pre: 4.6±0.9 puncta, n=19; WT-post: 66.6±13.5 puncta; n=19; p<0.001; IQ-pre: 67.5±7.4 puncta, n=17; IQ-post: 76.6±19.5 puncta; n=17; p=0.25; ΔN-pre: 0.0±0. puncta, n=18; ΔN-post: 13.8±3.8 puncta; n=19; p<0.005)). Asterisk indicates p ≤ 0.005 (Student’s t-test). (F) The N-terminus of BRAG1 is necessary for self-association. Myc-BRAG1-WT or myc-BRAG1-ΔN were co-expressed in Hela cells with GFP-BRAG1 WT. Cell lysates were then immunoprecipitated with anti-GFP, and the presence of bound myc-BRAG1 detected by immunoblotting with anti-myc. Scale bars = 10 μm.
This conformational change is completely reversible, as treatment with the cell permeable calcium chelator BAPTA-AM resulted in nearly complete dissolution of the ionomycin-induced puncta. This indicates that the redistribution of BRAG1 upon calcium influx is not simply due to protein degradation or denaturation, and likely involves a regulated change in BRAG1 conformation. Quantitation of this phenomenon indicated an approximately 15-fold increase in the number of BRAG1-WT puncta after ionomycin treatment, which was statistically indistinguishable from BRAG1-IQ in the absence of ionomycin (Figure 3D).
The N-terminus of BRAG1 mediates calcium-dependent self-association
Because coiled-coil domains commonly mediate homo-oligomerization or protein-protein interactions (Burkhard et al., 2001), we speculated that the N-terminal BRAG1 coiled-coil domain (residues 23–74) plays a role in its calcium-induced self-association. Deletion of this domain (BRAG1-ΔN) did not affect the steady-state distribution of BRAG1 in Hela cells (Figure 3E). However, unlike wild-type BRAG1, BRAG1-ΔN remained diffusely cytosolic upon addition of ionomycin (Figure 3E). This observation indicates that Ca2+-induced self-association of wild-type BRAG1 is dependent upon the N-terminal coiled-coil domain.
To support this hypothesis, we tested the ability of BRAG1 to oligomerize. For this purpose, GFP-tagged BRAG1-WT was expressed in Hela cells along with either myc-tagged BRAG1-WT or myc-BRAG1-ΔN. When GFP-BRAG1-WT was immunoprecipitated with anti-GFP antibody, we found that myc-BRAG1-WT co-precipitated efficiently while myc-BRAG1-ΔN did not (Figure 3F). This observation indicates that BRAG1 can oligomerize via its N-terminal coiled-coil domain, and suggests that regulated oligomerization, induced by CaM release, may have an important role in BRAG1 function in the synapse.
An influx of extracellular calcium is known to occur upon activation of NMDA-Rs. To determine if BRAG1 responds to physiological levels of calcium in the neuronal context, we expressed mCherry-tagged BRAG1-WT in cultured hippocampal neurons and followed its localization after NMDA stimulation using live cell imaging (Figure 4A). Prior to stimulation, BRAG1-WT was stably localized to the postsynaptic density. However, after the addition of 30 uM NMDA, small BRAG1 puncta appeared within spines and within the dendritic shaft, in addition to its normal synaptic localization. These smaller puncta were reminiscent of those seen in Hela cells after ionomycin stimulation, and are consistent with the idea of calcium-induced self-association of BRAG1. We also examined the effects of NMDA stimulation on the distribution of BRAG1-IQ and BRAG1-ΔN in hippocampal neurons (Figure 4B, C). Similar to our findings in Hela cells treated with ionomycin, we saw no detectable changes in the distribution of either mutant after NMDA stimulation (Figure 4D). This suggested that the NMDA-induced condensation of BRAG1 in hippocampal neurons requires both the IQ and the coiled-coil motifs.
Figure 4. Stimulation of NMDA-Rs in hippocampal neurons triggers a conformational change in BRAG1.

(A–C) Frames from time-lapse movies of DIV 12 hippocampal neurons expressing mCherry-tagged BRAG1 constructs and treated with 30 μM NMDA for the times shown. (A) BRAG1-WT, (B) BRAG1-IQ, (C) BRAG1-ΔN. Arrowheads indicate dendritic spines where single BRAG1 foci resolve into distinct puncta. Arrows indicate formation of new puncta in the dendritic shaft (D) Quantitation of puncta formation. Mean number of puncta per 100 μm2 pre- and post-NMDA stimulation (WT-pre: 17.5±1.4 puncta; WT-post: 46.1±6.6 puncta; n=3; p<0.05; IQ-pre: 20.0±3.4 puncta; IQ-post: 20.5±2.4 puncta; n=4; p=0.5; ΔN-pre: 6.0±4.1 puncta; ΔN-post: 7.0±3.9 puncta; n=3; p=0.5). Asterisk indicates p ≤ 0.05 (Wilcoxon). Scale bars = 10 μm.
Calmodulin binding is not required for BRAG1 catalytic activity
To test whether the IQ domain or the N terminal coiled coil domain regulates BRAG1 Arf-GEF activity, we measured their ability to activate Arf6 in Hela cells using a previously described GST-GGA3 pulldown assay (Santy and Casanova, 2001) to specifically precipitate GTP-bound Arf6. Coexpression of BRAG1-WT with Arf6 in Hela cells enhanced Arf6 activation 4-fold relative to cells expressing Arf6 alone (Figure 5A, B). As expected, the catalytically inactive mutant BRAG1-E849K failed to activate Arf6 above basal levels. Surprisingly, both BRAG1-IQ and BRAG1-ΔN mutants significantly stimulated Arf6 activity, although the BRAG1-ΔN-mediated activity was slightly lower than BRAG1-WT.
