

Abstract
Mutations in the gene encoding CREB-binding protein (CBP) cause deficits in long-term plasticity, learning, and memory. Here, long-term synaptic facilitation (LTF) at Aplysia sensorimotor synapses in cell culture was used as a model system to investigate methods for overcoming deficits in LTF produced by a CBP knockdown. Injecting CBP-siRNA into individual sensory neurons reduced CBP levels and impaired LTF produced by a Standard Protocol of five, 5-min pulses of serotonin (5-HT) delivered at 20 min interstimulus intervals. A computational model, which simulated molecular processes underlying LTF induction, identified a Rescue Protocol of five pulses of 5-HT at non-uniform interstimulus intervals that overcame the consequences of reduced CBP and restored LTF. These results suggest that complementary empirical and computational studies can identify methods for ameliorating impairments of learning due to molecular lesions.
INTRODUCTION
CBP is an essential co-activator for numerous transcription factors. It has a histone acetyltransferase (HAT) domain and interaction domains for different transcription factors, including phosphorylated CREB (pCREB) (Kalkhoven, 2004). Mutations of cbp that reduce activity of the HAT domain, and thus its ability to co-activate transcription, commonly result in Rubinstein-Taybi syndrome (RTS) (Rouax et al., 2004; Hallam and Bourtchouladze, 2006). Mouse cbp+/− models exhibit impaired LTP and LTM (Alarcon et al., 2004; Rouaux et al., 2004; Barco, 2007). Previously, we used a mathematical model of molecular processes that underlie serotonin (5-HT)-induced long-term facilitation (LTF), a form of synaptic plasticity that correlates with LTM (Frost et al., 1985; Cleary et al., 1998), to identify a training protocol that enhanced LTF and long-term memory (LTM) in normal animals (Zhang et al., 2012). Here, we investigated the ability of siRNA injections into individual neurons to reduce CBP levels in cultured sensory neurons from Aplysia, the consequences of reducing CBP levels for LTF at sensorimotor synapses, and whether a computational model can predict a training protocol that overcomes deficits in CBP to restore long-term synaptic plasticity.
MATERIALS AND METHODS
Animals and cell cultures
All experiments used primary cell cultures of identified cells from the marine mollusk Aplysia. Isolated sensory neurons (SNs) or sensory neuron-motor neuron (SN-MN) co-cultures were prepared according to conventional procedures (Zhang et al., 2012). Dishes of SN cultures were plated with 5–10 SNs. Dishes of SN-MN co-cultures were plated with a single SN and a single L7 MN.
siRNA knockdown of CBP
Before siRNA injection, cultures or co-cultures were allowed to grow for 2–3 d at 18°C. siRNA (5 µM final concentration) was injected into the cytoplasm of SNs following established procedures (Liu et al., 2011). Cultures and co-cultures were allowed to grow for another 96 h after siRNA injection. SN cultures were then processed for immunofluorescence and co-cultures were used for electrophysiological study.
CBP levels were measured using an anti-CBP antibody developed by Arias et al. (1994), designed to target the binding site for vertebrate phospho-CREB (Chrivia et al., 1993). An 80% identity exists between the mouse brain phospho-CREB binding sequence and the predicted sequence of CBP in Aplysia (Guan et al., 2002). To assess the usefulness of this antibody, nuclear extracts of Aplysia nervous system were prepared following the Invitrogen Protocol (www.invitrogen.com). Western blot analysis following our established procedures (Liu et al., 2008) indicated that the antibody recognized a single band with a molecular weight appropriate for Aplysia CBP, ~250 kDa (Arias et al., 1994; Guan et al., 2002) (Figure 1A1). For immunofluorescence, SNs were injected 96 h before fixation with control siRNA (Con-siRNA) or CBP-siRNA. Cells were incubated with anti-CBP antibody (1:500), followed by incubation with secondary antibody. The intensity of staining was quantitated in images obtained with a Zeiss LSM510 confocal microscope using a 63× oil immersion lens as described previously (Liu et al., 2011) (Figure 1A2). These experiments were done in a blind manner in that the investigator analyzing the images was unaware whether the neurons were injected with Con- or CBP-siRNA.
Figure 1. CBP-siRNA reduced the nuclear levels of CBP and impaired LTF.

