

Abstract
Adoptive cell therapy (ACT) employing ex vivo generated tumor-antigen specific CD8+ T cells shows tumor efficacy when the transferred cells possess both effector and memory functions. New strategies based on understanding of mechanisms that balance CD8+ T cell differentiation towards effector and memory responses are highly desirable. Emerging information confirms a central role for antigen-induced metabolic reprogramming in CD8+ T cell differentiation and clonal expansion. The mitochondrial protein uncoupling protein 2 (UCP2) is induced by antigen stimulation of CD8+ T cells; however its role in metabolic reprogramming underlying differentiation and clonal expansion has not been reported. Employing genetic (siRNA) and pharmacologic (Genipin) approaches, we note that antigen-induced UCP2 expression reduces glycolysis, fatty acid synthesis (FAS) and production of reactive oxygen species (ROS) to balance differentiation with survival of effector CD8+ T cells. Inhibition of UCP2 promotes CD8+ T cell terminal differentiation into short-lived effector cells (CD62LloKLRG1HiIFNγHi) that undergo clonal contraction. These findings are the first to reveal a role for antigen-induced UCP2 expression in balancing CD8+ T cell differentiation and survival. Targeting UCP2 to regulate metabolic reprogramming of CD8+ T cells is an attractive new approach to augment efficacy of tumor therapy by ACT.
Keywords: Uncoupling protein 2, metabolic reprogramming, CD8+ T cell fate, CITIM 2015
Introduction
Naïve CD8+ T cells integrate cell extrinsic signals from the T cell receptor (TCR) for antigen, co-stimulatory molecules such as B7.1 (CD28) and cytokines such as IL-12 (IL-12Rb) to activate mTORC1 kinase for growth, proliferation and differentiation. This provokes a metabolic shift predominantly from fatty acid oxidation (FAO) (catabolism) to aerobic glycolysis and high oxidative phosphorylation (OXPHOS) driven by pyruvate oxidation via the tricarboxylic acid cycle (TCA) (anabolism). Notably, concurrent enhancement of glycolysis and OXPHOS generates nicotinamide adenine dinucleotide/dehydrogenase (NAD+/NADH) and reactive oxygen species (ROS) via the electron transport chain (ETC) for maintaining the redox balance in antigen-stimulated CD8+ T cells [1–4]. Although ROS are required for T cell activation [5], they can also induce cell death and thus regulation of the redox state of antigen-stimulated CD8+ T cells is essential for optimal clonal expansion [6,7]. Metabolic reprogramming from catabolism to anabolism provides macromolecules and energy for growth, proliferation and differentiation of CD8+ T cells. A coordinated balance between the various metabolic pathways is therefore required for clonal expansion of functionally competent CD8+ T cells for tumor control [1].
Following clearance of antigen, the majority of effector cells undergoes profound contraction via apoptosis and only a small population of effector cells persist to form memory. A hallmark of memory CD8+ T cells is their ability to persist and mount a rapid recall response to antigen, enabling long-term immunity [8]. Transition of effector cells to memory is associated with reversal of metabolic re-programming (similar to naïve T cells); low glycolysis and OXPHOS maintained by FAO. However, memory cells demonstrate higher spare respiratory capacity (SRC), which is a benchmark of mitochondrial health, survival and rapid antigen recall response to enable durable immunity [9]. The successful implementation of ex vivo-generated antigen-specific CD8+ T cells for ACT of cancer requires developing a better understanding of mechanisms regulating metabolic reprogramming in antigen-stimulated CD8+ T cells and exploiting these insights to produce CD8+ effector cells with memory attributes for durable tumor efficacy.