Figure 5. The IQ motif and N-terminus are dispensable for BRAG1 catalytic activity in heterologous cells.

(A) Pulldown assay for Arf6 activity. Active GTP-bound Arf6 was precipitated with GST-GGA3 from lysates of HeLa cell co-expressing Arf6-HA and the indicated myc-tagged BRAG1 constructs. (B) Quantitation of Arf6 activation: Ctrl: 100.0±55.6%; WT: 362.0±99.4%; n=3; p<0.05; EK: 117.3±78.9%; n=3; p=0.5; IQ: 376.8±86.0%; n=3; p<0.05; ΔN: 340.6±131.3%; n=3; p<0.05. The relative values and standard errors were normalized to average amounts of Arf6-GTP from control cells expressing Arf6-HA alone. Asterisk indicates p<0.05 (Wilcoxon test).
Activation of BRAG1 depresses AMPA-R-mediated transmission in CA1 neurons
To further examine the synaptic functions of BRAG1, we used recombinant Sindbis virus to acutely over-express mCherry-BRAG1 in CA1 pyramidal neurons of rat hippocampal cultured slices. In expressing neurons, mCherry-BRAG1 was diffusely distributed and infiltrated in dendritic spines, the sites of excitatory synapses (Figure 6A). Electrophysiological recordings were obtained simultaneously from nearby expressing and non-expressing neurons. We found that expression of mCherry-BRAG1 had no effects on basic membrane properties, including resting membrane potentials, inputs resistance and membrane time constants (Fig. 6B–C). We then examined excitatory postsynaptic currents (EPSCs) in expressing neurons and nearby control non-expressing neurons by stimulating the afferent fibers. Neurons expressing wild type BRAG1 exhibited depressed (by ~30%) AMPA-R-mediated responses compared to nearby non-expressing controls (Figure 7AB), suggesting that activating BRAG1 depresses transmission. Interestingly, expression of BRAG1-ΔN did not suppress AMPA-R activity, but instead potentiated it (Figure 7A–B), suggesting a possible dominant-negative effect. No significant difference was observed in NMDA-R-mediated responses between BRAG1-expressing and non-expressing neurons (Figure 7A–B), suggesting a postsynaptic mechanism.
Figure 6. Expression of BRAG1 does not alter the basic membrane properties of CA1 neurons.

(A) Two-photon fluorescence imaging indicates that mCherry-BRAG1 is expressed in the dendrites and spines (insert) of CA1 pyramidal neurons. (B) Evoked responses to step depolarizing and hyperpolarizing pulses recorded from neighboring non-expressing (Ctrl) and BRAG1 expressing cells. (C) Resting membrane potentials (Ctrl: −58.1±1.1 mV; BRAG1: −57.2±0.9 mV; n=20; p=0.25), input resistances (Ctrl: 189.8±8.2 MΩ; BRAG1: 190.2±13.2 MΩ; n=20; p=0.50) and time constants (Ctrl: 27.3±1.7 ms; BRAG1: 28.5±2.1 ms; n=20; p=0.97) in mCherry-BRAG1-expressing cells are plotted against those obtained from control non-expressing cells. Blue dots indicate the average points.
Figure 7. BRAG1-WT and BRAG1 mutants differentially affect synaptic transmission.

(A) Evoked AMPA-R- (−60 mV) and NMDA-R- (+40 mV) mediated responses recorded from neighboring non-expressing (Ctrl) and mCherry-tagged BRAG1-WT or BRAG1-ΔN expressing neurons cultured in normal media or media containing 100 μM DL-APV. (B) Values for AMPA responses in neurons expressing BRAG1-WT and BRAG1-ΔN cultured in normal media (Ctrl: −31.6±2.5 pA; WT: −22.9±2.4 pA; n=38; p<0.05; Ctrl: −14.2±2.1 pA; ΔN: −20.1±3.2 pA; n=16; p<0.05), in media containing high Mg2+ (Ctrl: −26.8±2.6 pA; WT: −28.8±3.1 pA; n=22; p=0.94; Ctrl: −20.3±1.7 pA; ΔN: −21.4±2.3 pA; n=23; p=0.93), or in media containing DL-APV (Ctrl: −31.7±2.3 pA; WT: −30.1±2.4 pA; n=19; p=0.51; Ctrl: −22.3±3.8 pA; ΔN: −23.8±2.5 pA; n=19; p=0.69), and for NMDA responses in neurons expressing Brag1-WT and BRAG1-ΔN cultured in normal media (Ctrl: 79.3±6.5 pA; WT: 75.9±6.8 pA; n=38; p=0.60; Ctrl: 46.6±9.7 pA; ΔN: 41.3±8.5 pA; n=16; p=0.33), in media containing high Mg2+ (Ctrl: 66.4±11.6 pA; WT: 61.6±9.4 pA; n=22; p=0.78; Ctrl: 53.8±5.3 pA; ΔN: 54.3±7.4 pA; n=23; p=0.72), or in media containing DL-APV (Ctrl: 59.2±7.3 pA; WT: 59.3±6.7 pA; n=19; p=0.94; Ctrl: 47.4±6.8 pA; ΔN: 44.4±5.5 pA; n=19; p=0.57) related to control non-expressing neurons. (C) Evoked AMPA-R- (−60 mV) and NMDA-R- (+40 mV) mediated responses recorded from neighboring non-expressing (Ctrl) with either mCherry-tagged BRAG1-EK or BRAG1-IQ expressing neurons cultured in normal media or media containing 100 μM DL-APV. (D) Values for AMPA responses in neurons expressing BRAG1-EK and BRAG1-IQ cultured in normal media (Ctrl: -24.1±2.8 pA; EK: −26.5±2.5 pA; n=21; p=0.52; Ctrl: −25.4±2.6 pA; IQ: −19.4±1.9 pA; n=25; p<0.05), in neurons expressing BRAG1-IQ cultured in media containing high Mg2+ (Ctrl: −23.0±2.3 pA; IQ: −15.4±1.3 pA; n=25; p<0.005) or DL-APV (Ctrl: −26.7±2.8 pA; IQ: −19.9±1.7 pA; n=24; p<0.01), and for NMDA responses in neurons expressing BRAG1-EK or BRAG1-IQ cultured in normal media (Ctrl: 77.1±9.1 pA; EK: 74.1±8.0 pA; n=21; p=0.54; Ctrl: 52.5±5.2 pA; IQ: 50.8±6.6 pA; n=25; p=0.14), in neurons expressing BRAG1-IQ cultured in media containing high Mg2+ (Ctrl: 57.6±5.7 pA; IQ: 52.9±5.7 pA; n=25; p=0.20) or DL-APV (Ctrl: 68.9±6.8 pA; IQ: 64.4±5.8 pA; n=24; p=0.67) related to control non-expressing neurons. The relative values and standard errors were normalized to average amounts of AMPA and NMDA responses from control CA1 cells. Asterisk indicates p<0.05 (Wilcoxon test).
