

Abstract
Hsp90 is an attractive therapeutic target for the treatment of cancer. Extensive structural modifications to novobiocin, the first Hsp90 C-terminal inhibitor discovered, have produced a library of novobiocin analogues and revealed some structure–activity relationships. Based upon the most potent novobiocin analogues generated from prior studies, a three-dimensional quantitative structure-activity (3D-QSAR) model was built. In addition, a new set of novobiocin analogues containing various structural features supported by the 3D-QSAR model were synthesized and evaluated against two breast cancer cell lines. Several new inhibitors produced anti-proliferative activity at mid nano-molar concentrations, which results through Hsp90 inhibition.
Keywords: Heat shock protein 90, Hsp90 inhibitors, Novobiocin, 3D-QSAR, Breast cancer
The 90 kDa heat shock proteins (Hsp90) are responsible for the conformational maturation of numerous signaling proteins,1,2 many of which are directly associated with the six hallmarks of cancer.3 As a consequence of Hsp90 inhibition, a simultaneous attack occurs upon multiple signaling pathways that are essential for the proliferation of transformed cells. The clinical investigation of 17-AAG and 15 other Hsp90 N-terminal inhibitors has demonstrated Hsp90 as a promising cancer therapeutic target.4 Hsp90 C-terminal inhibitors may provide additional advantage for the pursuit of Hsp90 inhibitors, since no prosurvival heat shock response is observed at concentrations required to induce client protein degradation.5
Novobiocin, a natural product isolated from Streptomyces strains, was the first inhibitor of the Hsp90 C-terminal binding pocket identified, albeit with low efficacy.6 Structural modifications to benzamide side chain and coumarin core have elucidated preliminary structure-activity relationships and identified analogues that exhibit increased inhibitory activities (Figure 1).7–10 Investigation of the noviose appendage demonstrated that the stereochemically complex and rate-limiting synthon can be replaced with simplified mimics such as alkyl amines, while simultaneously maintaining or increasing solubility.11–13
Figure 1.
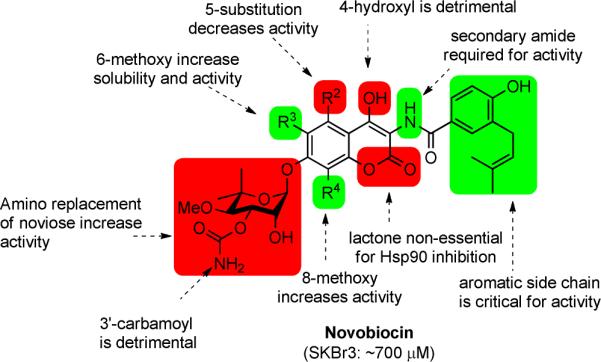
Structure-activity relationships generated from previous investigations.
In an effort to further understand the determinants of novobiocin binding to Hsp90 and to generate a set of rational, quantitative rules for the development of new derivatives that go beyond empirical observations, we set out to build a three-dimensional quantitative structure-activity model (3D-QSAR) of the most potent novobiocin-analogues identified in prior studies (Figure 2). Although this training set is of small size and may represent a limit within the model, our intention was to restrain the applicability of the model and to determine the chemical space for known compounds and to gain insights into the relative importance of distinct functional groups. Moreover, the activities of the compounds used in these studies represent a wide range of values that have been determined via the same experimental protocol.
Figure 2.
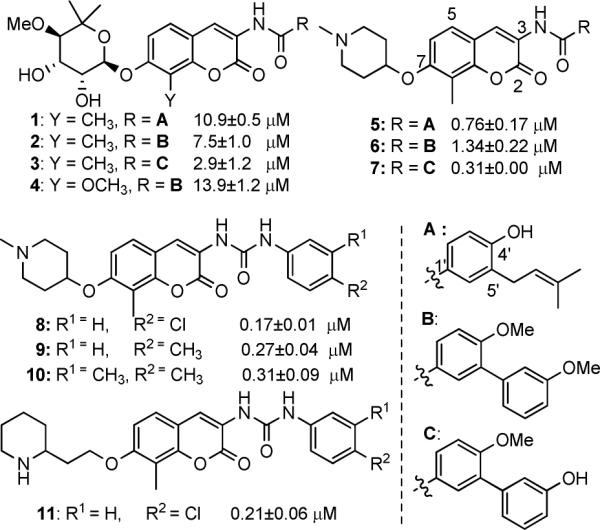
Strucures of novobiocin analogues used to build the 3D QSAR model and their experimental IC50 against SKBr3 cells.
