

Abstract
Venous neointimal hyperplasia (VNH) causes hemodialysis vascular access failure. Here we tested whether VNH formation occurs in part due to local vessel hypoxia caused by surgical trauma to the vasa vasorum of the outflow vein at the time of arteriovenous fistula placement. Selective targeting of the adventitia of the outflow vein at the time of fistula creation was performed using a lentivirus-delivered small hairpin RNA that inhibits VEGF-A expression. This resulted in significant increase in mean lumen vessel area, decreased media/adventitia area, and decreased constrictive remodeling with a significant increase in apoptosis (increase in caspase 3 activity and TUNEL staining) accompanied with decreased cellular proliferation and hypoxia inducible factor-1 alpha at the outflow vein. There was significant decrease in cells staining positive for α-smooth muscle actin (a myofibroblast marker) and VEGFR-1 expression with a decrease in MMP-2 and MMP-9. These results were confirmed in animals that were treated with humanized monoclonal antibody to VEGF-A with similar results. Since hypoxia can cause fibroblast to differentiate into myofibroblasts, we silenced VEGF-A gene expression in fibroblasts and subjected them to hypoxia. This decreased myofibroblast production, cellular proliferation, cell invasion, MMP-2 activity, and increased caspase 3. Thus, VEGF-A reduction at the time of arteriovenous fistula placement results in increased positive vascular remodeling.
Introduction
More than 400,000 patients have end-stage renal disease (ESRD) and require chronic hemodialysis in the United States 1. Vascular access through an arteriovenous fistula (AVF) is required for the optimal hemodialysis and clearance of uremic toxins. Arteriovenous fistula failure occurs frequently due to venous stenosis formation with the patency of AVFs at one year is estimated to be only 62% 2. Over a billion dollars are spent annually to maintain the function of hemodialysis AVFs and grafts 1. Angioplasty is the current treatment for treating venous stenosis formation and non-invasive treatments, which could reduce AVF stenosis, would be of advantageous to ESRD patients.
Venous neointimal hyperplasia (VNH) causes venous stenosis formation to occur in AVFs 3. When specimens removed from patients with failed AVFs are examined using histologic techniques, changes of angiogenesis are observed within the neointima and adventitia of the vessel. This is often accompanied with increased proliferation of cells which are α-smooth muscle actin (α-SMA) positive located within the neointima 4–6. There is emerging experimental data demonstrating that adventitial and medial cells which are hypothesized to be fibroblasts will convert to myofibroblasts (α-SMA positive cells) and thus contribute to the formation of VNH 7–9,10. In vitro, studies indicate that hypoxia causes increased differentiation of fibroblasts to myofibroblasts while in vivo, increased expression of hypoxia inducible factor-1 alpha (HIF-1α) has been observed in both animal models of hemodialysis AVF and graft failure as well as specimens removed from patients with hemodialysis AVF or graft failure 7, 11–14. One plausible explanation for these observations is that the vasa vasorum supplying the vessel wall become disrupted after the surgical placement of the AVF. Taken collectively, these observations suggest that hypoxia-driven angiogenic stimuli enhance matrix deposition while facilitating the conversion of adventitial fibroblasts to myofibroblasts 4. Therefore, therapy aimed at reducing hypoxia-driven VNH would be useful in reducing stenosis in AVFs. An ideal therapy to reduce venous stenosis formation would be one that can be delivered selectively to the adventitia of the vessel wall where it might target the resident fibroblasts. This directed approach to therapy might have several advantages over systemically delivered therapies because of higher local concentrations of drug due to direct delivery and reduced clearance by other organs, lower systemic side effects, and a reduced chance for non-target delivery to other vessels or organs.
Vascular endothelial growth factor-A (VEGF-A) is one of the most potent angiogenic factors and its action is mediated through its two receptors, VEGFR-1 (Flt-1) and VEGFR-2 (Flk-2). VEGF-A is important in vascular remodeling and has been shown to be involved in the pathogenesis of arterial stenosis, vein bypass grafts, and VNH associated with hemodialysis vascular access 6, 15–22. Previous work from our and other laboratories has demonstrated that there is increased expression of VEGF-A, VEGFR-1, and VEGFR-2 at the site of venous stenosis in murine and porcine models of hemodialysis AVF and graft failure. Recent work from our laboratory has shown that simvastatin treatment prior to the placement of arteriovenous fistulaplacement reduces VNH formation in a murine model of CKD with AVF by decreasing vascular endothelial growth factor-A and MMP-9 10. This study did not determine the mechanism through which VEGF-A plays a role in VNH formation as simvastatin has been implicated in reducing many different cytokines 13, 14, 23.
Experiments outlined in the present manuscript were performed in a murine model of CKD with AVF to test the hypothesis that reduction of VEGF-A gene expression by adventitial delivery to the outflow vein of the AVF at the time of placement would lead to a reduction in several important downstream matrix regulating genes such as MMP-2 and MMP-9, and reduced VNH and abnormal vascular remodeling. Gene expression for VEGF-A was reduced by adventitial delivery of a small hairpin RNA that inhibits VEGF-A expression. Gene, protein expression, and histomorphometric analyses were performed at the outflow vein after administration of anti-VEGF RNA therapy. We determined if Avastin, a humanized monoclonal antibody to VEGF-A (Bevacizumab, Genentech, San Francisco, CA) would decrease VNH formation. Finally, we investigated whether VEGF-A expression induced by hypoxia resulted in myofibroblast formation by silencing VEGF-A gene expression in fibroblasts and subjecting them to hypoxia for different time periods and subsequently determined the rates of protein expression, cell proliferation, and migration.
Results
Surgical outcomes
Two hundred and four male C57BL/6 mice weighing 25–30 grams underwent right nephrectomy and left upper pole occlusion surgery as described previously 10. One mouse was used to perform micro-CT analysis to evaluate the vasa vasorum. Seven mice died after nephrectomy, four after AVF fistula placement, and sixteen had significant arterial thickening and inflammation such that a new AV fistula could not be placed. One hundred and forty mice underwent placement of an AVF to connect the right carotid artery to the ipsilateral jugular vein 10. Next, either 1 × 106 PFU of LV-shRNA-VEGF-A (LV, n=72) or scrambled-shRNA-VEGF-A (control, C, n=68) was injected into the adventitia of the outflow vein where the stenosis forms in this model 23, 24. Animals were sacrificed for gene expression, protein, or histologic analyses at day 3 (D3), 7 (D7), 14 (D14), 21 (D21), and 28 (D28) after AVF placement. At time of sacrifice, the fistula was evaluated for patency. The fistula was considered patent if when the outflow vein was compressed, it became dilated under visual inspection. A separate group of thirty six animals underwent Avastin (5mg/kg) or IgG isotype matched antibody in equal volume amount of PBS administered I.P every other day which was started one week prior to fistula placement and continued to sacrifice. Animals were sacrificed at day 7 (D7) for gene expression of VEGF-A and 14 (D14), and 28 (D28) after AVF placement for histomorphometric analysis.
Serum BUN and creatinine after nephrectomy
In this model, we observed elevated creatinine and BUN levels similar to what is observed in the typical clinical scenario. The mean BUN and creatinine at baseline was 28 ± 5 mg/dL and 0.26 ± 0.1 mg/dL, respectively. The mean BUN increased significantly at 5, 6, and 8 weeks after nephrectomy when compared to baseline (P<0.05). In addition, there was no difference in the mean BUN and creatinine between the Avastin treated or control animals.
Adventitial transduction of LV-shRNA-VEGF-A to the outflow vein reduces gene expression of VEGF-A at days 3 and 7 and VEGF-A staining until day 14
The efficacy of reducing VEGF-A gene expression in vitro was first determined in AKR-2B (murine fibroblast cell line) cells, which were transduced with either LV-shRNA-VEGF-A (LV) or scrambled shRNA-VEGF-A (C, control). RT-PCR (Supplementary Fig. 1A) and Western blot analyses (Supplementary Fig. 1B) demonstrated greater than two fold decrease in VEGF-A expression in the LV-shRNA-VEGF-A transduced cells when compared to controls. To ascertain, if similar findings are present in vivo, experiments were designed and performed to determine the distribution of the lentivirus in the vasculature after delivery to the vessel wall.
The amount of reduction and localization of VEGF-A gene expression was determined in vivo using in situ hybridization for VEGF-A. By day 3, there was areduction of mRNA for VEGF-A (brown staining in cells) being localized to the media and adventitia and by day 7, it was localized to the media and intima (Fig. 1A, first to third column). In contrast, the vessels transduced with control shRNA showed increased mRNA expression of VEGF-A in the adventitia and media by day 3, and in the media and intima by day 7. Semiquantitative analysis of the in situ hybridization was performed and confirmed a significant reduction in the mRNA levels in the LV-shRNA-VEGF-A transduced vessels when compared to control vessels by day 7 (24.5 ± 3.3 vs. 65.3 ± 6, respectively, P<0.0001, Average reduction: 62%, Fig. 1B). Our next set of studies used RT-PCR analysis for VEGF-A on sections removed from the outflow vein at days 7, 14, and 28 after lentiviral transduction. By day 7, the mean gene expression of VEGF-A at the LV-shRNA-VEGF-A transduced vessels was significantly lower than the control vessels (1.31 ± 0.17 vs. 1.96 ± 0.15, respectively, P<0.0001, Average reduction: 44%, Fig. 1C), and by day 14 and 28, there was no difference in expression between the two groups (P=NS). Next, we determined the gene expression of VEGF-A in the Avastin treated vessels when compared to controls at day 7. By day 7, we observed a significant reduction in VEGF-A expression in the Avastin treated vessels when compared to controls 0.34 ± 0.05 vs. 0.67 ± 0.12, respectively, P<0.05, Average reduction: 49%).
Fig. 1. VEGF-A expression is reduced in LV-shRNA-VEGF-A transduced and Avastin treated vessels with decreased CD31 staining.