To determine whether BRAG1 signaling is stimulated by synaptic and NMDA-R activity, we included 12 mM MgCl2, which depresses synaptic transmission (Zhu et al., 2000), or DL-APV, a pharmacological blocker of NMDA-Rs, in culture media during expression of BRAG1. Both high Mg2+ and APV completely blocked the effects of both BRAG1-WT and BRAG1-ΔN expression on AMPA synaptic transmission (Figure 7A–B). These results indicate that spontaneous synaptic activity activates NMDA-Rs that in turn activate BRAG1, producing a tonic depression of AMPA-R-mediated transmission.
To examine how mutations in the catalytic or IQ domains might affect synaptic transmission, we expressed mCherry-tagged BRAG1-EK or BRAG1-IQ in CA1 neurons. In contrast to wild-type BRAG1, which depressed AMPA responses, neurons expressing the catalytically inactive BRAG1-EK mutant responded similarly to controls, indicating that BRAG1 catalytic activity is necessary for the observed depression observed upon expression of the wild-type protein (Figure 7C–D). The IQ domain mutant reduced AMPA responses to a similar extent as the wild-type protein, consistent with its retention of catalytic activity (Figure 7C–D). However unlike BRAG1-WT, which is entirely dependent upon NMDA-R signaling, the depressive effect of BRAG1-IQ was not blocked by high Mg2+ or APV. This observation suggests that the inability to interact with CaM abrogates the requirement for NMDA-R activation, and renders this mutant constitutively active.
BRAG1-Arf6 stimulates JNK-mediated synaptic GluA1 removal in CA1 neurons
To determine how BRAG1 depresses synaptic transmission, we examined whether BRAG1 mutants affect endogenous Arf6 signaling in CA1 neurons in hippocampal cultured slices using the GST-GGA3 pull-down assay (Santy and Casanova, 2001). As shown in Fig. 8, CA1 cells expressing BRAG1-IQ displayed increased levels of active Arf6-GTP, whereas those expressing BRAG1-ΔN had decreased levels of active Arf6-GTP compared to control non-expressing CA1 cells, indicating that BRAG1-IQ stimulates and BRAG1-ΔN inhibits endogenous Arf6 activity in neurons.
Figure 8. BRAG1 regulates endogenous Arf6 signaling.
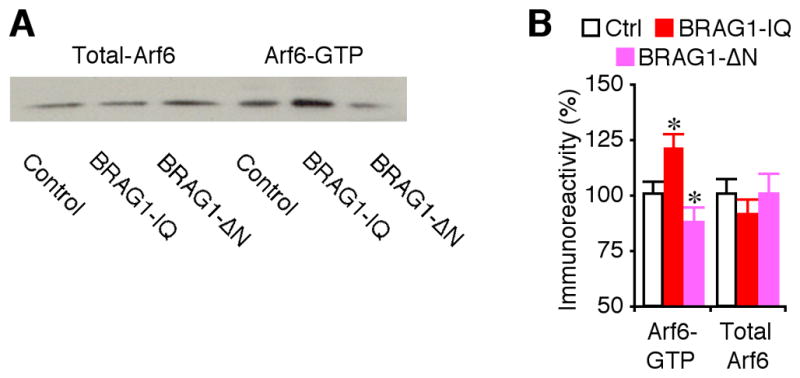
(A) Relative levels of GTP-bound Arf6 and total Arf6 in control hippocampal CA1 regions, and hippocampal CA1 regions expressing BRAG1-IQ or BRAG1-ΔN cultured in normal media. For each set of hippocampal CA1 cell lysate, 35 μg protein was used to precipitate and blot GTP-bound Arf6 and 7.5 μg protein was used to directly blot total Arf6. (B) Relative amounts of Arf6-GTP (Ctrl: 100.0±6.5%; IQ: 120.9±6.3%; n=22; p<0.005; ΔN: 87.1±7.7%; n=22; p<0.05) and total Arf6 (Ctrl: 100.0±7.1%; IQ: 90.7±7.5%; n=22; p=0.21; ΔN: 100.6±9.6%; n=22; p=0.86) in control hippocampal CA1 regions, and hippocampal CA1 regions expressing BRAG1-IQ or BRAG1-ΔN. The relative values and standard errors were normalized to average amounts of Arf6-GTP or total Arf6 from control untreated cells. Asterisk indicates p<0.05 (Wilcoxon test).