A1, Western blot analysis of nuclear extracts from Aplysia CNS revealed a single band with a molecular weight appropriate for CBP. A2, Representative confocal images of CBP immunofluorescence in SNs 96 h after injection with either control siRNA (Con-siRNA) or CBP-siRNA. Fluorescein dye was co-injected to verify that SN cytoplasm and processes (but not the nucleus) were filled. Scale bar, 20 µm. A3, Summary data. CBP levels in the nucleus of SNs were significantly reduced 96 h after injection with CBP-siRNA as compared to SNs injected with Con-siRNA. B1, Representative EPSPs recorded immediately before (Pretest) and 24 and 48 h after (Post-test) the Standard Protocol. Dashed lines represent the amplitude of the Pretest EPSP. B2, Summary data. Synaptic plasticity was measured as the ratio of post-test to pretest EPSPs (post/pre). Significant differences among treatment groups are indicated by * (indicates P < 0.05).
Electrophysiology
Cell culturing, treatment and electrophysiological procedures, and criteria for accepting viable preparations were identical to Zhang et al. (2012). Statistical analyses (Kruskal-Wallis one way analysis of variance on ranks) indicated that the pretest scores for each group were not significantly different (Figure 1B2: H = 7.428, df = 3; Figure 4B: H = 2.82, df = 2). Therefore, for statistical analysis, the amplitudes of the EPSPs at 24 and 48 h after treatment were normalized to the EPSPs measured before treatment (i.e., post/pre). This experiment was done in a blind manner in that the investigator performing the electrophysiology was unaware whether the neurons were injected with Con- or CBP-siRNA. In addition, different investigators performed the 5-HT treatments and the electrophysiology.
Figure 4. Rescue Protocol compensated for the CBP knockdown.

A, Representative EPSPs recorded immediately before (Pretest) and 24 and 48 h after (Post-test) either the Standard or Rescue Protocol. SNs were injected with either Con-siRNA or CBP-siRNA 96 h prior to the Pretest and treatment with 5-HT. Dashed lines represent the amplitude of the Pretest EPSP. B, Summary data. Significant differences among treatment groups are indicated by * for P < 0.05 and ** for P < 0.005.
Computational model
The model (Figure 2) was adapted from an earlier model (Zhang et al., 2012) that described activation of protein kinase A (PKA) and the ERK isoform(s) of MAP kinase. Descriptions of these processes remained as in (Zhang et al., 2012). That model was extended to include the phosphorylation of CREB1, CREB2 and synthesis of C/EBP (Eqs. 1–7). pCREB1 in conjunction with CBP activates transcription of c/ebp (Guan et al., 2002) and is required for LTF (Bartsch et al., 1998). Conversely, CREB2 represses transcription of c/ebp, but ERK-mediated phosphorylation of CREB2 relieves this repression (Bartsch et al., 1995; Guan et al., 2002). Phosphorylation of C/EBP by ERK decreases its degradation rate (Yamamoto et al., 1999). We assumed that new synthesis of CREB1 and CREB2 had limited effects on the induction of LTF. Therefore, the total amounts of CREB1 and CREB2 were conserved.
Figure 2. Model of a molecular network necessary for LTF.

A, Grey shading indicates components of the model originally developed by Zhang et al. (2012). The model was extended by incorporating CREB1, CREB2, C/EBP and CBP. Phosphorylation of CREB1 and its subsequent binding to CBP were assumed necessary for induction of c/ebp. Unphosphorylated CREB2 repressed c/ebp. Phosphorylation of CREB2 removed this repression. Arrows and circles indicate positive and negative regulation of transcription, respectively. B–C, Simulations of the Standard Protocol with normal CBP levels (B) and with the CBP knockdown (C). The Rescue Protocol with the CBP knockdown boosted pC/EBP levels beyond control levels (dashed/dotted line) (D).