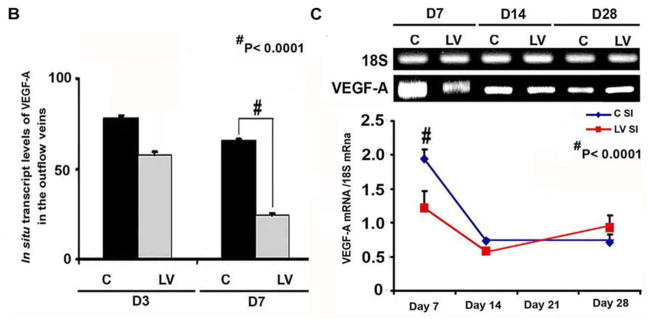

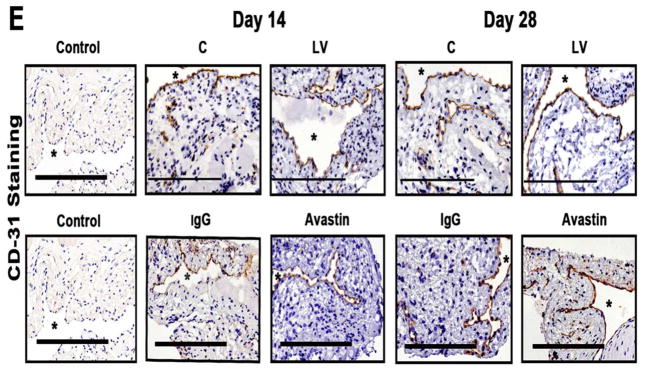
(A first to third columns) shows in situ hybridization for mRNA for VEGF-A in the LV transduced vessels when compared to C vessels with arrows on cells positive for VEGF-A mRNA expression (brown). By day 3, there was a reduction of mRNA for VEGF-A being localized to the media and adventitia and by day 7, it was localized to the media and intima. In contrast, the vessels transduced with C shRNA showed increased mRNA expression of VEGF-A in the adventitia and media by day 3, and in the media and intima by day 7. (B) is pooled data for the in situ transcript levels of VEGF-A in the outflow vein of the LV transduced vessels which is significantly reduced when compared to C vessels at day 7 (P<0.0001). (C) Upper panel shows the representative RT-PCR blots of the VEGF-A with 18S gene for loading with pooled data from the mean gene expression of VEGF-A using RT-PCR analysis. This demonstrates that there is significant reduction in the mean VEGF-A expression in the LV transduced vessels when compared to C vessels at days 7 (P<0.0001). D is representative sections from VEGF-A staining at the venous stenosis of the LV-shRNA-VEGF-A (LV) and scrambled-VEGF-A (C) transduced vessels or Avastin treated or IgG controls at day at day 14 and 28. At day 14, there is a significant reduction in the mean VEGF-A staining in the LV transduced vessels and Avastin treated vessels when compared to controls (P<0.0001). E is representative sections from CD31 staining at the venous stenosis of the LV-shRNA-VEGF-A (LV) andscrambled-VEGF-A (C) transduced vessels or Avastin treated or IgG controls at day at day 14 and 28. Cells staining brown are positive for CD-31. All are 40X. Bar is 200-μM. * Indicates the lumen. Qualitatively, in the C tissue when compared to the LV transduced vessels, there is increased CD31 staining localized to the neointima/media junction by day 14 to 28 in the control tissue. Similar results in the IgG control tissues when compared to Avastin treated vessels. Each bar shows mean ± SEM of 4–6 animals per group (B–D). Two-way ANOVA followed by Student t-test with post hoc Bonferroni’s correction was performed. Significant difference from control value was indicated by#P < 0.0001.
The decrease in expression of the protein lags behind the decrease in gene expression. We determined the expression of VEGF-A after silencing using immunohistochemistry at day 14, 21, and 28 after lentivirus silencing of VEGF-A. We observed a significant reduction in the average VEGF-A staining at day 14 in the LV-VEGF-A transduced vessels when compared to controls (22.5 ± 2.7 vs. 40.4 ± 1.4, respectively, P<0.0001, Average reduction: 44%, Fig. 1D). There was no difference at day 21 and 28 between the two groups. Finally, we determined the VEGF-A expression in the Avastin treated vessels when compared to controls and observed similar findings. By day 14, there was a significant reduction in the average VEGF-A staining in the Avastin treated vessels when compared to controls (3.8 ± 0.5 vs. 16.8 ± 1.8, respectively, P<0.0001, Average reduction: 77%, Fig. 1D)
VEGF-A is responsible for angiogenesis and it was assessed using CD31 staining. We observed qualitatively in the control vessels when compared to the LV-shRNA-VEGF-A transduced vessels, there is increased CD31 staining with microvessel formation localized to the adventitia/media junction by days 14 to 28 which was associated with a reduced luminal vessel area (Fig. 1E). Similar findings were observed in the Avastin treated vessels when compared to controls.
Collectively, these results indicate that the reduction in mRNA was reduced at adventitia and media at day 3 and by day 7, it was in the media and intima following a “top down effect.” Taken collectively, these results indicate that mRNA levels of VEGF-A can be reduced at the outflow vein using adventitial delivery of LV-shRNA-VEGF-A and the reduction in VEGF-A mRNA signal lasts for one week after delivery. Moreover, VEGF-A staining shows a reduction in the VEGF-A protein expression until 2-weeks after both lentiviral delivery and Avastin treatment. Therewas a decrease in the CD31 staining in both the LV-shRNA-VEGF-A transduced and Avastin treated vessels when compared to controls.
Adventitial transduction of LV-shRNA-VEGF-A to the outflow vein promotes positive vascular remodeling with a decrease in constrictive remodeling
We hypothesized that reducing mRNA levels of VEGF-A by adventitial transduction of LV-shRNA-VEGF-A to the outflow vein or systemic delivery of Avastin would result in positive vascular remodeling at the outflow vein (Fig. 2). We used hematoxylin and eosin staining (Fig. 2A first column) and picrosirius red staining (Fig. 2A second column) to assess the histomorphometric remodeling. A representative H and E stain is shown from day 14. At days 14–28, there was a significant increase in the average lumen area of the outflow vein of the LV-shRNA-VEGF-A transduced vessels when compared to the control vessels (P<0.001 for days 14, 21, and 28, average increase: 175–290%, Fig. 2B). In the Avastin treated vessels, we observed a significant increase in the average lumen vessel area when compared to controls (P<0.01 for days 14, average increase: 227%) with no difference by day 28.
Fig. 2. Hematoxylin and eosin (H and E) and picrosirius red staining of the LV-shRNA-VEGF-A transduced and Avastin treated vessels have increased lumen vessel area with decreased media and adventitia area and collagen expression.

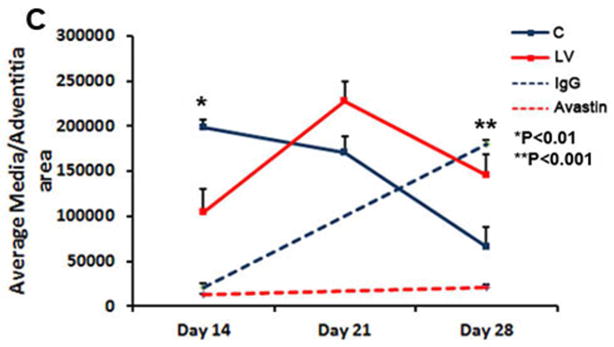
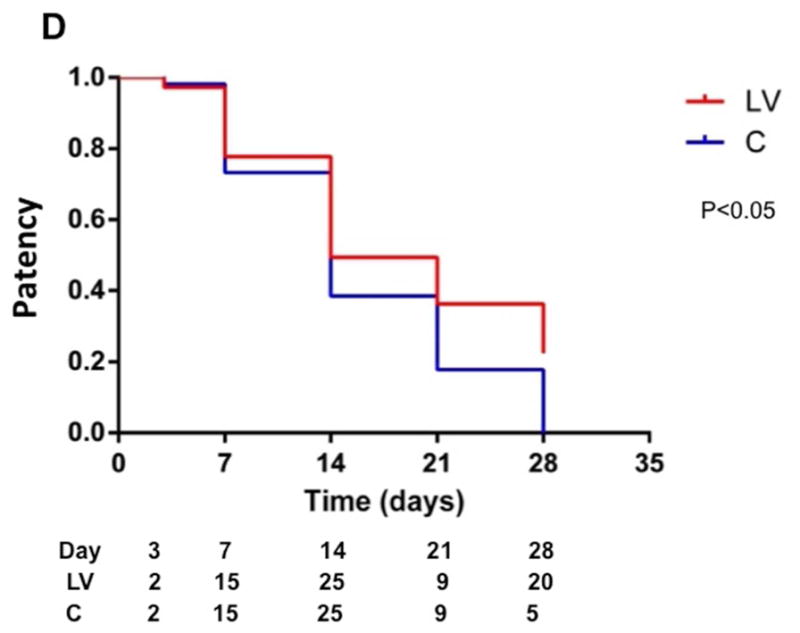
(A) First column is representative sections after hematoxylin and eosin (H and E) at the venous stenosis of the LV-shRNA-VEGF-A (LV) and scrambled-VEGF-A (C) transduced vessels or Avastin treated or IgG controls at day at day 14 showing increase in lumen vessel area. (A) Second column is representative polarized light microscopy of picrosirius red-stained venous stenosis showing decreased fibrosis (collagen fibers are bright yellow) of the LV-shRNA-VEGF-A (LV) and scrambled-VEGF-A (C) transduced vessels and Avastin treated vessels and controls. Qualitatively, there is a reduction in collagen staining by Sirius red by days 3–21. Bar is 200-μM. Pooled data for mean lumen vessel area LV and C groups and Avastintreated and control vessels are shown in B. There is a significant increase in the mean lumen vessel area in the LV transduced vessels when compared to C vessels for days 14–28 (all, P<0.001). By day 14, there was a significant increase in the mean lumen vessel area in the Avastin treated vessels when compared to IgG controls (P<0.001). Pooled data for mean media and adventitia area in LV and C groups and Avastin and control groups are shown in C. There is a significant decrease in the mean media and adventitia area in the LV transduced vessels when compared to C vessels for day 14 (P<0.01). By day 28, there is a significant reduction in the mean media and adventitia area in the Avastin treated vessels when compared to controls (P<0.001). D shows Kaplan-Meier estimates for LV transduced vessels (red) when compared to C vessels (blue). There is improved patency in the LV transduced vessels (red) when compared to C vessels (Log rank test: P<0.05). Each bar shows mean ± SEM of 4–6 animals per group (B–C). Two-way ANOVA followed by Student t-test with post hoc Bonferroni’s correction was performed. Significant difference from control value was indicated by **P < 0.001.