We then investigated whether BRAG1-Arf6 signals synaptic depression by stimulating the JNK and p38 MAPK signaling pathways, which depress transmission by stimulating synaptic removal of AMPA-Rs (Zhu et al., 2002; Zhu et al., 2005; Kielland et al., 2009). To test this idea, we included SP600125, an inhibitor of JNK (Bennett et al., 2001), or SB203580, which inhibits p38 MAPK (Lee et al., 1999), in culture media during expression of BRAG1-IQ and BRAG1-ΔN. SP600125, but not SB203580, completely blocked the depressive effect of BRAG1-IQ and the potentiative effect of BRAG1-ΔN in CA1 neurons (Fig. 9A–B), suggesting a selective involvement of JNK signaling. Consistent with this idea, Western blots showed that expression of BRAG1-IQ increased levels of phosphorylated JNK in CA1 cells, while expression of BRAG1-ΔN decreased JNK activation (Fig. 9C–D). Importantly neither construct affected the levels of p38 MAPK phosphorylation (Fig. 9CD). Expression of BRAG1-IQ or BRAG1-ΔN did not alter the levels of total JNK and p38 (Fig. 9C–D). Collectively, these results indicate that BRAG1-Arf6 signals synaptic depression via stimulating JNK signaling, but not p38 MAPK signaling.
Figure 9. BRAG1 signals synaptic depression via JNK.

(A) Evoked AMPA-R- (−60 mV) and NMDA-R- (+40 mV) mediated responses recorded from neighboring non-expressing (Ctrl) and mCherry-tagged BRAG1-IQ or BRAG1-ΔN expressing neurons cultured in media containing 5 μM SP600125 or 2 μM SB203580. (B) Values for AMPA responses in neurons expressing BRAG1-IQ and BRAG1-ΔN cultured in media containing SP600125 (Ctrl: −23.0±2.1 pA; IQ: −23.5±2.8 pA; n=26, p=0.81; Ctrl: −18.4±1.6 pA; ΔN: −19.7±2.0 pA; n=23, p=0.48), or SB203580 (Ctrl: −26.0±2.2 pA; IQ: −18.2±1.4 pA; n=28, p<0.005; Ctrl: −17.9±1.6 pA; ΔN: −25.5±1.9 pA; n=23, p<0.01), and for NMDA responses in neurons expressing BRAG1-IQ and BRAG1-ΔN cultured in media containing SP600125 (Ctrl: 55.4±4.7 pA; IQ: 55.4±6.0 pA; n=26, p=0.53; Ctrl: 60.7±10.6 pA; ΔN: 58.3±12.1 pA; n=23, p=0.38), or SB203580 (Ctrl: 51.3±6.7 pA; IQ: 54.0±6.2 pA; n=28, p=0.93; Ctrl: 60.6±6.3 pA; IQ: 56.4±7.7 pA; n=23, p=0.43) related to control nonexpressing neurons. (C) Western blots of phospho-p46/54 JNK or p38 MAPK in control hippocampal CA1 regions, hippocampal CA1 regions expressing BRAG1-IQ and hippocampal CA1 regions expressing BRAG1-ΔN. Each lane was loaded with the same amount of protein (45 μg). (D) Left, relative amounts of phospho-p46 (ctrl: 100.0±5.3%; IQ: 118.9±7.5%; n=12; p<0.005; ΔN: 89.6±5.8%; n=12; p<0.01) and phospho-p54 (ctrl: 100.0±11.0%; IQ: 117.4±11.6%; n=12; p<0.005; ΔN: 81.6±9.9%; n=12; p<0.005) JNK in hippocampal CA1 regions expressing BRAG1-IQ and BRAG1-ΔN. Relative amounts of total p46 (ctrl: 100.0±7.7%; IQ: 101.2±8.2%; n=12; p=0.64; ΔN: 98.3±8.1%; n=12; p=0.43) and total p54 (ctrl: 100.0±10.2%; IQ: 107.2±11.0%; n=12; p=0.14; ΔN: 101.3±10.2%; n=12; p=0.24) JNK in hippocampal CA1 regions expressing BRAG1-IQ and BRAG1-ΔN. Right, relative amounts of phospho-p38 MAPK (ctrl: 100.0±9.1%; IQ: 105.1±9.5%; n=12; p=0.31; ΔN: 107.9±8.7%; n=12; p=0.11) in hippocampal CA1 regions expressing BRAG1-IQ and BRAG1-ΔN. Relative amounts of total p38 MAPK (ctrl: 100.0±9.9%; IQ: 103.0±10.8%; n=12; p=0.64; ΔN: 106.0±10.7%; n=12; p=0.94) in hippocampal CA1 regions expressing BRAG1-IQ and BRAG1-ΔN. The relative values and standard errors were normalized to average amounts of AMPA and NMDA responses from control CA1 cells, phospho-JNK, phospho-p38 MAPK, total JNK or total p38 MAPK from control hippocampal CA1 regions. Asterisk indicates p<0.05 (Wilcoxon test).
JNK and p38MAPK depress transmission by signaling synaptic removal of GluA1- and GluA2-containing AMPA-Rs, respectively (Zhu et al., 2002; Zhu et al., 2005; Kielland et al., 2009). To test whether BRAG1-Arf6 regulates synaptic trafficking of GluA1- and/or GluA2-containg AMPA-Rs, we examined the effects of BRAG1 mutants in CA1 neurons prepared from GluA1 and GluA2 knockout mice. As shown in Fig. 10, the depressive effect of BRAG1-IQ and potentiative effect of BRAG1-ΔN were occluded or blocked in GluA1 but not GluA2 knockout CA1 neurons. These results suggest that BRAG1-Arf6 signals synaptic depression via stimulating JNK-mediated synaptic removal of GluA1-containing AMPA-Rs.