The equations for CREB1 were:
| (Eq. 1) |
| (Eq. 2) |
The parameter values were:
kPKA,CREB1 = 0.15 µM−1min−1, kpphos1 = 0.05 min−1, [CREB1]total = 0.05 µM
The dynamics of PKA and ERK were identical to the earlier model (Zhang et al., 2012). In that model and its present extension, basal levels of PKA and ERK activity, prior to stimulus, were set to zero as was the basal level of C/EBP. Basal levels of pCREB1 and pCREB2, which were dependent on basal PKA and ERK activities, were also zero. The equations for CREB2 were:
| (Eq. 3) |
| (Eq. 4) |
The parameter values were:
kERK,CREB2 = 3.5 µM−1min−1, kpphos2 = 0.5 min−1, [CREB2]total = 0.05 µM
For simplicity, induction of c/ebp was modeled as an increase in the synthesis of C/EBP. The following three equations describe C/EBP dynamics. In these equations C/EBP is denoted “CEBP.”
| (Eq. 5) |
| (Eq. 6) |
| (Eq. 7) |
The parameter values were:
kd,CEBP = 0.075 min−1, kERK,CEBP = 0.25 µM−1min−1, kpphos3 = 0.5 min−1, νmax_synthesis,CEBP = 20 µM/min, K5 = 0.009 µM, K6 = 0.003 µM, kd,pCEBP = 0.0015 min−1, KCBP = 20 µM.
Fourth-order Runge-Kutta integration was used for integration of differential equations, with a time step of 3 s. The model was programmed in XPP version 6.1 (www.math.pitt.edu/~bard/xpp/xpp.html). Source codes are available upon request.
The parameters were adjusted by trial and error so that: 1) CREB1 and CREB2 were transiently phosphorylated (Hagiwara et al., 1993; Bartsch et al., 1995, 1998; McLean et al., 1999); 2) the increase in C/EBP expression occurred within 15 min after 5-HT treatment (Alberini et al., 1994); and 3) in the absence of ERK activity, C/EBP was degraded within 2 h (Yamamoto et al., 1999).
In our previous model (Zhang et al., 2012), the output was a phenomenological variable termed inducer, which represented the convergence of the PKA and ERK pathways onto an undefined molecular process necessary for LTF. In the present model, inducer was replaced by pC/EBP as the output, representing a point of convergence among several second-messenger and transcriptional pathways. The Enhanced Protocol of Zhang et al. (2012) increased the peak level of inducer by ~50%. Thus, in the extended model, the parameters governing C/EBP expression and phosphorylation were adjusted such that the Enhanced Protocol produced a peak pC/EBP level ~50% greater than that in the Standard protocol.
RESULTS
siRNA decreased CBP levels and LTF
CBP protein levels were assayed by immunofluorescence 96 h after injection of either CBP-siRNA (n = 11) or scrambled-siRNA (Con-siRNA) (n = 12). CBP levels in nuclei of SNs injected with CBP-siRNA averaged 35% less than those in Con-siRNA injected SNs (Figure 1A3, Mann-Whitney Rank Sum Test, U11,12 = 16, P = 0.002). We next examined the effects of CBP knockdown on LTF using four groups of SN-MN co-cultures (Figure 1B). SNs were injected with either CBP-siRNA or Con-siRNA. 96 h after injection, cultures were treated with five, 5-min pulses of either 5-HT (CBP-siRNA + 5-HT, n = 9; Con-siRNA + 5-HT, n = 7) or control saline (CBP-siRNA + Veh, n = 4; Con-siRNA + Veh, n = 4), which were delivered with a uniform interstimulus interval (ISI) of 20 min. This 5-HT treatment has been commonly used (Montarolo et al., 1986) and was termed the Standard Protocol. In each group, EPSPs were measured immediately prior to the Standard Protocol (pretest) and 24 and 48 h after treatment (post-test). Statistical analyses (two-way, repeated measures ANOVA) indicated significant overall differences among the treatment groups (F(3,20) = 5.712, P = 0.005). The knockdown of CBP attenuated LTF. Post hoc comparisons (Student-Newman-Keuls tests, SNK) revealed a significant difference between the Con-siRNA + 5-HT group and the other three groups (Con-siRNA + 5-HT vs. Con-siRNA + Veh, q2 = 4.448, P = 0.014; Con-siRNA + 5-HT vs. CBP-siRNA + Veh, q2 = 4.548, P = 0.021; Con-siRNA + 5-HT vs. CBP-siRNA + 5-HT, q2 = 4.776, P = 0.003). Moreover, no significant differences were observed among the Con-siRNA + Veh, CBP-siRNA + Veh, and CBP-siRNA + 5-HT groups (Con-siRNA + Veh vs. CBP-siRNA + Veh, q2 = 0.088; Con-siRNA + Veh vs. CBP-siRNA + 5-HT, q2 = 0.083; CBP-siRNA + Veh vs. CBP-siRNA + 5-HT, q2 = 0.097).