Adventitial remodeling has been seen in experimental models of venous neointimal hyperplasia associated with hemodialysis vascular access 7–9. We determined the area of the media and adventitia. By day 14, we observed a significant reduction in the average area of the media and adventitia of the lentivirus transduced vesselswhen compared to controls (P<0.01 for days 14, average decrease: 47%, Fig. 2C) with gradual increase by days 21 and 28. In the Avastin treated vessels when compared to controls, there was a trend to a significant decrease in the average area of the media and adventitia by day 28 (P<0.001, average decrease: 89%). Interestingly, we did not observe a difference in the average area of the neointima of either treatment (lentiviral VEGF-A or Avastin) at any time point, suggesting that the positive remodeling may be related to the favorable adventitial and medial remodeling possibly due to a decrease in the migration of fibroblasts and smooth muscle cells.
We next determined if the increase in positive vascular remodeling was due to a decrease in the collagen 1 and 3 content using picrosirius staining. Yellow color is positive for collagen 1 or 3 staining (Fig. 2A and Supplementary Fig. 2). As shown, picrosirius staining was decreased at day 3, 7, 14, and 21 with recovery by day 28. We also observed at day 3 and 7, there was reduction of the collagen expression in the adventitia and media with expression of collagen localized in the intimal area, which persisted at day 14 and 21 (Supplementary Fig. 2).
Patency of the LV-shRNA-VEGF-A and control shRNA transfected vessels at days 3, 7, 14, and 28
Patency of the fistula was determined visually by occluding the outflow vein prior to harvesting the tissue. In a patent fistula, occluding the outflow vein would allow thefistula to dilate while an occluded fistula would not dilate. This was performed several times to be certain that the fistula was occluded or patent. A Kaplan-Meier estimate was performed for the LV-shRNA-VEGF-A transduced vessels compared to controls as shown in D. This showed a significant difference in the patency between the LV-shRNA-VEGF-A transduced vessels when compared to controls (Log rank test: P<0.05).
Adventitial transduction of LV-shRNA-VEGF-A to the outflow vein increases apoptosis at day 14
Current literature suggests that VEGF-A is needed for maintaining cellular homeostasis and therefore we hypothesized that decreasing VEGF-A would result in increased apoptosis 25. Apoptosis was first assessed using TUNEL staining performed on sections removed after transduction with either LV-shRNA-VEGF-A or control shRNA (Fig. 3 upper panel). By day 14, the average TUNEL index (number of TUNEL positive cells (brown)/total number of cells X 100) at the outflow vein of the LV-shRNA-VEGF-A group was significantly higher than the control group (40.4 ± 1.4 vs. 20.6 ± 2.9, respectively, average increase: 196%, P<0.0001, Fig. 3 lower panel) with no significant differences by days 21 and 28. Similar observations were observed in the Avastin treated vessels when compared to controls. By day 14, there was a significant increase in the TUNEL index in the Avastin treated vessels when compared to controls (3.8 ± 0.5 vs. 16.8 ± 1.8, respectively, average increase: 442%, P<0.0001, Fig. 3 lower panel)
Fig. 3. Apoptosis is increased in the LV-shRNA VEGF-A transduced and Avastin treated vessels.

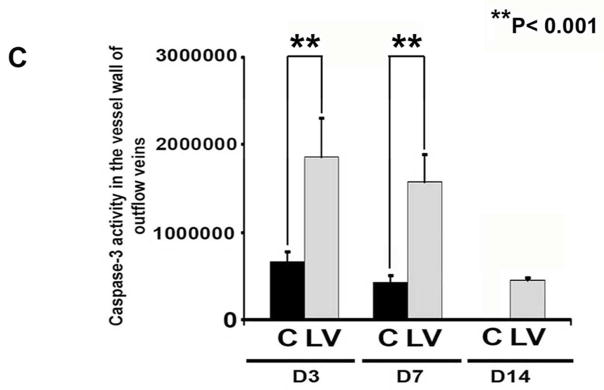
A is representative sections from TUNEL staining at the venous stenosis of the LV-shRNA-VEGF-A (LV) with scrambled-VEGF-A (C) transduced control vessels or Avastin treated with IgG controls at day 14 and 28. Nuclei staining brown are positive for TUNEL. Negative control is shown where the recombinant terminal deoxynucleotidyl transferase enzyme was omitted. All are 40X. Bar is 200-μM. * Indicates the lumen. Pooled data for LV and C transduced vessels or Avastin treated or IgG controls are shown in B. This demonstrates a significant increase in the mean TUNEL index at day 14 (P<0.0001) in the LV group when compared to C. Similar results is seen in Avastin treated vessels when compared to IgG controls at day 14 (P<0.0001). Pooled data for the LV and C transduced vessels for caspase 3 activity at days 3, 7, and 14 is shown in C. This demonstrates a significant increase in the mean caspase 3 activities at days 3 to 14 (all P<0.001). Each bar shows mean ± SEM of 3–6 animals per group (B–C). Two-way ANOVA followed by Student t-test with post hoc Bonferroni’s correction was performed. Significant difference from control value was indicated by **P < 0.001, # P< 0.0001.
Caspase 3 is an effector of apoptosis. We hypothesized that increased caspase 3 activity would be present in sections removed from outflow vein after transduction with either LV-shRNA-VEGF-A or control shRNA. By days 3–14, the average caspase 3 activity was significantly higher in the LV-shRNA-VEGF-A transduced vessels when compared to controls (average increase: 281% at day 3, 368% at day 7, and 365% at day 14, all P<0.001, Fig. 3B). Overall, these results indicate that adventitial delivery of LV-shRNA-VEGF-A results in a significant increase in caspase 3 activity with accompanying increased TUNEL index at the outflow vein suggesting that there is increased cell death in LV-shRNA-VEGF-A transduced vessels when compared to controls. Similar findings were observed in Avastin treated vessels when compared to controls.
Adventitial transduction of LV-shRNA-VEGF-A to the outflow vein decreases cellular proliferation
VEGF-A is needed for cells to proliferate and we determined cell proliferation by performing Ki-67 staining on sections from the outflow vein after transduction with either LV-shRNA-VEGF-A or control shRNA (Fig. 4A upper panel). The average Ki-67 index (number of Ki-67 positive cells (brown)/total number of cells X 100) in the LV-shRNA-VEGF-A group was significantly lower than the control group by day 14 (7.9 ± 1 vs. 24.6 ± 2.9, respectively, average reduction: 68%, P<0.01, Fig. 4A lower panel) and by day 21 (25.5 ± 1.3 vs. 37.2 ± 2.3, respectively, average reduction: 35%, P<0.01). In the Avastin treated vessels when compared to controls, similar findings were observed at day 14 (9.6 ± 0.8 vs. 31.7 ± 6.1, respectively, average reduction: 70%, P<0.01). In both experimental groups, by day 28, there was no difference between (LV-shRNA-VEGF-A or Avastin and controls). Overall, these results indicate that cellular proliferation in the scrambled group is increasing with length of time of fistula and decreasing in the LV-shRNA-VEGF-A group with reduction in both LV-shRNA-VEGF-A and Avastin treated vessels (Table 2).
Fig. 4. Cellular proliferation, MMP-2, and MMP-9 are decreased in LV-shRNA-VEGF-A transduced vessels.


A upper panel is representative sections after Ki-67 staining at the venous stenosis of the LV-shRNA-VEGF-A (LV) and scrambled shRNA (C) or Avastin treated or IgG controls at day at day 14 and 28. Nuclei staining brown are positive for Ki-67. IgG antibody staining was performed to serve as negative control. All are 40X. Bar is 200-μM. * indicates the lumen. Pooled data for the LV and C groups or Avastin treated or IgG controls are shown in lower panel. This demonstrates a significant decrease in the mean Ki-67 index in the LV transduced vessels when compared to C at day 14 (P<0.01). Similar results are seen with Avastin treated vessels when compared to control at day 14 (P<0.01). Significant difference from control value was indicated by **P < 0.001. Each bar shows mean ± SEM of 4 animals per group. Pooled data for RT- PCR analysis for MMP-2 (B) and MMP-9 (C) expression after transduction with either LV or C are shown. This demonstrates a significant reduction in the average amount of MMP-2 (B) and MMP-9 (C) in the LV transduced vessels when compared to C at day 7 (P<0.0001) with a significant increase in MMP-2 by day 28 (P<0.01). Significant difference from control value was indicated by *P<0.01 and #P < 0.0001. Each bar shows mean ± SEM of 4 animals per group (B–C). A representative zymogram for MMP-2 (D) and MMP-9 (E) is shown in the upper panel and the pooled data on the lower panels. By zymography, pro-MMP-2 expression (D) was significantly decreased in the LV transduced vessels when compared to C at day 7 (P<0.0001) and by day 14, both pro and active MMP-2 was significantly decreased (both P<0.0001). By zymography, active-MMP-9 expression (E) was significantly decreased in the LV transduced vessels when compared to C at day 14 (P<0.001). Each bar shows mean ± SEM of 3–6 animals per group (D-E). Two-way ANOVA followed by Student t-test with post hoc Bonferroni’s correction was performed. Significant difference from control value was indicated by *P < 0.01, **P < 0.001 or #P < 0.0001.