Figure 10. BRAG1 signaling regulates synaptic trafficking of GluA1-containing AMPA-Rs.
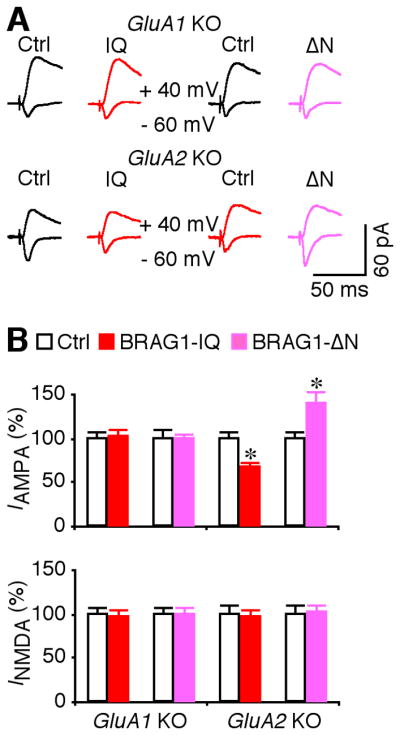
(A) Evoked AMPA-R- (−60 mV) and NMDA-R- (+40 mV) mediated responses recorded from neighboring non-expressing (Ctrl) and mCherry-tagged BRAG1-IQ or BRAG1-ΔN expressing neurons prepared from GluA1 or GluA2 knockout mice. (B) Values for AMPA responses in BRAG1-IQ and BRAG1-ΔN expressing neurons prepared from GluA1 (Ctrl: −23.5±1.8 pA; IQ: −24.1±2.0 pA; n=20, p=0.88; Ctrl: −26.2±2.5 pA; ΔN: −26.2±1.5 pA; n=20, p=0.82), or GluA2 (Ctrl: −30.9±2.1 pA; IQ: −20.9±1.6 pA; n=20, p<0.005; Ctrl: −24.5±1.7 pA; ΔN: − 31.5±2.9 pA; n=24, p<0.005) knockout mice, and for NMDA responses in BRAG1-IQ and BRAG1-ΔN expressing neurons prepared from GluA1 (Ctrl: 94.9±7.6 pA; IQ: 91.9±8.4 pA; n=20, p=0.91; Ctrl: 113.6±8.7 pA; ΔN: 111.9±9.8 pA; n=20, p=0.97), or GluA2 (Ctrl: 54.5±5.0 pA; IQ: 54.8±5.2 pA; n=20, p=0.46; Ctrl: 60.1±5.2 pA; ΔN: 61.8±4.4 pA; n=24, p=0.63) knockout mice. The relative values and standard errors were normalized to average amounts of AMPA and NMDA responses from control CA1 cells. Asterisk indicates p<0.05 (Wilcoxon test).
Discussion
The Arf GEFs BRAG1, BRAG2 and BRAG3 are highly enriched in the brain, where they are concentrated in postsynaptic densities. While all three BRAG family proteins are expressed in hippocampal neurons, BRAG3 localizes specifically to the PSDs of inhibitory synapses, whereas both BRAG1 and BRAG2 are found at excitatory synapses (Sakagami et al., 2008; Scholz et al., 2010; Fukaya et al., 2011). While BRAG2 was recently shown to regulate mGluR-dependent synaptic removal of GluA2-containing AMPA-Rs (Scholz et al., 2010), the synaptic function of BRAG1, which is implicated in nonsyndromic X-linked intellectual disabilility (Shoubridge et al., 2010), had not been investigated. Here we report that BRAG1 signals synaptic depression of AMPA transmission in response to synaptic activation of NMDA-Rs. We further show that diseaseassociated mutations, which affect either catalytic activity or CaM binding, result in either inhibition or constitutive activation of Arf6 signaling, respectively. Moreover, while BRAG2 acts on GluA2-containing AMPA-Rs (Scholz et al., 2010), BRAG1 appears to selectively modulate GluA1-containing AMPA-R-mediated transmission through a mechanism that involves the downstream activation of JNK. These observations provide new insight into the machinery controlling AMPA-R trafficking, and provide a mechanistic basis for the defects in learning and memory exhibited by patients with X-linked intellectual disability.
Mechanisms of BRAG1-Arf6 signaling
Synaptic NMDA-R activation induces a rapid local increase in Ca2+ levels that is critical for the induction of synaptic plasticity (Malenka et al., 1988). The IQ motif is evolutionarily conserved among the BRAG family Arf-GEFs, and although it has been assumed to bind CaM, this had not been previously demonstrated. Here we provide the first evidence that the BRAG1 IQ motif does indeed bind calmodulin, and that it preferentially interacts with the calcium-free form. We also show that CaM dissociation triggered by Ca2+ influx induces a conformational change in BRAG1 resulting in a change in subcellular distribution. However, while CaM binding clearly impacts conformation, its relationship to BRAG1 function is complex. In heterologous cells, BRAG1 catalytic activity appears to be constitutive and is not affected by mutations in the IQ motif that abrogate CaM binding. Similarly, disruption of the catalytic domain, but not the IQ motif, of the single Drosophila BRAG gene Loner was found to cause defects in myoblast fusion (Chen et al., 2003). However, our results show that in hippocampal neurons BRAG1 activity is tightly regulated, requiring upstream NMDA-R activity. Mutation of the IQ motif relieves this constraint, allowing AMPA-R downregulation in the absence of NMDA-R activity. These observations suggest a model in which NMDA-R-mediated Ca2+ influx triggers the release of CaM from BRAG1, which then stimulates AMPA-R endocytosis via its activation of Arf6 (Fig. 11). They also provide a mechanistic explanation for how mutation of the IQ motif found in one family with X-linked intellectual disability may cause disease: failure to bind CaM leads to constitutive BRAG1 activity, resulting in chronic downregulation of AMPA-R signaling.