The attenuation of LTF by the knockdown of CBP was not associated with differences among the pretest EPSPs of the four groups. A separate series of experiments examined the potential effects of knocking down CBP on short-term synaptic facilitation (STF) which was induced by a single, 5-min pulse of 5-HT 96 h after CBP-siRNA injection. Comparison of STF in cells injected with Con-siRNA (n = 6) vs. cells injected with CBP-siRNA (n = 6) revealed no differences in STF (STF in Con-siRNA group = 123 ± 7 % of pretest vs. STF in CBP-siRNA group 135 ± 14 % of pretest; t(10) = 0.463). These results indicated that CBP knockdown did not affect basal synaptic transmission or short-term plasticity.
Prediction of a stimulus protocol to rescue LTF
To predict a stimulus protocol that could rescue LTF, the model of Zhang et al. (2012) was extended to describe phosphorylation of the transcription factors CREB1, CREB2, and C/EBP, and induction of c/ebp (Figure 2A; Eqs. 1–7). Induction of c/ebp and phosphorylation of C/EBP by ERK are both necessary for LTF (Alberini et al., 1994; Yamamoto et al., 1999). Therefore, the peak level of phosphorylated C/EBP (pC/EBP) was taken as the output of the model. The simulated response to a Standard Protocol with control parameter values is illustrated in Figure 2B. Reduction of CBP by 35% (equivalent to the average empirical siRNA-mediated knockdown) reduced pC/EBP (Figure 2C), which agreed with the empirical attenuation of LTF.
Following our previous strategy (Zhang et al., 2012), we sought a stimulus protocol that would compensate for the reduced CBP levels and restore peak pC/EBP to or above the peak produced by the Standard Protocol with normal CBP levels. Each protocol included five, 5-min 5-HT stimuli, but the ISIs were chosen as multiples of 5 min, in the range from 5–50 min. Combining all of these permutations yields 104 total protocols, one of which was the original Standard Protocol. After simulated CBP knockdown, the 104 protocols were ranked according to the peak levels of pC/EBP they elicited. Of these 104 protocols, 421 restored the simulated peak level of pC/EBP to or above the level without C/EBP knockdown. One was the previously described Enhanced Protocol (Zhang et al., 2012). These protocols have a general property that the durations of the first two ISIs are ≤ 15 min. These relatively brief ISIs induced an earlier activation of ERK, which tended to maximize the overlap of the elevation in pCREB2 levels with that of pCREB1 levels. Maximizing this overlap, in turn, strongly induced C/EBP synthesis and coincident C/EBP phosphorylation, yielding high peak pC/EBP. Among these 421 potential rescue protocols, the protocol that produced the highest peak pC/EBP was selected for further analysis in empirical studies (Figures 2D and 3E). This Rescue Protocol had ISIs of 10, 10, 20, and 10 min (Figures 2D and 3E).
Figure 3. Simulating CBP knockdown and rescue.