Table 2.
Synopsis of major findings
| Day 7 | Day 14 | Day 21 | Day 28 | |
|---|---|---|---|---|
| VEGF-A | ⬇ | NC | NC | |
| LVA | ⬆ | ⬆ | ⬆ | |
| CD31 | ⬇ | ⬇ | ⬇ | |
| Sirius Red | ⬇ | ⬇ | ⬇ | NC |
| TUNEL | ⬆ | NC | NC | |
| Caspase 3 | ⬆ | ⬆ | ||
| Ki-67 | ⬇ | ⬇ | NC | |
| RT-PCR MMP-2 | ⬇ | NC | ⬆ | |
| Pro-MMP-2 | ⬇ | ⬇ | ||
| Active-MMP-2 | NC | ⬇ | ||
| RT-PCR MMP-9 | ⬇ | NC | NC | |
| MMP-9 | NC | ⬇ | ||
| α-SMA | NC | ⬇ | ⬇ | |
| VEGFR1 | ⬇ | NC | NC | |
| HIF-1α | NC | ⬇ | ||
| VEGF-A staining | ⬇ | ⬇ |
LVA = lumen vessel area
NC = no change between the LV-shRNA VEGF-A transduced specimens and controls
⬆ = significant increase between the LV-shRNA VEGF-A transduced specimens and controls
⬇ = significant decrease between the LV-shRNA VEGF-A transduced specimens and controls
Adventitial transduction of LV-shRNA-VEGF-A to the outflow vein reduces expression of MMP-2 and MMP-9 at the outflow vein
Several studies have shown that there is increased expression of MMP-2 and MMP-9 in animal models of hemodialysis AVF and graft failure as well as clinical samples 7, 11–14. MMP-2 and MMP-9 gene products are thought to be responsible for cellular proliferation and cell migration resulting in VNH formation 7, 11–14. To test this hypothesis we designed a set of experiments to ascertain if reducing VEGF-A expression would lead to a reduction in MMP-2 and MMP-9 expression 26. Gene expression of MMP-2 and MMP-9 was determined by RT-PCR analysis on specimens removed from the outflow vein transduced with either LV-shRNA-VEGF-A or control shRNA. By day 7, the average gene expression of both MMP-2 and MMP-9 was significantly lower in the LV-shRNA-VEGF-A transduced vessels when compared to control shRNA (MMP-2: 2.04 ± 0.23 vs. 2.87 ± 0.25, respectively, average reduction: 29%, P<0.0001, Fig. 4B and MMP-9: 0.98 ± 0.13 vs. 2.63 ± 0.17, respectively, average reduction: 63%, P<0.0001, Fig. 4C). By day 14, there was no difference in the mean gene expression of MMP-2 and MMP-9 between both groups (P=NS), however, by day 28, there was a significant increase in MMP-2 expression at the LV-shRNA transduced vessels when compared to controls (0.96 ± 0.3 vs. 0.39 ± 0.13, respectively, average increase: 246%, P<0.01).
Because the translation of the protein lags behind the gene changes, we assessed the protein activity of MMP-2 and MMP-9 using zymography performed on the outflow vein transduced with either LV-shRNA-VEGF-A or control shRNA at day 7 and 14. By day 7, the average pro-MMP-2 activity was significantly lower in the LV-shRNA-VEGF-A transduced vessels when compared to control vessels (4309376 ± 150167 vs. 5788726 ± 12611, respectively, average reduction: 26%, P<0.0001, Fig. 4D) and by day 14, both pro and active MMP-2 was significantly lower in the LV-shRNA-VEGF-A treated vessels when compared to controls (pro-MMP-2: 3124526 ± 122611 vs. 2056144 ± 150167, respectively, average reduction: 52%, P<0.0001, active MMP-2: 2056149 ± 150167 vs. 4309376 ± 150168, respectively, average reduction: 52%, P<0.0001). By day 14, the average active MMP-9 activity was significantly lower in the LV-shRNA-VEGF-A transduced vessels when compared to control vessels (2542569.5 ± 183929 vs. 3424355 ± 150178, respectively, average reduction: 26%, P<0.001, Fig. 4E).
Adventitial transduction of LV-shRNA-VEGF-A to the outflow vein decreases α-SMA expression
The majority of the cells comprising VNH are positive for α-SMA. Outflow sections from LV-shRNA-VEGF-A and control groups were stained for α-SMA (Fig. 5A) The average SMA index (number of SMA positive cells (brown)/total number of cells X 100) at the outflow vein of the LV-shRNA-VEGF-A group transduced vessels was significantly lower than the control group by day 21 (36.9 ± 2.3 vs. 63.3 ± 5.8 respectively, average reduction: 42%, P<0.001, Fig. 5B) and day 28 (39.8 ± 6.9 vs. 65.3 ± 6.9, respectively, average reduction: 37%, P<0.01). We performed vimentin staining and observed no difference between the two groups (Supplementary Fig. 3). By day 14, there was a significant reduction in the SMA index in the Avastin treated vessels when compared to control (32.8 ± 2.1 vs. 53.1 ± 4.1 respectively, average reduction: 38%, P<0.001).
Fig. 5. Smooth muscle cell index and VEGFR-1 expression are reduced in LV-shRNA-VEGF-A transduced vessels.


(A upper panel) is representative sections after α-SMA staining at the venous stenosis of the LV-shRNA-VEGF-A (LV) and scrambled shRNA (C) and Avastin treated with control vessels at day 14 and 28. Cells staining brown are positive for α-SMA. IgG antibody staining was performed to serve as negative control. All are 40X. Bar is 200-μM. * indicates the lumen. Pooled data for the LV and C groups and Avastin treated and control vessels are shown in A lower panel. This demonstrates a significant reduction in the average α-SMA index in LV transduced vessels when compared to C by day 21 (P<0.001) and day 28 (P<0.01). There is also a significant decrease in the average α-SMA index in Avastin treated vessels when compared to controls by day 14 (P<0.001). (C) is pooled data from RT-PCR analysis for VEGFR-1 expression after transduction from the LV and C groups. A typical blot is shown in the upper panel and the pooled data on the lower panel (C). This demonstrates a significant reduction in the mean VEGFR-1 expression in the LV transduced vessels when compared to C at day 7 (P<0.001). Each bar shows mean ± SEM of 4–6 animals per group (B–C). Two-way ANOVA followed by Student t-test with post hoc Bonferroni’s correction was performed. Significant difference from control value was indicated by *P < 0.05, **P < 0.01, or #P < 0.001.
Smooth muscle cells express VEGFR-1. Smooth muscle cell expression was performed using RT-PCR analysis for VEGFR-1. The mean gene expression for VEGFR-1 by day 7 at the LV-shRNA-VEGF-A transduced vessels was significantly lower than the control group (0.69 ± 0.06 vs. 1.03 ± 0.06, respectively, average reduction: 34%, P<0.001, Fig. 5C). By days 14 and 28, there was no difference between the two groups (P=NS).
VEGF-A silencing is associated with decreased mRNA levels and immunostaining for HIF-1α
Because increased expression of HIF-1α has been observed in animal models of hemodialysis graft failure and in clinical specimens from patients with chronic graft failure, we determined the expression levels for HIF-1α in outflow vein sections transduced with either LV-shRNA-VEGF-A or control shRNA. The mean gene expression of HIF-1α (Fig. 6A) at the LV-shRNA-VEGF-A transduced vessels was significantly lower than the control vessels by day 7 (1.54 ± 0.11 vs. 2.1 ± 0.17, respectively, average reduction: 27%, P<0.001) and day 14 (0.64 ± 0.1 vs. 1.04 ± 0.06, respectively, average reduction: 39%, P<0.01).
Fig. 6. There is decreased HIF-1α expression and staining in LV-shRNA-VEGF-A transduced and Avastin treated vessels.


(A) is RT- PCR analysis for HIF-1α expression after transduction with LV and C. A typical blot is shown in the upper panel and the pooled data on the lower panel. This demonstrates a significant reduction in average HIF-1α expression in the LV transduced vessels when compared to C at days 7 (P<0.001) and 14 (P<0.01). (B) is representative sections after HIF-1α staining at the venous stenosis of the LV-shRNA-VEGF-A (LV) and control shRNA (C) and Avastin and IgG treated vessels atday 14 and 28. Brown staining cells are positive for HIF-1α. IgG antibody staining was performed to serve as negative control. All are 40X. Bar is 200-μM. * indicates the lumen. Pooled data for the LV transduced and C vessels and Avastin treated and control vessels are shown in B. This demonstrates a significant reduction in the average HIF-1α index in the LV transduced vessels when compared to C at day 14 (P<0.001) and 21 (P<0.01). There is a significant reduction in the average HIF-1α index in the LV transduced vessels when compared to C at day 14 (P<0.001) and 21 (P<0.01). There is no difference in the Avastin treated vessels compared to controls at either time point. Each bar shows mean ± SEM of 4–6 animals per group. Two-way ANOVA followed by Student t-test with post hoc Bonferroni’s correction was performed. Significant difference from control value was indicated by #P < 0.001.
We next determined the expression of HIF-1α by immunostaining (Fig. 6B). The average HIF-1α index (number of HIF-1α positive cells (brown)/total number of cellsX100) of LV-shRNA-VEGF-A transduced vessels when compared to controls at day 14 (35.3 ± 3.4 vs. 63.1 ± 6.6, respectively, average reduction: 44%, P<0.001, Fig. 6B) and day 21 (52.9 ± 2.5 vs. 74.2 ± 4.5, respectively, average reduction: 29%, P<0.01). There was no difference by day 28 in the LV-shRNA-VEGF-A transduced vessels when compared to controls. In the Avastin treated vessels when compared to controls, there was no difference in the HIF-1α index at day 14 or 28.