Figure 11. Model for BRAG1-mediated synaptic trafficking of GluA1-containing AMPA-Rs.

BRAG1 is anchored at postsynaptic densities via its interaction with PSD95, which also interacts with NMDA receptors. Calcium influx triggered by NMDAR activation causes calmodulin (CaM) release from BRAG1, which causes a conformational change leading to enhanced catalytic activity. BRAG1 activates Arf6 which, through downstream activation of JNK, triggers internalization of GluA1-containing AMPA receptors.
The responsiveness of BRAG1 to Ca2+ in the neuronal context is presumably due to the presence of neuron-specific binding partners that help anchor it in the PSD or mediate interactions with other proteins involved in AMPA-R trafficking. In this regard it is interesting that a BRAG1 mutant lacking the N-terminal coiled-coil domain actually potentiates AMPA responses, suggesting that it acts as a dominant negative to inhibit the function of endogenous BRAG1. This hypothesis is supported by the observation that both endogenous Arf6 activity and JNK activity are reduced in the presence of BRAG1-ΔN. Because BRAG1-ΔN is more diffusely distributed within the dendritic shaft and spines, it may bind and sequester components that are limiting for receptor internalization, JNK activation or both.
Signaling interaction of BRAG1-Arf6 and Rap2-JNK-PP2B
In this study, we provide the first evidence that BRAG1-Arf6 signaling intersects the Rap2-MINK-JNK-PP2B signaling pathway at synapses. Previous studies have shown that synaptic activation of NMDA-Rs increases Rap2 signaling, which controls dephosphorylation and synaptic removal of GluA1-containing AMPA-Rs during depotentiation via stimulating the MINK-JNK-PP2B signaling pathway (Zhu et al., 2005; Kielland et al., 2009; Hussain et al., 2010; Yang et al., 2011). We show here that synaptic activity also stimulates BRAG1-Arf6 activity. Interestingly, activation of BRAG1-Arf6 depresses synaptic transmission via stimulating JNK, and blocking JNK activity blocks BRAG1-Arf6-mediated synaptic depression. These results are consistent with previous observations that Arf6 can signal downstream via a neuronal scaffolding protein JIP3 (Montagnac et al., 2009; Suzuki et al., 2010), and that JIP3 regulates JNK signaling(Kelkar et al., 2000; Ongusaha et al., 2008). Moreover, the BRAG1-mediated synaptic depression, which requires Arf6 activation, is mediated by synaptic trafficking of GluA1-containing AMPA-Rs. Together, these results suggest that BRAG1-Arf6 depresses synaptic transmission via regulating Rap2-JNK-PP2B signaling.
Altered BRAG1 signaling in X-linked mental disability
Our results suggest a novel synaptic signaling mechanism whose dysregulation results in X-linked mental retardation. Previous studies have examined the signaling and synaptic mechanisms for two other X-linked mental disorders, oligophrenin-1-associated X-linked mental retardation and fragile X syndrome. Loss of function of oligophrenin-1 is believed to be responsible for the cognitive impairment associated with X-linked mental retardation (Billuart et al., 1998), and recent evidence shows that oligophrenin-1 signals synaptic removal of GluA2-containing AMPA-Rs in a synaptic activity-dependent manner (Nadif Kasri et al., 2009; Nadif Kasri et al., 2011). In FMR1 knockout mice, a mouse model for fragile X syndrome (Bakker and Dutch-Belgian Fragile X Consortium, 1994), mGluA-dependent LTD is modestly up-regulated by ~10–15%, whereas NMDA-R-dependent LTP is considerably reduced (by ~50%) in the knockout animals (Pfeiffer and Huber, 2009; Stornetta and Zhu, 2011). The increased mGluA-dependent LTD is mediated by enhanced Arc signaling (Waung et al., 2008), which controls p38 MAPK-mediated synaptic removal of GluA2-containing AMPA-Rs (Bolshakov et al., 2000; Rial Verde et al., 2006). Exaggerated mGluR signaling seems responsible for several syndromic features of fragile X, including the altered ocular dominance plasticity, seizure and passive avoidance (Krueger and Bear, 2011). The defect in LTP is due to the selective impairment of signal transduction between Ras and PI3K that abolishes synaptic delivery of GluA1-containing AMPA-Rs (Hu et al., 2008). This deficient LTP is responsible for the impaired active, high-level associative learning associated with fragile X (Zhao et al., 2005; Hayashi et al., 2007), which is consistent with the finding that synaptic trafficking of GluA1-containing AMPA-Rs is essential for experience-dependent synaptic plasticity and associative learning (Qin et al., 2005; Rumpel et al., 2005; McCormack et al., 2006; Hu et al., 2007; Matsuo et al., 2008; Kielland et al., 2009; Zhu, 2009). Here, we report that BRAG1-Arf6 regulates the JNK-mediated synaptic removal of GluA1-containing AMPA-Rs (Fig. 11). Moreover, BRAG1 mutations associated with nonsyndromic X-linked mental retardation impair both JNK signaling and synaptic trafficking of GluA1-, but not GluA2-containing AMPA-Rs. These results thus provide the first evidence that dysregulation of JNK signaling and synaptic removal of GluA1-containing AMPA-Rs may also lead to X-linked mental retardation, and provide a new mechanistic explanation for how mutations that either inhibit or enhance Arf6 activity may all result in nonsyndromic X-linked mental disability.
Acknowledgments
The authors would like to thank Bettina Winckler and David Castle for careful reading of the manuscript. This work was supported by NIH grants GM078585 to J.E.C and NS053570 to J.J.Z.