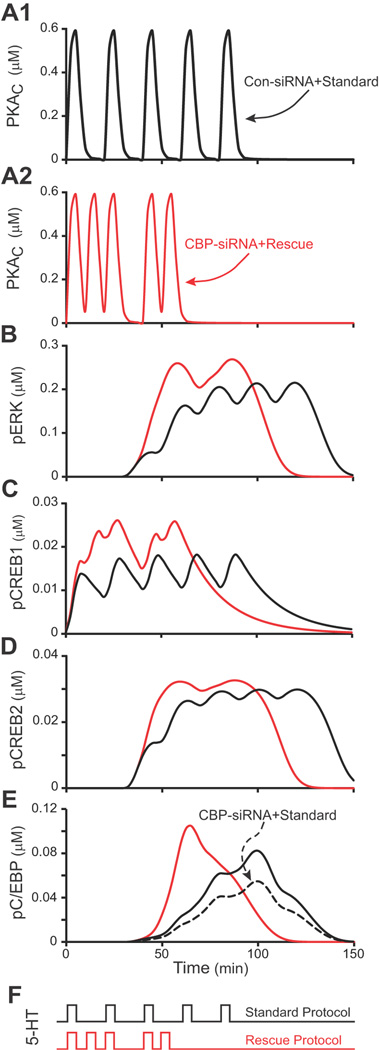
In Panels A–E, the black traces illustrate the responses of the model to the Standard Protocol with control levels of CBP, whereas red traces illustrate the responses to the Rescue Protocol following knockdown of CBP. A1 and A2, Time courses of PKA activity (PKAC). B, Time courses of ERK activity (pERK). C, Time courses of active CREB1 (pCREB1). D, Time courses of inactive CREB2 (pCREB2). E, Time courses of active C/EBP (pC/EBP). The black solid and black dashed time courses are for the Standard Protocol with and without CBP knockdown, respectively. Panels A–D do not illustrate responses to the CBP knockdown because in the model, CBP only affects the expression of C/EBP, and not the dynamics of phosphorylation of PKA, ERK, CREB1 or CREB2. F, The 5-HT pulses and ISIs for the Standard Protocol (black) and Rescue Protocol (red). The Standard Protocol had uniform ISIs of 20 min, whereas the Rescue Protocol had non-uniform ISIs of 10, 10, 20, and 10 min.
In Figure 3 (panels A–E), the black traces illustrate the response of the model to the Standard Protocol with control levels of CBP, whereas the red traces illustrate the response of the model to the Rescue Protocol after knockdown of CBP. The ISIs of the Standard (black trace) and Rescue (red trace) Protocols are illustrated in panel F. Each 5-HT pulse briefly activated PKA (panel A, increased PKAC). Accumulation of active ERK (pERK) was delayed (panel B, as discussed in Zhang et al., 2012). pCREB1, which was phosphorylated by PKA, accumulated rapidly (panel C), whereas accumulation of pCREB2, which was phosphorylated by ERK, was delayed (panel D). Equations 5 and 6 describe regulation of the synthesis of C/EBP by CBP/pCREB1 and CREB2. To induce C/EBP synthesis, the stimulus-induced increases in pCREB1 and pCREB2 must overlap. During overlap, pCREB1 is available to activate c/ebp expression during the same times at which CREB2 is largely converted to pCREB2, relieving its repression of c/ebp expression. By Equation 6, both activation and relief from repression are needed for high levels of C/EBP synthesis to occur. Panel E illustrates the time course of pC/EBP, which is largely driven by increased synthesis of C/EBP. The solid black trace is with normal [CBP] and the dashed trace is with [CBP] reduced by 35%. The red traces in A – E show, in contrast, the response to the Rescue Protocol with [CBP] reduced by 35%. The non-uniform ISIs resulted in greater activation of PKA within the first 40 min, and consequently more rapid accumulation of pCREB1. Activation of ERK was similarly more rapid. Accumulation of pCREB2 coincided with a second surge in PKA activity (due to pulses 4 and 5 of the Rescue Protocol), which in turn elevated pCREB1. In this way, the Rescue Protocol acted to maximize the overlap of pCREB2 and pCREB1, the consequent synthesis of C/EBP, and the peak of pC/EBP. Thus, simulations predicted that the Rescue Protocol would restore and possibly enhance LTF despite the CBP knockdown.
Validation of the Rescue Protocol in vitro
Three groups of SN-MN co-cultures were treated with 5-HT. In group 1, SNs were injected with CBP-siRNA and treated with the Standard Protocol (CBP-siRNA + Standard, n = 7) to assess the knockdown. In group 2, SNs were injected with CBP-siRNA but treated with the Rescue Protocol (CBP-siRNA + Rescue, n = 7) to assess the effectiveness of the Rescue Protocol. In group 3, SNs were injected with Control siRNA and treated with the Standard Protocol (Con-siRNA + Standard, n = 8) to assess normal LTF. As before, siRNA was injected 96 h before 5-HT treatment. EPSPs were measured immediately before (pretest) 5-HT treatment and 24 and 48 h later (post-test) (Figure 4A). A two way ANOVA (Figure 4B) indicated significant overall differences among the groups (F(2,19) = 7.143, P = 0.005). Replicating the knockdown results of Figure 1, LTF in the CBP-siRNA + Standard group was significantly less than that in the Con-siRNA + Standard group (q2 = 3,511, P = 0.023). Importantly, LTF in the CBP-siRNA + Rescue group was significantly greater than in the CBP-siRNA + Standard group (q2 = 5.265, P = 0.004). Moreover, no significant difference in LTF was observed between the Con-siRNA + Standard and CBP-siRNA + Rescue groups (q2 = 1.927). Therefore, the Rescue Protocol compensated for the deficit of CBP and restored LTF.