VEGF-A silencing in hypoxic fibroblasts reduces α-SMA production at 24 and 72 hours
Previous studies have demonstrated that HIF-1α gene expression is increased in failed hemodialysis graft specimens removed from patients and in animal models of graft failure and other studies indicate that hypoxia can cause an increase in fibroblast to myofibroblast differentiation 11, 24, 27–29. To investigate this possibility we determined whether reducing VEGF-A gene expression in fibroblasts and then subjecting them to hypoxia would cause a decrease in α-SMA production. Murine AKR-2B cells transduced with either LV-shRNA-VEGF-A (LV) or control shRNA-VEGF-A (C) were subjected to 24 (24h) or 72 hours (72h) of hypoxia. Expression of α-SMA in the cell lysate was determined using Western blot analysis. Our results indicate a significant reduction in α-SMA production at 24 hours (17.7 ± 1.5 vs. 45.9 ± 4.6, LV-shRNA-VEGF-A vs. control shRNA, respectively, Average reduction: 61%, P<0.0001) and 72 hours (17 ± 3 vs. 66.3 ± 9.2, LV-shRNA-VEGF-A vs. control shRNA, respectively, average reduction: 74%, P<0.0001) of hypoxia when compared to control (Fig. 7A). Confocal microscopy for α-SMA staining was performed on AKR-2B cells transduced with either LV-shRNA-VEGF-A or control shRNA that had been subjected to 24 or 72 hours of hypoxia demonstrated similar results (Fig. 7B). Semiquantitative analysis for cells staining positive for α-SMA (red) demonstrated a significant decrease in the intensity of the α-SMA staining in the LV-shRNA-VEGF-A transduced cells when compared to controls at 24 (132 ± 5.8 vs. 179 ± 3.4, respectively, average reduction: 27%, P<0.0001) and 72 (18.7 ± 9.42 vs. 100 ± 9.5, respectively, average reduction: 71%, P<0.0001) hours (Fig. 7C).
Fig. 7. There is decreased proliferation, invasion, α-SMA, and MMP-2 expression with increased caspase 3 activities in the LV-shRNA-VEGF-A transduced cells subjected to hypoxia.


Western blot for α-SMA after transduction LV-shRNA-VEGF-A (LV) and scrambled shRNA-VEGF-A (C) in AKR-2B Fibroblasts subjected to hypoxia at 24 (24 h) and 72 hours (72 h). A typical Western blot is shown in the upper panel and the pooled data on the lower panel (A). This demonstrates a significant reduction in the mean α-SMA expression in the LV transduced cells when compared to C at 24 (P<0.0001) and 72 hours (P<0.0001). B shows staining for α-SMA (red positive cells) at 24 and 72 hours. C is the pooled data for the average intensity of cells staining positive forα-SMA demonstrating a significant decrease in the LV transduced cells when compared to C at both 24 and 72 hours (both P<0.001). D is the pooled data for proliferation using the thymidine incorporation assay for the LV transduced cells when compared to C showing a significant decrease in proliferation for the normoxic groups at 48 and 72 hours (P<0.0001) and for 24 (P<0.05), 48 and 72 hours (P<0.0001) of hypoxia. E shows the invasion assay for LV transduced cells when compared to C for normoxia and hypoxia. F is the pooled data for invasion for the LV transduced cells when compared to C showing a significant decrease in both the normoxic (P<0.0001) and hypoxic groups (P<0.0001). G upper panel is a representative zymogram of LV transduced cells when compared to C subjected to hypoxia at 24 and 72 h and the lower panel is the pooled data demonstrating a significant reduction in both pro and active MMP-2 activity at 24 hours for LV transduced cells when compared to C (P<0.05). H is pooled caspase 3 activity in LV transduced cells when compared to C subjected to hypoxia at 24 and 72 hours showing a significant increase in the mean caspase 3 activity in the LV transduced cells when compared to C at 24 and 72 hours (both P<0.0001). Each bar shows mean ± SEM of 3–6 different experiments per group (C). Two-way ANOVA followed by Student t-test with post hoc Bonferroni’s correction was performed. Significant difference from control value was indicated by *P < 0.05, **P < 0.01, or #P < 0.001.
VEGF-A silencing in hypoxic fibroblasts reduces proliferation and invasion
We next determined if reducing VEGF-A gene expression in fibroblasts and subsequently subjecting them to hypoxia would decrease the proliferative potential (thymidine incorporation assay) of fibroblasts when compared to controls. Murine AKR-2B cells transduced with either LV-shRNA-VEGF-A or control shRNA were subjected to normoxia and hypoxia and proliferation assay was performed. This demonstrated a significant reduction in LV-shRNA-VEGF-A transduced cells when compared to control for normoxia at 48 hours (85552 ± 5428 vs. 50498 ± 3346, control shRNA vs. LV-shRNA-VEGF-A, respectively, average reduction: 41%, P<0.05, Fig. 7D) with significant reduction in hypoxia at 48 (75658 ± 2915 vs. 50243 ± 1851, control shRNA vs. LV-shRNA-VEGF-A, respectively, average reduction: 34%, P<0.0001) and 72 hours (110250 ± 1843 vs. 85895 ± 1492, control shRNA vs. LV-shRNA-VEGF-A, respectively, average reduction: 22%, P<0.0001).
Since the proliferative ability of these cells was reduced, we determined if the invasive capacity of these cells was reduced under the same conditions. Murine AKR-2B cells transduced with either LV-shRNA-VEGF-A or control shRNA were subjected to normoxia and hypoxia and a matrigel invasion assay was performed. This demonstrated a significant reduction in the invasive potential of LV-shRNA-VEGF-A transduced cells when compared to control (Normoxia: 600 ± 10 vs. 400 ± 10, control shRNA vs. LV-shRNA-VEGF-A, respectively, average reduction: 33%, P<0.05; Hypoxia: 1400 ± 10 vs. 400 ± 10, control shRNA vs. LV-shRNA-VEGF-A, respectively, average decrease: 350%, P<0.0001, Figs. 7E and F).
VEGF-A silencing in hypoxic fibroblasts decrease MMP-2 activity and increases caspase 3 activity
Because there was a decrease in proliferation and invasion in cells transduced with LV-shRNA-VEGF-A, we determined if there was a decrease in MMP-2 expression using zymography. We observed a significant decrease in the pro MMP-2 (122 ± 5.4 vs. 180 ± 11, LV-shRNA-VEGF-A vs. control shRNA, respectively, average reduction: 33%, P<0.05) and active MMP-2 activity (103 ± 24 vs. 171 ± 28, LV-shRNA-VEGF-A vs. control shRNA, respectively, average reduction: 33%, P<0.05) at 24 hours and with no difference by 72 hours (P=NS, Fig. 7G).
Since VEGF-A is involved in cellular homeostasis, we determined if reducing VEGF-A expression would result in an increase in caspase 3 activity (Fig. 7H). We observed a significant increase in caspase 3 activity in LV-shRNA-VEGF-A transduced cells when compared to controls at 24 (405725 ± 1013 vs. 292723 ± 558, respectively, average increase: 160%, P<0.0001) and 72 (254277 ± 5870 vs. 137980 ± 2810, respectively, average increase: 184%, P<0.0001) hours of hypoxia.
Discussion
It is estimated that the number of patients with ESRD who use hemodialysis as the preferred mode of therapy will double in the next decade 1. For patients to receive adequate hemodialysis, a well functioning vascular access is needed. The “Achilles heel” of hemodialysis AVF or grafts is stenosis formation caused by VNH which typically occurs in the proximal outflow vein 6. Recent studies have shown that one year primary patency of an AVF is 61% and that the 6-month patency rates after angioplasty of venous stenosis range from 0–23% 30, 31. The mechanisms underlying the formation of VNH associated with hemodialysis AVF or graft failure remain poorly understood.
VEGF-A is a multifunctional cytokine which has been shown to be involved in cell proliferation, migration, and cell death 25. Previous studies from our laboratory using experimental animal models of VNH formation in hemodialysis AVF or graft failure have demonstrated that there is increased expression of VEGF-A and VEGFR-1 at the venous stenosis when compared to the control vein (P<0.05) 11–13. Increased expression of VEGF-A has been localized to the adventitia and neointima in samples removed from patients with failed hemodialysis vascular access 6. A recent study from our laboratory demonstrated that simvastatin treatment prior to the placement of AVF in a murine model of CKD reduced VNH formation by decreasing VEGF-A and MMP-9 expression 10. In the present study, we targeted the adventitia using a LV-shRNA-VEGF-A to reduce VEGF-A gene expression, which resulted in a significant increase in luminal vessel area, significant decrease in the media/adventitia area, and a decrease in collagen expression. We further observed similar results when we used systemic delivery of Avastin. In both forms of therapy, a significant reduction in VEGF-A staining was observed as well.
The result of silencing VEGF-A had two very important effects. First VEGF-A is needed for cell survival and in the present study; the decrease in VEGF-A resulted in increased cell death as demonstrated by increased caspase 3 activity and TUNEL staining. Furthermore, we observed a decrease in cells staining positive for α-SMA with a decrease in VEGFR-1 expression. VEGFR-1 is needed for cell proliferation and migration and a recent study showed that it might influence cell death of smooth muscle cells 16. These results are consistent with reports in animal models of arterial stenosis which showed that blockade of VEGF-A using soluble VEGF receptor 1 (sFlt-1) reduces intimal hyperplasia by blocking recruitment of monocytes 21, 22. In contrast, increased expression of VEGFR-1 has been associated with increased proliferation of mesenchymal cells such as vascular smooth muscle and allied synthetic phenotypes 19.