3D-QSARs were generated based upon cellular anti-proliferative activities against SKBr3 cells and the 3D properties of novobiocin analogues enlisting GRIND descriptors.14 These relationships were calculated, analyzed and interpreted using the program PENTACLE (version 1.0.6).14–16 Since no co-crystal structure with Hsp90-C-terminal inhibitors exists, the bioactive conformation of these molecules remains unknown. The Monte Carlo based conformational search was used to identify the most probable conformation. GRIND descriptors, including the O probe (carbonyl oxygen as a hydrogen bond acceptor), the N1 probe (amide nitrogen as a hydrogen bond donor), the TIP17 probe (the shape between the ligand and protein) and the DRY probe (hydrophobic interactions), were calculated from molecular interaction fields (MIFs),18 which provide an array of interactive energy values between a molecule of known structure and a probe group calculated at each node of the grid surrounding the molecule of interest. Descriptors obtained from this analysis can be graphically represented in diagrams called “correlogram plots”, wherein the products from these interaction energies are plotted versus the distance between them. Four auto-correlograms, including (DRY-DRY, O-O, N1-N1 and TIP-TIP) belonging to the same MIF, and six cross-correlograms (DRY-O, DRY-N1, DRY-TIP, O-N1, O-TIP, and N1-TIP) from different MIFs were obtained using interaction energy values. The GRIND descriptors obtained from this analysis were used to obtain a multivariable model and the partial least-squares regression (PLS) method was used to correlate descriptors with activity.19 The quality of the model was evaluated by the predictive correlation coefficient (q2), obtained by the leave-one-out (LOO) scheme or by a 5-random groups (5RG) procedure.
Since not all of the calculated interactions correlated with activity, the fractional factorial designs (FFD)19 variable selection procedure implemented in PENTACLE was applied to exclude variables that increased the standard deviation of prediction errors. Optimal predictive abilities for the 3D-QSAR model were obtained with a model dimensionality of 4 latent variables. The analysis produced ten correlograms of 68 variables each. The prediction correlation coefficient using the LOO scheme was q2LOO=0.93, while using the 5 random group scheme was q2-RG=0.91 (Table 1).
Table 1.
LOO and 5RG cross-validation of the PLS 3D QSAR regression model.
| Cmpd | IC50 exp (μM) | IC50 pred (LOO, μM) | IC50 pred (RG, μM) |
|---|---|---|---|
| 4 | 13.9 | 10.1 | 9.4 |
| 1 | 10.9 | 11.2 | 11.1 |
| 2 | 7.5 | 7.8 | 5.9 |
| 3 | 2.9 | 3.9 | 4.0 |
| 6 | 1.34 | 1.5 | 1.3 |
| 5 | 0.76 | 2.3 | 2.2 |
| 7 | 0.31 | 1.1 | 0.8 |
| 10 | 0.31 | 0.4 | 0.5 |
| 9 | 0.27 | 0.2 | 0.1 |
| 11 | 0.21 | 1.6 | 1.9 |
| 8 | 0.17 | 1.6 | 1.6 |
The model that summarizes contributions of the original variables can be interpreted with the aid of PLS coefficients. The positive variables represent features found in the most active compounds while negative ones represent features correlated with diminished activity. The PLS coefficients are plotted in Figure 3 and the most influential variables are indicated alongside the variable number.
Figure 3.

PLS coefficients plot of the GRIND variables used in the model. Different correlograms are separated by dotted line and the pair probes are define at the bottom. The most relevant variables are indicated with the variable number.
Not all variables are able to efficiently separate active from non-active molecules. An active molecule may feature an unfavorable functionality that is overcome by that of another group that correlates with favorable activity. In order to provide effective suggestions for lead optimization, only the variables that have PLS coefficients of high absolute value and those that clearly distinguish active and less active compounds are considered, which arise from the O-O, O-N1 and DRY-DRY correlograms (interpreted in Supporting Information, Table S1). Such variables, and DRY-DRY in particular, were selected since hydrogen bonding and hydrophobic/aromatic interactions represent the most common intermolecular forces that determine host/guest recognition in drugs.