Footnotes
Conflicts of interest: none.
References
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Current opinion in neurobiology. 2012 doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker C Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billuart P, Bienvenu T, Ronce N, des Portes V, Vinet MC, Zemni R, Roest Crollius H, Carrie A, Fauchereau F, Cherry M, Briault S, Hamel B, Fryns JP, Beldjord C, Kahn A, Moraine C, Chelly J. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature. 1998;392:923–926. doi: 10.1038/31940. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F. Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nature neuroscience. 2000;3:1107–1112. doi: 10.1038/80624. [DOI] [PubMed] [Google Scholar]
- Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends in cell biology. 2001;11:82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- Casanova JE. Regulation of Arf activation: the Sec7 family of guanine nucleotide exchange factors. Traffic. 2007;8:1476–1485. doi: 10.1111/j.1600-0854.2007.00634.x. [DOI] [PubMed] [Google Scholar]
- Chen EH, Pryce BA, Tzeng JA, Gonzalez GA, Olson EN. Control of myoblast fusion by a guanine nucleotide exchange factor, loner, and its effector ARF6. Cell. 2003;114:751–762. doi: 10.1016/s0092-8674(03)00720-7. [DOI] [PubMed] [Google Scholar]
- Choi S, Ko J, Lee JR, Lee HW, Kim K, Chung HS, Kim H, Kim E. ARF6 and EFA6A regulate the development and maintenance of dendritic spines. J Neurosci. 2006;26:4811–4819. doi: 10.1523/JNEUROSCI.4182-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Silva AJ. Mouse models of neurofibromatosis type I: bridging the GAP. Trends in molecular medicine. 2003;9:19–23. doi: 10.1016/s1471-4914(02)00008-4. [DOI] [PubMed] [Google Scholar]
- D’Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nature reviews Molecular cell biology. 2006;7:347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Jackson CL. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nature reviews Molecular cell biology. 2011;12:362–375. doi: 10.1038/nrm3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A, Makusky AJ, Jankowska-Stephens E, Yang X, Slotta DJ, Markey SP. Composition of the synaptic PSD-95 complex. Molecular & cellular proteomics : MCP. 2007;6:1749–1760. doi: 10.1074/mcp.M700040-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya M, Kamata A, Hara Y, Tamaki H, Katsumata O, Ito N, Takeda S, Hata Y, Suzuki T, Watanabe M, Harvey RJ, Sakagami H. SynArfGEF is a guanine nucleotide exchange factor for Arf6 and localizes preferentially at post-synaptic specializations of inhibitory synapses. Journal of neurochemistry. 2011;116:1122–1137. doi: 10.1111/j.1471-4159.2010.07167.x. [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11489–11494. doi: 10.1073/pnas.0705003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Deviez DJ, Casanova JE, Wilson JM. Regulation of dendritic development by the ARF exchange factor ARNO. Nature neuroscience. 2002;5:623–624. doi: 10.1038/nn865. [DOI] [PubMed] [Google Scholar]
- Hernandez-Deviez DJ, Roth MG, Casanova JE, Wilson JM. ARNO and ARF6 regulate axonal elongation and branching through downstream activation of phosphatidylinositol 4-phosphate 5-kinase alpha. Molecular biology of the cell. 2004;15:111–120. doi: 10.1091/mbc.E03-06-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Qin Y, Bochorishvili G, Zhu Y, van Aelst L, Zhu JJ. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J Neurosci. 2008;28:7847–7862. doi: 10.1523/JNEUROSCI.1496-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Hussain NK, Hsin H, Huganir RL, Sheng M. MINK and TNIK differentially act on Rap2-mediated signal transduction to regulate neuronal structure and AMPA receptor function. J Neurosci. 2010;30:14786–14794. doi: 10.1523/JNEUROSCI.4124-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, Neubert TA. Identification and verification of novel rodent postsynaptic density proteins. Molecular & cellular proteomics : MCP. 2004;3:857–871. doi: 10.1074/mcp.M400045-MCP200. [DOI] [PubMed] [Google Scholar]
- Kelkar N, Gupta S, Dickens M, Davis RJ. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Molecular and cellular biology. 2000;20:1030–1043. doi: 10.1128/mcb.20.3.1030-1043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nature reviews Neuroscience. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielland A, Bochorishvili G, Corson J, Zhang L, Rosin DL, Heggelund P, Zhu JJ. Activity patterns govern synapse-specific AMPA-R trafficking between deliverable and synaptic pools. Neuron. 2009;62:84–101. doi: 10.1016/j.neuron.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolleker A, Zhu JJ, Schupp BJ, Qin Y, Mack V, Borchardt T, Kohr G, Malinow R, Seeburg PH, Osten P. Glutamatergic plasticity by synaptic delivery of GluR-B(long)-containing AMPA receptors. Neuron. 2003;40:1199–1212. doi: 10.1016/s0896-6273(03)00722-0. [DOI] [PubMed] [Google Scholar]
- Krauss M, Kinuta M, Wenk MR, De Camilli P, Takei K, Haucke V. ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Igamma. The Journal of cell biology. 2003;162:113–124. doi: 10.1083/jcb.200301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DD, Bear MF. Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annual review of medicine. 2011;62:411–429. doi: 10.1146/annurev-med-061109-134644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Kassis S, Kumar S, Badger A, Adams JL. p38 mitogen-activated protein kinase inhibitors--mechanisms and therapeutic potentials. Pharmacol Ther. 1999;82:389–397. doi: 10.1016/s0163-7258(99)00008-x. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Reijmers L, Mayford M. Spine-type-specific recruitment of newly synthesized AMPA receptors with learning. Science. 2008;319:1104–1107. doi: 10.1126/science.1149967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack SG, Stornetta RL, Zhu JJ. Synaptic AMPA receptor exchange maintains bidirectional plasticity. Neuron. 2006;50:75–88. doi: 10.1016/j.neuron.2006.02.027. [DOI] [PubMed] [Google Scholar]