A second important effect of reducing mRNA for VEGF-A was a decrease in cellular proliferation and reduction of Ki-67 staining in both LV-shRNA-VEGF-A transduced and Avastin treated vessels when compared to controls. This reduction in cellular proliferation was associated with reduction of several important genes responsible for cellular migration including MMP-2 and MMP-9. In patients with failed vascularaccess as well as experimental animal models of hemodialysis AVF or graft failure, increased expression of MMP-2 with adventitial localization of MMP-2 at the venous stenosis formation has been observed 11–13. These results are consistent with other studies that have shown MMP inhibition in a rat and porcine model of arteriovenous hemodialysis graft failure model has been shown to reduce VNH formation 32, 33.
Adventitial delivery of LV-shRNA-VEGF-A demonstrated a “top down” effect by showing that silencing mRNA expression for VEGF-A was localized to the adventitia and media at day 3 and the whole vessel wall by day 7 after transduction. The temporal and spatial localization of silencing VEGF-A corresponds with previous studies that have demonstrated that adventitial fibroblasts and medial cells can contribute to VNH formation 7–9. In addition, adventitial remodeling has been observed to occur in stenosis associated with arterial injury and lately and that associated with hemodialysis graft failure 7–9. We observed a significant reduction in the average area of the media and adventitia. Taken collectively, these findings lead us to believe that adventitial and medial remodeling is an important contributor to the positive vascular remodeling that was observed.
Hypoxic injury is known to accelerate conversion of fibroblast to myofibroblasts (α-SMA positive cells) and increased HIF-1α has been observed in animal models and in clinical specimens of AVF or graft failure 11, 24, 27–29. VEGF-A mediated activation of fibroblasts to myofibroblast phenotype is often accompanied by activation of matrix regulatory signaling moieties including MMP-2 and MMP-9 34. Fibroblaststransduced with either LV-shRNA-VEGF-A and subjected to hypoxia had a significant decrease in α-SMA production, decreased invasion, proliferation, MMP-2 activity and an increase in caspase 3 offering a potential cellular target for the in vivo observations. We demonstrated that mRNA for HIF-1α in the outflow vein vessels of the LV-shRNA-VEGF-A transduced vessels was significantly lower than control vessels which was confirmed with HIF-1α staining. In Avastin treated vessels, we observed no difference in HIF-1α index at either time point.
In vivo, we observed a significant reduction in both mRNA and staining for HIF-1α in LV-shRNA-VEGF-A transduced vessels when compared to controls. We speculate that the paradoxical reduction in HIF-1α associated with inhibition of VEGF maybe related to the decrease in metabolic demand for oxygen secondary to a reduction in proliferation, migration, and cell death. Taken in aggregate, these results suggest that anti VEGF-A therapy at the time of fistula placement results in positive vascular remodeling in AVFs due to multiple mechanisms that were mentioned.
Previous experimental models to study hemodialysis AVF or graft failure have used animals with normal kidney function. A recent study demonstrated that animal models with abnormal kidney function have increased VNH formation when compared to animals with normal kidney function 35. Furthermore, increased expression of VEGF-A, MMPs, and other important proteins has been found to be elevated in patients on hemodialysis and therefore would be consistent with the data obtained in our animal model 36–38.
One limitation of the current study is that the cells such as lymphocytes and macrophages may have also been transduced with LV-shRNA-VEGF-A and may also be contributing in part to the observed effects of positive vascular remodeling besides the fibroblast. Another is the choice of mouse strain used to perform the remnant kidney model. It is well know that C57BL/6 mice are resistant to glomerulosclerosis while other animals such as the BALB/c are more prone 39, 40. These genetic differences may influence the outcomes observed in the current study. The fistulas were placed in central veins, which may behave, different than those placed in peripheral veins. We used Avastin, which is a humanized antibody to VEGF-A, and there are reports that it may weakly inhibit murine VEGF-A 41.
In conclusion, our central hypothesis is that VNH formation occurs in part due to local vessel hypoxia caused by surgical trauma to the vasa vasorum supplying the outflow vein at the time of AVF placement (Supplementary Fig. 4). This hypoxia in turn leads to an increase in gene expression of VEGF-A, MMP-2 and MMP-9 resulting in activation of adventitial fibroblast which undergo conversion to myofibroblasts with increased proliferative and migratory capacity thereby resulting in VNH formation (3, 7, 11, 19, 26) (Fig. 8B). The normal vein is shown for comparison in figure 8A. The present study demonstrates that adventitial delivery of LV-shRNA-VEGF-A decreases expression of several important pro-migratory cytokines such as VEGFR-1, MMP-2, and MMP-9 (Fig. 8C). The net result is an overall decrease in cell proliferation, decrease in α-SMA positive cells, withincreased apoptosis resulting in positive vascular remodeling, decreased media/adventitia area with decreased constrictive remodeling. However, a significant decrease in the average area of the neointima was not observed. The results of these findings were extended to Avastin, which demonstrated similar findings. The clinical significance of this study is that it provides rationale for using anti-VEGF-A therapies at the time of fistula creation to reduce VNH formation.
Fig. 8. Cartoon of proposed mechanism.

Schematic showing normal vein (A), vein after AFV placement (B), and outflow vein after fistula placement with LV-shRNA-VEGF-A silencing and its different mechanisms (C).
Methods and materials
Experimental animals
Appropriate Institutional Animal Care and Use Committee approval was obtained prior to performing any procedures. The housing and handling of the animals was performed in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals revised in 2000 42. Animals were housed at 22°C temperature, 41% relative humidity, and 12-/12-hour light/dark cycles. Animals were allowed access to water and food ad libitum. Anesthesia was achieved with intraperitoneal injection of a mixture of ketamine hydrochloride (0.20 mg/g) and xylazine (0.02 mg/g) and maintained with intraperitoneal pentobarbital (20–40 mg/kg). Two hundred and four male C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) weighing 25–30 grams were used for the present study as depicted. One normal kidney function mouse was used for micro-CT analysis. Chronic renal insufficiency was created by surgical removal of the right kidney accompanied by ligation of the arterial blood supply to the upper pole of the left kidney as described previously 14. Four weeks after nephrectomy, connecting the right carotid artery to the ipsilateral jugular vein created an AVF 14, 23. Five million particle forming units (PFU) of either lentivirus-shRNA-VEGF-A (LV-shRNA-VEGF-A) or scrambled-shRNA-VEGF-A (control shRNA, non targeting empty vector) in 5-μL of PBS were injected using a 30-guage needle, into the adventitia of the proximal outflow vein at the time of AVF creation at the time of fistula creation 43. In this animal model, venous stenosis forms reproducibly at this location. Animals were sacrificed at day 3 (D3), day 7 (D7), day 14 (D14), day 21 (D21), and day 28 (D28) following AV fistula placement. Real time polymerase chain reaction (RT-PCR), protein, and histologic analyses were obtained. Serum BUN and creatinine were measured by removing blood from the tail vein at baseline (before nephrectomy), at AV-fistula creation, and at the time of sacrifice. In a separate group of animals Avastin (5mg/kg) IP or isotype matched IgG was given every other day one week before fistula placement and continued to sacrifice at either day 14 or 28. Specimens were removed for histomorphometric analysis only.
Vector constructs
The shRNA for VEGF-A was obtained from Open Biosystems, Huntsville, AL (www.openbiosystems.com, RMM4534-NM_001025250) and the lentivirus was prepared according to the manufacturer’s protocol.
Tissue harvesting
At euthanasia, all mice were anesthetized as described previously and the fistula was dissected free of the surrounding tissue. Animals were euthanized by CO2 asphyxiation and the outflow veins harvested for RT-PCR, histologic, or protein analyses. For histologic analysis, all vessels were perfusion fixed prior to removal.
Cell culture
To determine the efficacy and efficiency of lentiviral silencing on VEGF-A expression, murine fibroblasts (AKR-2B) cells were transduced with either lentivirus-shRNA-VEGF-A (LV-shRNA-VEGF-A) or scrambled-shRNA-VEGF-A and the expression analyzed using RT-PCR or Western blotting as described previously 10.
Hypoxia chamber
AKR-2B transduced with LV-shRNA-VEGF-A or control shRNA were made hypoxic for 24 or 72 hours as previously described 27, 10.
RNA isolation
The tissue was stored in RNA stabilizing reagent (Qiagen, Gaithersburg, MD) as per the manufactures guidelines. To isolate the RNA, the specimens were homogenized and total RNA isolated using RNeasy mini kit (Qiagen) 14, 23.
Real time polymerase chain reaction (RT-PCR) analysis
Expression for the gene of interest was determined using RT-PCR analysis as described previously 23, 10. Primers used are shown in table 1.
Table 1.
PCR primers used in current study
| Gene | Sequence | Amplicon Length | Cycles |
|---|---|---|---|
|
| |||
| HIF-1α | 5′ –agtgatgaaagaattact– 3′ (sense) | 2759 | 35 |
| 5′ –aataataccacttacaaca– 3′ (antisense) | |||
|
| |||
| VEGF-A | 5′ –atgaagtgatcaagttcatgg– 3′ (sense) | 360 | 35 |
| 5′ –ggatcttggacaaacaaatgc– 3′ (antisense) | |||
|
| |||
| VEGFR-1 | 5′ –tttccatttgatactcttac– 3′ (sense) | 310 | 35 |
| 5′ –tcttagttgctttaccaggg– 3′ (antisense) | |||
|
| |||
| MMP-2 | 5′ – agatcttcttcttcaaggaccggtt– 3′ (sense) | 225 | 35 |
| 5′ – ggctggtcagtggcttggggta– 3′ (antisense) | |||
|
| |||
| MMP-9 | 5′ – gtttttgatgctattgctgagatcca– 3′ (sense) | 208 | 35 |
| 5′ – cccacatttgacgtccagagaagaa3′(antisense) | |||
|
| |||
| 18S | 5′ –agctaggaataatggaatag-3′ (sense) | 150 | 19 |
| 5′-aatcaagaacgaaagtcggag- 3′(antisense) | |||
In situ hybridization for VEGF-A
In situ hybridization for VEGF-A was performed as described elsewhere 44. Briefly, the digoxigenin (DIG) labeled complementary RNA probe was made with plasmid pBS-164-VEGF (Gift from Andreas Nagy, Toronto, Canada) using DIG RNA labeling kit with T7 RNA polymerase for antisense (complementary to VEGF mRNA) probe and T3 RNA polymerase for sense (control) probe (Roche Applied Science, USA). The probe hybridization was performed as per guidelines from Roche applied science. The probe hybridization was visualized by using anti-DIG-alkaline phosphatase antibody and NBT-BCIP solution as substrate (Roche Applied Science, Indianapolis, IN).
