
Figure 5. Activating mutations in FGFRs in heritable and acquired disease.
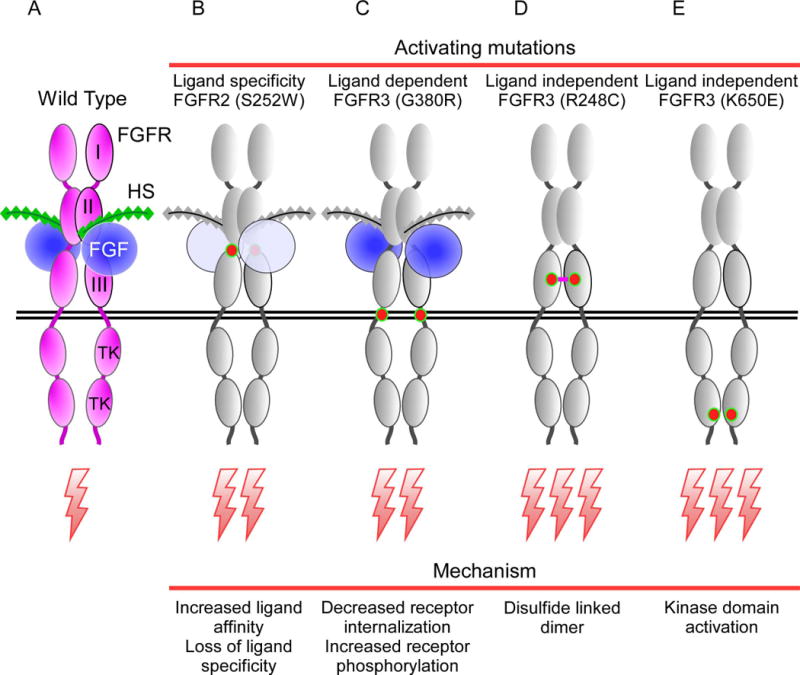
(A) Wild type FGFR-FGF-HS complex. (B) Missense mutations in the linker between immunoglobulin-like domain II and III affect the affinity and specificity of the receptor. The Apert syndrome mutation, S252W, allows FGF10 to activate the IIIc splice variant of FGFR2. (C) Missense mutations in the transmembrane domain, as seen in the G380R Achondroplasia mutation, weakly activates the receptor in a ligand dependent manner by impeding receptor internalization. (D) The strongly activating ligand independent mutation, R248C, in Thanatophoric dysplasia, type I, causes constitutively active disulfide linked receptor dimers. (E) Mutations in the tyrosine kinase domain, as seen in the K640E Thanatophoric dysplasia, type II mutation, are often ligand independent and result in receptor autophosphorylation and signaling from intracellular sites such as the endoplasmic reticulum.