The DRY auto-correlogram (DRY-DRY-43) indicated that hydrophobic interactions correlated poorly with biological activity, which may result from the fact that all of these molecules bear similar hydrophobic groups. However, it is worth noting that the most relevant variable resulting from this correlogram suggests that the hydrophobic character of the piperidine ring and the first aromatic ring on the amide/carbamide substituent correlate positively with activity. The most important variable on the O-N1 cross-correlogram (ON1-529) exhibits a higher value for three of the most potent compounds (5–7). This variable describes the interaction between the NH of the protonated piperidine ring and the oxygen atom in 4'-position with the N1 probe, indicating these interactions are important for activity. The importance of the NH group on the piperidine ring is further supported by variable OO-120, which produced a higher value for compounds 5 and 7. Moreover, this variable suggested that a hydrogen bond donor on the amide side chain can further enhance inhibitory activity. In the same correlogram, variable OO-102 produced a high value for the most potent compounds, all of which bear the piperidine ring (5–11). This variable suggested a positive effect on inhibitory activity given by the amino group on the piperidine ring and the amide nitrogen.
In contrast, variables with a high negative impact on inhibitory activity were identified, in particular, variables OO-80 and N1N1-141, which correlate with the presence of multiple hydrogen bond donor and acceptor atoms on the sugar moiety of compounds 1–4. This data suggested that hydrophobic groups produce favorable activities. Accordingly, molecules with lower IC50 values (5 and 7–11) bear the (N-methyl) piperidine as a replacement for noviose.
The model can be summarized as: (i) The side chain of the coumarin core should be an amide, while the noviose sugar should be replaced by an amine, (ii) the amide side chain should exhibit hydrophobic character, (iii) the oxygen atom in the 4'-position is important for binding, and (iv) the presence of a hydrogen bond donor on this side chain can bind more tightly. Figure 4 illustrates the most relevant variables that produce a direct impact on activities for the most active compounds.
Figure 4.

Graphical representation of the four most relevant GRIND variables with direct impact on activity for the most active compounds. MIFs are shown in colored spheres (DRY probe, yellow; O probe, red; N1 probe, blue). The most important variables in each correlogram correspond to the pairs of grid nodes DRY-DRY, O-O, O-N1 linked in this MIFs representation.
On the basis of these computationally-derived SARs, compound 5 was chosen as a lead molecule for the development of a new series of derivatives that contain various structural features supported by the 3D-QSAR model. As noted by this model, the benzamide side chain plays a beneficial role towards inhibitory activity. Thus, the initial investigation commenced with modification of the benzamide side chain.
Analogues containing substituted benzamide side chains were assembled in a modular fashion (see Supporting Information, Scheme S1), and utilized a Mitsunobu coupling reaction between 1-methyl-4-hydroxypeperidine 14 and coumarin phenol 15,13 followed by coupling of amine 12 with various aryl acid chlorides, which were prepared from 13 and provide direct access to a number of functionally distinct analogues.
Preparation of these analogues is described in Scheme 1. Methylation of the benzamide 4'-phenol occurred upon treatment of 2 with sodium hydride followed by iodomethane to give methylether 16 in good yield. Similar etherifications occurred by Mitsunobu reactions between 2 and n-butanol or prop-2-yn-1-ol to afford 17 or 18, respectively. Hydrogenation of 2 gave the saturated prenyl group found in 19, while exposure to acid resulted in cyclization to give ether 20. Amide coupling between aniline 12 and carboxylic acid chlorides prepared from 13a–e afforded compounds 21a–e, which upon solvolysis provided 22a, 22b and 22e, respectively.
Scheme 1.

Structural modifications to the benzamide side chain
The anti-proliferative activity manifested by these compounds was determined using two breast cancer cell lines, SKBr3 (ER-, Her2 over-expressing) and MCF-7 (ER+). As shown in Table 2, alkylation of the 4'-phenol (16–18) resulted in decreased anti-proliferative activity against the SKBr3 cell line, but maintained similar activity against MCF-7 cells, suggesting that the 4'-phenol may be binding Hsp90 as an H-bond acceptor. Saturation of the double bond (19) produced no effect on inhibitory activity; while intramolecular cyclization (20), the removal of two terminal methyl groups on the prenyl chain (21a and 22a), and removal of the prenyl group (21b and 22b) resulted in decreased anti-proliferative activities. Thus, the prenyl side chain appears important and may establish hydrophobic interactions with the protein binding site. Consistent with the 3D QSAR studies, removal of the 4'-phenol (21c) resulted in a two-fold lower activity, while removal of both the phenol and prenyl group (21d) resulted in significantly decreased activity, confirming the 4'-phenol is important for binding. Likewise, replacement of the prenyl group with electron-withdrawing substituents (21e and 22e vs 21b and 22b) resulted in diminished activity against the SKBr3 cell line, suggesting the potential for interactions between the 4'-phenol and the surrounding environment.