DISCUSSION
This study provided evidence that a detailed understanding of biochemical signaling pathways underlying synaptic plasticity, as represented in a computational model, facilitated the development of a training protocol that rescued deficits resulting from a molecular lesion. The rescue protocol with the highest simulated efficacy was validated empirically. The model predicted numerous other rescue protocols with different ISI patterns. This potential diversity bears a similarity to a previous modeling study of neuronal activity patterns in the stomatogastric ganglion, which demonstrated that numerous combinations of parameter values can simulate a specific pyloric rhythm (Prinz et al., 2004). In the present study, the goal was to find stimulus protocols that restored or improved the function of a molecular network in the presence of a molecular lesion. We believe this methodology, demonstrated here, could, in principle, guide the development of therapeutic interventions. For example, RTS leads to serious cognitive deficits. Candidate pharmacotherapies for this disorder have been tested in CBP deficient mouse models. Phosphodiesterase inhibitors and histone deacetylase inhibitors were found to reverse deficits in long-term potentiation (LTP) and memory (Bourtchouladze et al., 2003; Alarcon et al., 2004). This strategy is promising. However, drugs often have undesirable side effects, which can limit their therapeutic value. Therefore, we believe that a complementary strategy of optimizing durations and intervals of training, based in part on simulations of signaling pathways that underlie LTP, may help to improve clinical outcomes and learning for humans affected with RTS or other molecular lesions that impair learning and memory.
A detailed understanding of the signaling pathways that underlie mechanisms of LTM and of the molecular lesions that underlie memory deficits are both prerequisites for applying this optimization strategy. A limitation is the currently incomplete understanding of these processes. Moreover, optimal training protocols will likely differ depending on the molecular lesion(s). With knowledge of the molecular lesion and a detailed understanding of the signaling pathways that underlie a form of memory for which a deficit exists, it may be possible to develop a model and evaluate many thousands of training protocols, a process that is impractical using empirical studies alone. Considering the many different vertebrate memory systems, this may seem like a daunting task. However, molecular mechanisms of memory are highly conserved. For example, mammalian LTP requires the PKA and ERK signaling pathways (English and Sweatt, 1997; Abel and Nguyen, 2008), and CREB activation and CBP function are required for at least some forms of mammalian LTP and learning (Pittenger et al., 2002; Peters et al., 2009). These pathways are conserved in Aplysia, Drosophila and mammals. Progress is being made in developing models of the molecular pathways that underlie mammalian LTP and learning (e.g., Bhalla and Iyengar, 1999; Smolen et al., 2006). Therefore, it is likely that progress in many systems will facilitate development of a predictive model describing human LTP and learning.
In addition, models might help design therapies that optimize the combination of training protocols with drug treatments. Combining these two could enhance the effectiveness of the latter while compensating, at least in part, for any limitations or undesirable side effects of drugs. These two approaches are likely to be more effective together than separately, and may have broad generality in treating individuals with learning and memory deficits.
ACKNOWLEDGMENTS
This study was supported by NIH grants NS019895 and NS073974. The authors thank M.R. Montminy (Salk Institute for Biological Sciences) for his generous gift of CBP antibody, J. Liu and E. Kartikaningrum for preparing cultures, and L. Zhou for assistance with 5-HT treatment.
