
Figure 1. Genomic variants.
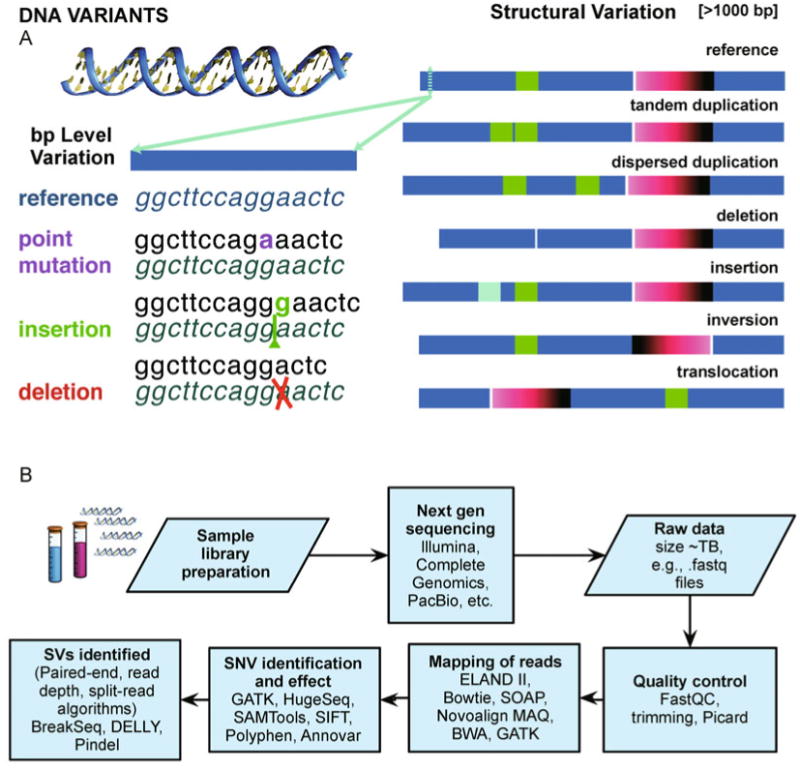
(A) Variation in the human genome. The personal genomic code can differ from the published reference genome. Basic examples of variation are shown on a single or few base variants (e.g., point mutations, insertions and deletions), or a larger scale for structural variants (>1000 bp, e.g., large insertions, deletions, inversions, tandem repeats, translocations). (B) Sample variant analysis workflow. In a genomic variant analysis, for example, after sample preparation and sequencing the raw files can be passed through quality control (e.g., using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/) and removing PCR artifacts using tools as Picard (http://picard.sourceforge.net)). Reads are mapped to the genome and variants are assessed, e.g., mapping with several algorithms, including ELAND II (Illumina), SOAP [221], MAQ and Burrows-Wheeler Aligner (BWA) [222] and Novoalign by © Novocraft Technologies (http://www.novocraft.com). Read re-alignment can be performed, e.g., using Genome Analysis Toolkit (GATK) [223], or HugeSeq [211], to call variants, including implementations with Sequence Alignment Map format Tools (SAMtools) [224], annotation using Annovar [225], SIFT [226] and Polyphen [227] for determining variant effects on proteomic translation [228]. Furthermore, using a variety of methods the structural variants can be determined. For example the paired-end mapping method considers how paired-end reads mapped to the reference to assign deletions and insertions, from reads whose mapped span is longer or shorter than the average span; inversions, from position and relative orientations of the ends of reads [39,40]. The read depth method allows the possibility to identify the proportional genomic copy number variation. In the approach of Abyzov et al. [229] the read depth considered as an image is analyzed using image processing techniques, viz. mean-shift-theory [230]. Programs such as Pindel [231] and BreakSeq [232] consider split-read analysis to determine breakpoints of insertions and deletions. DELLY [233] by Rausch et al. takes into account paired-end and split-read methods for determining structural variants. Many packages for analysis are available through the Bioconductor [234] project as implemented in the freely available R statistical analysis platform (http://www.R-project.org).