Table 2.
Experimental and predicted anti-proliferative activity of analogues derived from 2
| SKBr3 (μM) | Predicted (μM) | MCF-7 (μM) | |
|---|---|---|---|
| 16 | 1.41±0.20a | 0.99 | 1.58±0.09 |
| 17 | 1.40± 0.07 | 1.63 | 1.44± 0.05 |
| 18 | 1.40± 0.11 | 0.73 | 1.12± 0.16 |
| 19 | 0.60±0.01 | 0.58 | 1.25±0.39 |
| 20 | 3.46±0.02 | 1.74 | 5.01±0.01 |
| 21a | 2.26±0.12 | 2.00 | 2.00±0.18 |
| 21b | 1.67±0.20 | 1.42 | 1.62±0.04 |
| 21c | 1.68±0.12 | 1.83 | 2.10±0.08 |
| 21d | 11.5± 2.0 | 2.90 | 11.7±1.0 |
| 21e | 3.50±0.01 | 0.97 | 1.61±0.16 |
| 22a | 2.43±0.04 | 1.15 | 2.17±0.11 |
| 22b | 0.84±0.18 | 0.96 | 1.43±0.08 |
| 22e | 2.30±0.57 | 0.33 | 1.48±0.14 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Since the 3D-QSAR model suggests the coexistence of a hydrogen bond donor and a hydrogen bond acceptor on the benzamide side chain that can increase activity, alkylation of the 4'-phenol with an alkyl tertiary amine was explored to determine whether this modification could establish favorable interactions with the binding pocket. Three amines (2-(dimethylamino)ethanol, 3-(dimethylamino)propan-1-ol and 1-methylpiperidin-4-ol) were attached to the 4'-phenol benzamide side chain of 5 via Mitsunobu etherification to afford compounds 23–25 (Scheme 2). Prior studies demonstrated that replacement of the flexible prenyl side-chain with an aromatic ring leads to improved inhibitory activity.9 Therefore, installation of two additional functionalities was pursued by the use of biaryl analogues, 33a and 33b. Accordingly, the phenol was placed on both phenyl rings of the biaryl system, and 33b was expected to align with the spatial requirements identified in the 3D QSAR model, while 33a was not. The syntheses of these two compounds were achieved by coupling of 12 with acyl chloride 31 (see Supporting Information Scehme S2) and subsequent basic solvolysis.
Scheme 2.
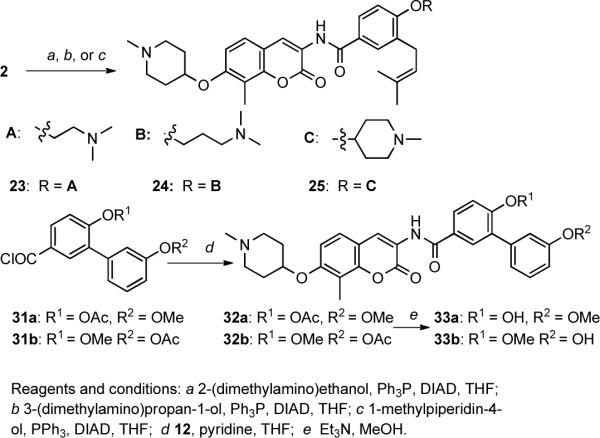
Synthesis of novobiocin analogues with an amine tether on the benzamide side chain.
Evaluation of these benzamide analogues against breast cancer cell lines generated the data in Table 3. These data demonstrated that tethering the phenol to alkyl amines (23–25) leads to increased anti-proliferative activity, supporting the formation of stronger interactions with the binding site. For biaryl analogues (32 and 33), replacement of the prenyl group with a substituted phenyl ring maintained inhibitory activity (32a and 33a) as previously suggested, however, switching the phenol from the first to the second benzene ring increased activity almost three-fold (32b and 33b). This latter result suggests that placement of a hydrogen bond donor at the second ring is more favorable.
Table 3.