SDS PAGE Zymography for MMP-2 and MMP-9
MMP-2 and MMP-9 protein activities were determined using zymographic analysis. This was performed on homogenates from cultured cells or outflow veins transduced with either LV-shRNA-VEGF-A or control shRNA as described previously 7, 13.
Western Blot of α-SMA
We assessed the differentiation of fibroblasts to myofibroblasts by performing Western blot analysis for α-SMA. The cultured cells were processed for Western analysis using rabbit polyclonal antibody as described previously 13.
Caspase 3 Activity
Apoptosis was assessed using an ELISA assay for caspase 3. Cellular protein was extracted from cultured cells and mouse tissue as described previously. The enzymatic activity of caspase 3 was accessed by Caspase Glo assay (G811C, Promega, Madison, WI) as described earlier 10.
Proliferation Assay
Twenty thousand AKR-2B cells transduced with LV-shRNA-VEGF-A or control shRNA were seeded in 24-well plates and cultured for 24, 48, 72 hours in DMEM medium. After 20, 44 and 68 hours 1 mCi of (3H) thymidine was added to each well; and 4 hours later, cells were washed with chilled PBS, fixed with 100% cold methanol and collected for measurement of trichloroacetic acid-precipitable for radioactivity. Experiments were repeated at least three times for each time point.
Invasion and Cell Migration Assay
Five thousand AKR-2B cells transduced with LV-shRNA-VEGF-A or control shRNA were seeded in 8-micron trans-wells that were pre-coated with low growth factor matrigel in a serum free media. The complete media was supplemented under the trans-well and incubated for 6 hours at 37°C. After 6h, trans-wells were washed with PBS and fixed with paraformaldehyde (4% v/v). Finally trans-wells were stained withbromophenol (0.1%) solution. The cells from upper well were removed with cotton tip applicators. The cells from the bottom well were counted for analysis.
Tissue processing and immunohistochemistry
Each outflow vein from each animal was embedded in paraffin length-wise so that the sections would be orthogonal to the long axis of the vessel as described previously 10. Typically, 80 to 120, 4-mm sections were obtained and the cuff used to make the anastomosis could be visualized. Every 40-μm, 2–4 sections were stained with hematoxylin and eosin, Ki-67, α-SMA, CD31, HIF-1α, and VEGF-A were performed on paraffin-embedded sections from the outflow vein after transduction with either LV-shRNA-VEGF-A or control shRNA or Avastin with control using the EnVision(DAKO, Carpinteria, CA) method with a heat-induced antigen retrieval step 7, 13. The following antibodies were used: mouse monoclonal antibody Ki-67 (DAKO, 1:400), CD31 (Abcam, Cambridge, MA; 1:400), rabbit polyclonal antibody to mouse for VEGF-A (Abcam, 1:600), rabbit polyclonal antibody to mouse for HIF-1α (Novus Biologics, 1:200), or rabbit polyclonal antibody to mouse for α-SMA (Abcam, Cambridge, MA; 1:400). IgG antibody staining was performed to serve as controls.
TUNEL staining 10
TUNEL staining was performed on paraffin-embedded sections from the outflow vein after transduction with either LV-shRNA-VEGF-A or control shRNA or Avastin or control vessels as specified by the manufacturer (DeadEnd Colorimetric tunnel assay system, G7360, Promega). Negative control is shown where the recombinant terminal deoxynucleotidyl transferase enzyme was omitted.
Picrosirius red staining 10
Picrosirius red staining was performed on unstained sections from LV-shRNA-VEGF-A and scrambled shRNA VEGF-A or Avastin treated or control vessels at different time points as described previously 10.
Morphometry and image analysis 10
Four-μm paraffin embedded sections immunostained for hematoxylin and eosin stains were viewed with an Axioplan 2 Microscope (Zeiss, Oberkochen, Germany) equipped with a Neo-Fluor × 20/0.50 objective and digitized to capture a minimum of 3090 × 3900 pixels using a Axiocam camera (Zeiss) 7, 13. Images covering one entire cross-section from each section of the outflow vein transduced with either LV-shRNA-VEGF-A or control shRNA or Avastin treated with control vessels were acquired and analyzed using KS 400 Image Analysis software (Zeiss). Formorphometric analysis used to quantify the lumen vessel area and wall vessel area, we used 6 to 10, 4-μm paraffin embedded sections removed from the outflow vein for each animal at each time point. Sections were subsequently viewed with an Axioplan 2 Microscope (Zeiss) equipped with a Neo-Fluor × 20/0.50 objective and digitized to capture at least 1030 × 1300 pixels and cell density determined along with the vessel wall and luminal vessel areas. The area was measured by tracing the vessel wall using an automated program 13. Ki-67 (brown), α-SMA positive (brown), TUNEL positive (brown), HIF-1α positive (brown) or in situ hybridization positive (brown) were highlighted, in turn, by selecting the appropriate RGB (red-green-blue) color intensity range and then counted. The Ki67, α-SMA, TUNEL, HIF-1α indices were calculated by counting the number of brown positive cells/total number of cells X 100. This was performed manually. This was repeated twice to ensure intraobserver variability was less than 10%.
Micro-CT analysis for the vasa vasorum of the jugular vein
Three-dimensional microscopic computed tomography was performed to determine the presence of the vasa vasorum in the jugular vein of a C57Bl/6 mouse as previously described 45. A representative figure is provided in Supplemental Fig. 4.
Statistical methods
Data are expressed as mean ± SEM. Multiple comparisons were performed with two-way ANOVA followed by Student t-test with post hoc Bonferroni’s correction. We employed a Kaplan-Meier survival model to assess for differences in patency between groups. A log rank test was performed. Significant difference from control value was indicated by *P < 0.05, **P < 0.001, or #P < 0.0001 for in vitro experiments and *P < 0.01, **P < 0.001, or #P < 0.0001 for in vivo experiments. For comparison of gene expression for VEGF-A in Avastin treated vessels compared to controls only, a paired Student t-test was performed with significance at P<0.05. JMP version 9 (SAS Institute Inc., Cary, N.C.) was used for statistical analyses.
Supplementary Material
Acknowledgments
We are grateful to Dr. Nagy (University of Toronto) for providing us the probe for the in situ hybridization experiments. This work was funded by a R01HL098967 (SM) from the National Heart, Lung, And Blood Institute.