REFERENCES
- Abel T, Nguyen PV. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res. 2008;169:97–115. doi: 10.1016/S0079-6123(07)00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Alberini CM, Ghirardi M, Metz R, Kandel ER. C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell. 1994;76:1–20. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M, Feramisco J, Montminy MR. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature. 1994;370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- Barco A. The Rubinstein-Taybi syndrome: modeling mental impairment in the mouse. Genes Brain Behav. 2007;6(Suppl 1):32–39. doi: 10.1111/j.1601-183X.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- Bartsch D, Casadio A, Karl KA, Serodio P, Kandel ER. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- Bartsch D, Casadio A, Karl KA, Serodio P, Kandel ER. CREB1 encodes a nuclear activator, a repressor, and a cytoplasmic modulator that form a regulatory unit critical for long-term facilitation. Cell. 1998;95:211–223. doi: 10.1016/s0092-8674(00)81752-3. [DOI] [PubMed] [Google Scholar]
- Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, Tully T. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci USA. 2003;100:10518–10522. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Cleary LJ, Lee WL, Byrne JH. Cellular correlates of long-term sensitization in Aplysia. J Neurosci. 1998;18:5988–5998. doi: 10.1523/JNEUROSCI.18-15-05988.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Frost WN, Castellucci VF, Hawkins RD, Kandel ER. Monosynaptic connections made by the sensory neurons of the gill- and siphon-withdrawal reflex in Aplysia participate in the storage of long-term memory for sensitization. Proc Natl Acad Sci USA. 1985;82:8266–8269. doi: 10.1073/pnas.82.23.8266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim J-H, Miniaci MC, Schwartz JH, Thanos D, Kandel ER. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;11:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Hagiwara H, Brindel P, Harootunian A, Armstrong R, Rivier J, Vale W, Tsien R, Montminy MR. Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol Cell Biol. 1993;13:4852–4859. doi: 10.1128/mcb.13.8.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam TM, Bourtchouladze R. Rubinstein-Taybi syndrome: molecular findings and therapeutic approaches to improve cognitive dysfunction. Cell Mol Life Sci. 2006;63:1725–1735. doi: 10.1007/s00018-005-5555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145–1155. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Liu R-Y, Floravante D, Shah S, Byrne JH. cAMP response element-binding protein 1 feedback loop is necessary for consolidation of long-term facilitation in Aplysia. J Neurosci. 2008;28:1970–1976. doi: 10.1523/JNEUROSCI.3848-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R-Y, Cleary LJ, Byrne JH. The requirement for enhanced CREB1 expression in consolidation of long-term synaptic facilitation and long-term excitability in sensory neurons of Aplysia. J Neurosci. 2011;31:6871–6879. doi: 10.1523/JNEUROSCI.5071-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean JH, Harley CW, Darby-King A, Yuan Q. pCREB in the neonate rat olfactory bulb is selectively and transiently increased by odor preference-conditioned training. Learn Mem. 1999;6:608–618. doi: 10.1101/lm.6.6.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarolo PG, Goelet P, Castellucci VF, Moran J, Kandel ER, Schacher S. A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 1986;234:1249–1251. doi: 10.1126/science.3775383. [DOI] [PubMed] [Google Scholar]
- Peters M, Bletsch M, Catapano R, Zhang X, Tully T, Bourtchouladze R. RNA interference in hippocampus demonstrates opposing roles for CREB and PP1α in contextual and temporal long-term memory. Genes Brain Behav. 2009;8:320–329. doi: 10.1111/j.1601-183X.2009.00474.x. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34:447–462. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- Prinz AA, Bucher D, Marder E. Similar network activity from disparate circuit parameters. Nat Neurosci. 2004;7:1345–1352. doi: 10.1038/nn1352. [DOI] [PubMed] [Google Scholar]
- Rouaux C, Loeffler JP, Boutillier A-L. Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem Pharmacol. 2004;68:1157–1164. doi: 10.1016/j.bcp.2004.05.035. [DOI] [PubMed] [Google Scholar]
- Smolen P, Baxter DA, Byrne JH. A model of the roles of essential kinases in the induction and expression of late long-term potentiation. Biophys J. 2006;90:2760–2775. doi: 10.1529/biophysj.105.072470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, Hegde AN, Chain DG, Schwartz JH. Activation and degradation of the transcription factor C/EBP during long-term facilitation in Aplysia. J Neurochem. 1999;73:2415–2423. doi: 10.1046/j.1471-4159.1999.0732415.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu R-Y, Heberton GA, Smolen P, Baxter DA, Cleary LJ, Byrne JH. Computational design of enhanced learning protocols. Nat Neurosci. 2012;15:294–297. doi: 10.1038/nn.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]