Experimental and predicted anti-proliferative activity of novobiocin analogues containing a modified benzamide side chain
| SKBr3 (μM) | Predicted (μM) | MCF-7 (μM) | |
|---|---|---|---|
| 23 | 0.33±0.00a | 0.33 | 0.72±0.08 |
| 24 | 0.25±0.02 | 1.75 | 0.46±0.05 |
| 25 | 0.21±0.00 | 1.16 | 0.47±0.081 |
| 32a | 0.92±0.06 | 0.88 | 0.91±0.00 |
| 32b | 0.29±0.01 | 0.25 | 0.36± 0.06 |
| 33a | 0.86± 0.16 | 0.63 | 0.81± 0.14 |
| 33b | 0.31± 0.04 | 0.50 | 0.38± 0.08 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Predictions for the new compounds (Tables 3 and 4, and Supporting Information Figure S1) show that the QSAR model is able to predict most of the activities within two sigma, yielding a correlation coefficient r2 of 0.68. Except for 24 and 25, the activities of derivatives with IC50 values lower than 1.5 μM are well predicted, and the best performance was obtained for compounds that manifest an IC50 < 1 μM. The activities for less active compounds are underestimated. This result is a reflection of the properties used for training, which utilized compounds that manifest activities of less than 1 μM. A potential source of error may be related to the dependence of the model on the 3D structures of the compounds used to calculate the interaction energy maps. The minimum energy solution conformation used for the calculations can clearly influence the prediction capabilities in this model, in particular, if the molecules used to build the model are similar as in this case, as small differences in the position of functional groups could yield different activities. The activities of compounds 24 and 25, e.g, are overestimated because their conformations are highly similar to the conformation of compound 1, and the position of the amino group in the amide side chains is not optimally described by the model. However, despite these limitations, the model shows a non-trivial agreement between the predicted and experimental activities.
To confirm that the anti-proliferative activities observed by these modified novobiocin analogues resulted from Hsp90 inhibition, western blot analyses of cell lysates following administration of 24 was performed. Figure 5 shows that in MCF-7 cells, the Hsp90-dependent client proteins Her2, Raf, and Akt underwent degradation in a concentration-dependent manner upon treatment with 24 at the same concentration needed to manifest anti-proliferative activity, thereby linking Hsp90 inhibition to cell viability. The non-Hsp90-dependent protein, actin, was not affected, and indicates the selective degradation of Hsp90-dependent proteins. In addition, Hsp90 levels remained constant at all concentrations tested, which is consistent with prior studies involving Hsp90 C-terminal inhibitors.20,21
Figure 5.

Western blot analyses of MCF-7 cell lysates for Hsp90 client protein degradation after 24 h incubation. Concentrations (in μM) of 24 are indicated above each lane. Geldanamycin (G, 500 nM) and DMSO (D) were respectively employed as positive and negative controls.
In summary, a set of potent novobiocin analogues was designed with the assistance of a 3D-QSAR model and shown to exhibit promising anti-proliferative activities between 200~400 nM against two breast cancer cell lines. Western blot analysis confirmed that the anti-proliferative activity manifested by 24 resulted from Hsp90 inhibition. Overall, these results support the potential to use quantitative structure-based rational studies to investigate the chemical space of molecules targeting the Hsp90 C-terminus. The designed compounds produced anti-proliferative activity and blocked Hsp90 chaperoning activity. Together, these features may make this model an attractive starting point for the rational development of new Hsp90 inhibitors.
Supplementary Material
Acknowledgement
The authors gratefully acknowledge support of this project by the NIH/NCI (CA120458) and the DOD Prostate Cancer Research Program (QH815179). GC gratefully acknowledges AIRC (Associazione Italiana Ricerca sul Cancro) for support through the grant IG.11775 and the Flagship “INTEROMICS” project (PB.P05) funded by MIUR and CNR.