- Miyazaki H, Yamazaki M, Watanabe H, Maehama T, Yokozeki T, Kanaho Y. The small GTPase ADP-ribosylation factor 6 negatively regulates dendritic spine formation. FEBS letters. 2005;579:6834–6838. doi: 10.1016/j.febslet.2005.11.022. [DOI] [PubMed] [Google Scholar]
- Montagnac G, Sibarita JB, Loubery S, Daviet L, Romao M, Raposo G, Chavrier P. ARF6 Interacts with JIP4 to control a motor switch mechanism regulating endosome traffic in cytokinesis. Current biology : CB. 2009;19:184–195. doi: 10.1016/j.cub.2008.12.043. [DOI] [PubMed] [Google Scholar]
- Murphy JA, Jensen ON, Walikonis RS. BRAG1, a Sec7 domain-containing protein, is a component of the postsynaptic density of excitatory synapses. Brain research. 2006;1120:35–45. doi: 10.1016/j.brainres.2006.08.096. [DOI] [PubMed] [Google Scholar]
- Myers KR, Casanova JE. Regulation of actin cytoskeleton dynamics by Arf-family GTPases. Trends in cell biology. 2008;18:184–192. doi: 10.1016/j.tcb.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadif Kasri N, Nakano-Kobayashi A, Van Aelst L. Rapid synthesis of the X-linked mental retardation protein OPHN1 mediates mGluR-dependent LTD through interaction with the endocytic machinery. Neuron. 2011;72:300–315. doi: 10.1016/j.neuron.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadif Kasri N, Nakano-Kobayashi A, Malinow R, Li B, Van Aelst L. The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes & development. 2009;23:1289–1302. doi: 10.1101/gad.1783809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongusaha PP, Qi HH, Raj L, Kim YB, Aaronson SA, Davis RJ, Shi Y, Liao JK, Lee SW. Identification of ROCK1 as an upstream activator of the JIP-3 to JNK signaling axis in response to UVB damage. Science signaling. 2008;1:ra14. doi: 10.1126/scisignal.1161938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Kim MJ, Cheng D, Duong DM, Gygi SP, Sheng M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J Biol Chem. 2004;279:21003–21011. doi: 10.1074/jbc.M400103200. [DOI] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. The state of synapses in fragile X syndrome. Neuroscientist. 2009;15:549–567. doi: 10.1177/1073858409333075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Zhu Y, Baumgart JP, Stornetta RL, Seidenman K, Mack V, van Aelst L, Zhu JJ. State-dependent Ras signaling and AMPA receptor trafficking. Genes & development. 2005;19:2000–2015. doi: 10.1101/gad.342205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- Rial Verde EM, Lee-Osbourne J, Worley PF, Malinow R, Cline HT. Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron. 2006;52:461–474. doi: 10.1016/j.neuron.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Sakagami H, Sanda M, Fukaya M, Miyazaki T, Sukegawa J, Yanagisawa T, Suzuki T, Fukunaga K, Watanabe M, Kondo H. IQ-ArfGEF/BRAG1 is a guanine nucleotide exchange factor for Arf6 that interacts with PSD-95 at postsynaptic density of excitatory synapses. Neuroscience research. 2008;60:199–212. doi: 10.1016/j.neures.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Santy LC, Casanova JE. Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. The Journal of cell biology. 2001;154:599–610. doi: 10.1083/jcb.200104019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz R, Berberich S, Rathgeber L, Kolleker A, Kohr G, Kornau HC. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron. 2010;66:768–780. doi: 10.1016/j.neuron.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Shoubridge C, et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nature genetics. 2010;42:486–488. doi: 10.1038/ng.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Zhu JJ. Ras and Rap signaling in synaptic plasticity and mental disorders. Neuroscientist. 2011;17:54–78. doi: 10.1177/1073858410365562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Arikawa C, Kuwahara Y, Itoh K, Watanabe M, Watanabe H, Suzuki T, Funakoshi Y, Hasegawa H, Kanaho Y. The scaffold protein JIP3 functions as a downstream effector of the small GTPase ARF6 to regulate neurite morphogenesis of cortical neurons. FEBS letters. 2010;584:2801–2806. doi: 10.1016/j.febslet.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nature reviews Neuroscience. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological reviews. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Courtney MJ, Martinsson P, Manahan-Vaughan D. Hippocampal long-term depression is enhanced, depotentiation is inhibited and long-term potentiation is unaffected by the application of a selective c-Jun N-terminal kinase inhibitor to freely behaving rats. The European journal of neuroscience. 2011;33:1647–1655. doi: 10.1111/j.1460-9568.2011.07661.x. [DOI] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Horwitz AF. Synapse formation is regulated by the signaling adaptor GIT1. The Journal of cell biology. 2003;161:131–142. doi: 10.1083/jcb.200211002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005;25:7385–7392. doi: 10.1523/JNEUROSCI.1520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ. Activity level-dependent synapse-specific AMPA receptor trafficking regulates transmission kinetics. J Neurosci. 2009;29:6320–6335. doi: 10.1523/JNEUROSCI.4630-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nature neuroscience. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pak D, Qin Y, McCormack SG, Kim MJ, Baumgart JP, Velamoor V, Auberson YP, Osten P, van Aelst L, Sheng M, Zhu JJ. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron. 2005;46:905–916. doi: 10.1016/j.neuron.2005.04.037. [DOI] [PubMed] [Google Scholar]