Footnotes
Disclosures none
References
- 1.Collins AJ, Kasiske B, Herzog C, et al. Excerpts from the United States Renal Data System 2003 Annual Data Report: atlas of end-stage renal disease in the United States. Am J Kidney Dis. 2003;42:A5–7. [PubMed] [Google Scholar]
- 2.Rooijens PPGM, Tordoir JHM, Stijnen T, et al. Radiocephalic Wrist Arteriovenous Fistula for Hemodialysis: Meta-analysis Indicates a High Primary Failure Rate. European Journal of Vascular and Endovascular Surgery. 2004;28:583–589. doi: 10.1016/j.ejvs.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan KL, Besarab A, Bonn J, et al. Hemodynamics of failing dialysis grafts. Radiology. 1993;186:867–872. doi: 10.1148/radiology.186.3.8430200. [DOI] [PubMed] [Google Scholar]
- 4.Rekhter M, Nicholls S, Ferguson M, et al. Cell proliferation in human arteriovenous fistulas used for hemodialysis. Arterioscler Thromb. 1993;13:609–617. doi: 10.1161/01.atv.13.4.609. [DOI] [PubMed] [Google Scholar]
- 5.Swedberg SH, Brown BG, Sigley R, et al. Intimal fibromuscular hyperplasia at the venous anastomosis of PTFE grafts in hemodialysis patients. Clinical, immunocytochemical, light and electron microscopic assessment. Circulation. 1989;80:1726–1736. doi: 10.1161/01.cir.80.6.1726. [DOI] [PubMed] [Google Scholar]
- 6.Roy-Chaudhury P, Kelly BS, Miller MA, et al. Venous neointimal hyperplasia in polytetrafluoroethylene dialysis grafts. Kidney Int. 2001;59:2325–2334. doi: 10.1046/j.1523-1755.2001.00750.x. [DOI] [PubMed] [Google Scholar]
- 7.Misra S, Doherty MG, Woodrum D, et al. Adventitial remodeling with increased matrix metalloproteinase-2 activity in a porcine arteriovenous polytetrafluoroethylene grafts. Kidney Int. 2005;68:2890–2900. doi: 10.1111/j.1523-1755.2005.00763.x. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Terry CM, Blumenthal DK, et al. Cellular and morphological changes during neointimal hyperplasia development in a porcine arteriovenous graft model. Nephrol Dial Transplant. 2007;22:3139–3146. doi: 10.1093/ndt/gfm415. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Krishnamoorthy M, Banerjee R, et al. Venous stenosis in a pig arteriovenous fistula model anatomy, mechanisms and cellular phenotypes. Nephrol Dial Transplant. 2007;22:3139–3146. doi: 10.1093/ndt/gfm547. [DOI] [PubMed] [Google Scholar]
- 10.Janardhanan R, Yang B, Vohra P, et al. Simvastatin Reduces Venous Stenosis Formation in a Murine Hemodialysis Vascular Access Model. Kidney International. 2013 doi: 10.1038/ki.2013.112. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misra S, Fu A, Rajan D, et al. Expression of hypoxia inducible factor-1 alpha, macrophage migration inhibition factor, matrix metalloproteinase-2 and -9, and their inhibitors in hemodialysis grafts and arteriovenous fistulas. J Vasc Interv Radiol. 2008;19:252–259. doi: 10.1016/j.jvir.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 12.Misra S, Fu A, Anderson J, et al. The rat femoral arteriovenous fistula model: Increased expression of MMP-2 and MMP-9 at the venous stenosis. J Vasc Interv Radiol. 2008;19:587–594. doi: 10.1016/j.jvir.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Misra S, Fu AA, Puggioni A, et al. Increased shear stress with up regulation of VEGF-A and its receptors and MMP-2, MMP-9, and TIMP-1 in venous stenosis of hemodialysis grafts. Am J Physiol Heart Circ Physiol. 2008;294:H2219–2230. doi: 10.1152/ajpheart.00650.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Misra S, Shergill U, Yang B, et al. Increased expression of HIF-1α, VEGF-A and its receptors, MMP-2, TIMP-1, and ADAMTS-1 at the venous stenosis of arteriovenous fistula in a mouse model with renal insufficiency. J Vasc Interv Radiol. 2010;21:1255–61. doi: 10.1016/j.jvir.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhardwaj S, Roy H, Heikura T, et al. VEGF-A, VEGF-D and VEGF-D induced intimal hyperplasia in carotid arteries. Eur J Clin Invest. 2005;35:669–676. doi: 10.1111/j.1365-2362.2005.01555.x. [DOI] [PubMed] [Google Scholar]
- 16.Di Marco GS, Reuter S, Hillebrand U, et al. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J Am Soc Nephrol. 2009;20:2235–2245. doi: 10.1681/ASN.2009010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hutter R, Carrick FE, Valdiviezo C, et al. Vascular endothelial growth factor regulates reendothelialization and neointima formation in a mouse model of arterial injury. Circulation. 2004;110:2430–2435. doi: 10.1161/01.CIR.0000145120.37891.8A. [DOI] [PubMed] [Google Scholar]
- 18.Inoue M, Itoh H, Ueda M, et al. Vascular Endothelial Growth Factor (VEGF) Expression in Human Coronary Atherosclerotic Lesions : Possible Pathophysiological Significance of VEGF in Progression of Atherosclerosis. Circulation. 1998;98:2108–2116. doi: 10.1161/01.cir.98.20.2108. [DOI] [PubMed] [Google Scholar]
- 19.Simons M. VEGF and restenosis: the rest of the story. Arterioscler Thromb Vasc Biol. 2009;29:439–440. doi: 10.1161/ATVBAHA.109.183970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiojima I, Walsh K. The Role of Vascular Endothelial Growth Factor in Restenosis: The Controversy Continues. Circulation. 2004;110:2283–2286. doi: 10.1161/01.CIR.0000146723.23523.47. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Q, Egashira K, Hiasa K, et al. Essential role of vascular endothelial growth factor and Flt-1 signals in neointimal formation after periadventitial injury. Arterioscler Thromb Vasc Biol. 2004;24:2284–2289. doi: 10.1161/01.ATV.0000147161.42956.80. [DOI] [PubMed] [Google Scholar]
- 22.Ohtani K, Egashira K, Hiasa K-i, et al. Blockade of Vascular Endothelial Growth Factor Suppresses Experimental Restenosis After Intraluminal Injury by Inhibiting Recruitment of Monocyte Lineage Cells. Circulation. 2004;110:2444–2452. doi: 10.1161/01.CIR.0000145123.85083.66. [DOI] [PubMed] [Google Scholar]
- 23.Yang B, Shergill U, Fu AA, et al. The mouse arteriovenous fistula model. J Vasc Interv Radiol. 2009;20:946–950. doi: 10.1016/j.jvir.2009.03.044. [DOI] [PubMed] [Google Scholar]
- 24.Misra S, Shergill U, Yang B, et al. Increased expression of HIF-1alpha, VEGF-A and its receptors, MMP-2, TIMP-1, and ADAMTS-1 at the venous stenosis of arteriovenous fistula in a mouse model with renal insufficiency. J Vasc Interv Radiol. 2010;21:1255–1261. doi: 10.1016/j.jvir.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shay-Salit A, Shushy M, Wolfovitz E, et al. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. PNAS. 2002;99:9462–9467. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Keiser JA. Vascular endothelial growth factor upregulates the expression of matrix metalloproteinases in vascular smooth muscle cells: role of flt-1. Circ Res. 1998;83:832–840. doi: 10.1161/01.res.83.8.832. [DOI] [PubMed] [Google Scholar]
- 27.Misra S, Fu AA, Misra KD, et al. Hypoxia-induced phenotypic switch of fibroblasts to myofibroblasts through a matrix metalloproteinase 2/tissue inhibitor of metalloproteinase-mediated pathway: implications for venous neointimal hyperplasia in hemodialysis access. J Vasc Interv Radiol. 2010;21:896–902. doi: 10.1016/j.jvir.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das M, Burns N, Wilson SJ, et al. Hypoxia exposure induces the emergence of fibroblasts lacking replication repressor signals of PKC{zeta} in the pulmonary artery adventitia. Cardiovasc Res. 2008 doi: 10.1093/cvr/cvn014. [DOI] [PubMed] [Google Scholar]
- 29.Misra S, Fu A, Pugionni A, et al. Increased expression of Hypoxia inducible factor-1α in a porcine model of chronic renal insufficiency with arteriovenous polytetrafluoroethylene grafts. J Vasc Interv Radiol. 2008;19:260–265. doi: 10.1016/j.jvir.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 30.Misra S, Bonan R, Pflederer T, et al. BRAVO I: A pilot study of vascular brachytherapy in polytetrafluoroethylene dialysis access grafts. Kid Int. 2006;70:2006–2013. doi: 10.1038/sj.ki.5001869. [DOI] [PubMed] [Google Scholar]
- 31.Haskal ZJ, Trerotola S, Dolmatch B, et al. Stent graft versus balloon angioplasty for failing dialysis-access grafts. N Engl J Med. 2010;362:494–503. doi: 10.1056/NEJMoa0902045. [DOI] [PubMed] [Google Scholar]
- 32.Abbruzzese TA, Guzman RJ, Martin RL, et al. Matrix metalloproteinase inhibition limits arterial enlargements in a rodent arteriovenous fistula model. Surgery. 1998;124:328–334. discussion 334–325. [PubMed] [Google Scholar]
- 33.Rotmans JI, Velema E, Verhagen HJ, et al. Matrix metalloproteinase inhibition reduces intimal hyperplasia in a porcine arteriovenous-graft model. J Vasc Surg. 2004;39:432–439. doi: 10.1016/j.jvs.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Zhang M, Bianchi M, et al. Fetuin (alpha 2-HS-glycoprotein) opsonizes cationic macrophagedeactivating molecules. PNAS. 1998;95:14429–14434. doi: 10.1073/pnas.95.24.14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kokubo T, Ishikawa N, Uchida H, et al. CKD accelerates development of neointimal hyperplasia in arteriovenous fistulas. J Am Soc Nephrol. 2009;20:1236–1245. doi: 10.1681/ASN.2007121312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pawlak K, Pawlak D, Mysliwiec M. Circulating beta-chemokines and matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 system in hemodialyzed patients--role of oxidative stress. Cytokine. 2005;31:18–24. doi: 10.1016/j.cyto.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 37.Pawlak K, Pawlak D, Mysliwiec M. Possible association between circulating vascular endothelial growth factor and oxidative stress markers in hemodialysis patients. Med Sci Monit 2006. 2006 Apr;12(4):CR181–5. [PubMed] [Google Scholar]
- 38.Pawlak K, Pawlak D, Mysliwiec M. Serum matrix metalloproteinase-2 and increased oxidative stress are associated with carotid atherosclerosis in hemodialyzed patients. Atherosclerosis. 2007;190:199–204. doi: 10.1016/j.atherosclerosis.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 39.Esposito C, He CJ, Striker GE, et al. Nature and severity of the glomerular response to nephron reduction is strain-dependent in mice. Am J Pathol. 1999;154:891–897. doi: 10.1016/S0002-9440(10)65336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Wang YP, Tay YC, et al. Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 2000;58:1797–1804. doi: 10.1046/j.1523-1755.2000.00342.x. [DOI] [PubMed] [Google Scholar]
- 41.Bock F, Onderka J, Dietrich T, et al. Bevacizumab as a potent inhibitor of inflammatory corneal angiogenesis and lymphangiogenesis. Investigative ophthalmology & visual science. 2007;48:2545–2552. doi: 10.1167/iovs.06-0570. [DOI] [PubMed] [Google Scholar]
- 42.Committee on care and use of laboratory animals of the institute of laboratory animal resources. Government print office; Washington, DC: 1996. [Google Scholar]
- 43.Turunen MP, Lehtola T, Heinonen SE, et al. Efficient regulation of VEGF expression by promoter-targeted lentiviral shRNAs based on epigenetic mechanism: a novel example of epigenetherapy. Circ Res. 2009;105:604–609. doi: 10.1161/CIRCRESAHA.109.200774. [DOI] [PubMed] [Google Scholar]
- 44.Basu S, Nagy JA, Pal S, et al. The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat Med. 2001;7:569–574. doi: 10.1038/87895. [DOI] [PubMed] [Google Scholar]
- 45.Hughes D, Fu AA, Puggioni A, et al. Adventitial transplantation of blood outgrowth endothelial cells in porcine haemodialysis grafts alleviates hypoxiaand decreases neointimal proliferation through a matrix metalloproteinase-9-mediated pathway--a pilot study. Nephrol Dial Transplant. 2009;24:85–96. doi: 10.1093/ndt/gfn433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.