Footnotes
Supporting Information Experimental procedures for the synthesis and characterization of new compounds (1H and 13C NMR, HRMS). This information is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Blagg BSJ, Kerr TA. Hsp90 inhibitors: small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Medicinal Research Reviews. 2006;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- (2).Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. Journal of Molecular Medicine (Heidelberg, Germany) 2004;82:488–499. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- (3).Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. Cell 2011, 144, 646-674. [DOI] [PubMed] [Google Scholar]
- (4).Biamonte MA, Van de Water R, Arndt JW, Scannevin RH, Perret D, Lee WC. Heat Shock Protein 90: Inhibitors in Clinical Trials. Journal of Medicinal Chemistry. 2010;53:3–17. doi: 10.1021/jm9004708. [DOI] [PubMed] [Google Scholar]
- (5).Zhao HP, Michaelis ML, Blagg BSJ. Hsp90 modulation for the treatment of Alzheimer's disease. Advances in pharmacology. (San Diego, Calif.) 2012;64:1–25. doi: 10.1016/B978-0-12-394816-8.00001-5. [DOI] [PubMed] [Google Scholar]
- (6).Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- (7).Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. Hsp90 inhibitors identified from a library of novobiocin analogues. Journal of the American Chemical Society. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- (8).Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. Novobiocin: redesigning a DNA gyrase inhibitor for selective inhibition of Hsp90. Journal of the American Chemical Society. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- (9).Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. Development of novobiocin analogues that manifest anti-proliferative activity against several cancer cell lines. Journal of Organic Chemistry. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- (10).Donnelly AC, Mays JR, Burlison JA, Nelson JT, Vielhauer G, Holzbeierlein J, Blagg BSJ. The design, synthesis, and evaluation of coumarin ring derivatives of the novobiocin scaffold that exhibit antiproliferative activity. Journal of Organic Chemistry. 2008;73:8901–8920. doi: 10.1021/jo801312r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhao HP, Kusuma BR, Blagg BSJ. Synthesis and evaluation of noviose replacements on novobiocin that manifest antiproliferative activity. Acs Medicinal Chemistry Letters. 2010;1:311–315. doi: 10.1021/ml100070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Donnelly AC, Zhao HP, Kusuma BR, Blagg BSJ. Cytotoxic sugar analogues of an optimized novobiocin scaffold. Medchemcomm. 2010;1:165–170. [Google Scholar]
- (13).Zhao HP, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. Engineering an Antibiotic to Fight Cancer: Optimization of the Novobiocin Scaffold to Produce Anti-proliferative Agents. J. Med. Chem. 2011;54:3839–3853. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pastor M, Cruciani G, McLay I, Pickett S, Clementi S. GRid-INdependent Descriptors (GRIND): A novel class of alignment-independent three-dimensional molecular descriptors. Journal of Medicinal Chemistry. 2000;43:3233–3243. doi: 10.1021/jm000941m. [DOI] [PubMed] [Google Scholar]
- (15).Duran A, Martinez GC, Pastor M. Development and validation of AMANDA, a new algorithm for selecting highly relevant regions in molecular interaction fields. Journal of Chemical Information and Modeling. 2008;48:1813–1823. doi: 10.1021/ci800037t. [DOI] [PubMed] [Google Scholar]
- (16).Duran A, Zamora I, Pastor M. Suitability of GRIND-based principal properties for the description of molecular similarity and ligand-based virtual screening. Journal of Chemical Information and Modeling. 2009;49:2129–2138. doi: 10.1021/ci900228x. [DOI] [PubMed] [Google Scholar]
- (17).Fontaine F, Pastor M, Sanz F. Incorporating molecular shape into the alignment free GRid-INdependent descriptors. Journal of Medicinal Chemistry. 2004;47:2805–2815. doi: 10.1021/jm0311240. [DOI] [PubMed] [Google Scholar]
- (18).Goodford PJ. Computational procedure for determining energetically favorable binding sites on biologically important macromolecules. Journal of Medicinal Chemistry. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- (19).Baroni M, Costantino G, Cruciani G, Riganelli D, Valigi R, Clementi S. Generating optimal linear PLS estimations (GOLPE): An advanced chemometric tool for handling 3D-QSAR problems. Quantitative Structure-Activity Relationships. 1993;12:9–20. [Google Scholar]
- (20).Zhao HP, Brandt GE, Galam L, Matts RL, Blagg BSJ. Identification and initial SAR of silybin: An Hsp90 inhibitor. Bioorganic & Medicinal Chemistry Letters. 2011;21:2659–2664. doi: 10.1016/j.bmcl.2010.12.088. [DOI] [PubMed] [Google Scholar]
- (21).Zhao HP, Yan B, Peterson LB, Blagg BSJ. 3-Arylcoumarin derivatives manifest anti-proliferative activity through Hsp90 inhibition. Acs Medicinal Chemistry Letters. 2012;3:327–331. doi: 10.1021/ml300018e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.