UCP proteins regulate metabolic activity in CD8+ T cells
UCP2 is a mitochondrial protein predominantly expressed by hematopoietic cells and it shares 60% sequence homology with UCP1; an isoform expressed primarily in adipose tissue to regulate thermogenesis [10]. Mitochondrial respiration couples glycolysis and ETC by utilizing pyruvate generated from glycolysis to drive TCA. Electron transport through the ETC at the inner mitochondrial membrane results in transfer of protons out of the mitochondrial matrix. This generates an electrochemical gradient that drives ATP synthesis as well as ROS, both essential for T cell differentiation and clonal expansion [5,7]. The ability of UCP family of proteins to reflux protons and reduce inner mitochondrial membrane potential can moderate ROS generation [10–12]. In addition, recent studies employing genetic and pharmacologic means have demonstrated a role for UCP2 in reducing glycolysis and pyruvate utilization for mitochondrial respiration (OXPHOS) [12]. Because metabolic reprogramming induced by antigen stimulation enhances both glycolysis (extracellular acidification rate - ECAR), as well as mitochondrial respiration (oxygen consumption rate - OCR) [9], it is conjectured that cytoplasmic glycolysis, mitochondrial TCA cycle and the ETC are coupled to generate macromolecule intermediates required for T cell responses. However, the molecular basis for the integration of metabolic pathways in the T cell cytoplasm and mitochondria remain somewhat enigmatic. Since antigen stimulation induces UCP2 expression as well as metabolic reprogramming, we predicted that UCP2 serves to regulate metabolic activity and ROS production to enhance clonal expansion of effector CD8+ T cells. Surprisingly, loss of UCP2 engenders greater immunity against intracellular challenges like T. gondii as well as L. monocytogenes infections [13–15]. This was primarily due to higher ROS generated by innate cells, but the requirement for IFNγ to mediate protection strongly implicates a role for T cells. Based on these contradictory observations we hypothesized that antigen-induced UCP2 plays a regulatory role in moderating terminal differentiation of CD8+ T cells thereby limiting attrition to promote clonal expansion of effector CD8+ cells for tumor immunity.
To date a single study has shown that anti-TCR/CD28 stimulation of CD4+ and CD8+ T cells increased UCP2 expression at 24h, which was further enhanced during re-stimulation [16]. However, that study did not characterize the function of increased UCP2 expression in antigen-stimulated T cells. Because UCP2 protein is present primarily in tissues with a high immune cell content (11) and because the UCP2 protein has a short half-life; around 30 minutes, it can be envisioned that UCP2 acts as a regulator of rapid biological responses typically ascribed to antigen-stimulated T cells [11]. To understand the role of antigen-induced UCP2 expression in metabolic regulation of CD8+ T cell responses, we employed a reductionist in vitro approach to conduct molecular and physiological investigations. Naïve CD8+ T cells obtained from TCR transgenic mice (OT-1/Rag −/−) mice were reacted with latex microspheres immobilized with MHC Class I (H-2Kb) dimers bearing 10 nM of cognate peptide (SIINFEKL) (Ag) along with 1 μg/ml of recombinant murine B7.1 (co-stimulation) in the presence of 2 ng/ml of rmIL-12 (cytokine). The stimulated CD8+ T cells were harvested at various time points and evaluated for molecular, physiologic and phenotypic characteristics by standard methodologies like flow cytometry, western blot, polymerase chain reaction (PCR) and metabolic flux analysis (Fig. 1). Our observations show that stimulation of naïve CD8+ T cells with antigen induced UCP2 (mRNA and protein) expression, optimally at 24 h. The expression of UCP2 in CD8+ T cells was regulated by the strength of antigen signal (dose-dependent; 1 to 10 nM) and required activation of mTORC1 kinase activity.
Fig. 1.
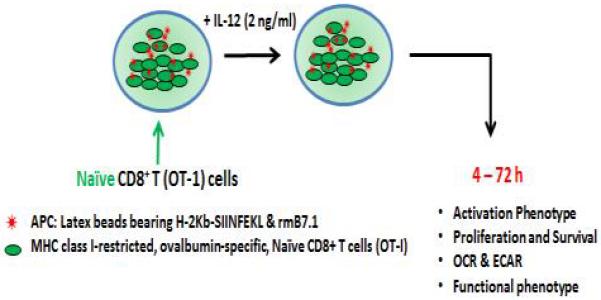
In vitro stimulation of naïve cells isolated from TCR transgenic mice (OT-1/Rag −/−). The CD8+ T cells obtained from naïve TCR transgenic mice (OT-1/Rag −/−) mice are stimulated in vitro with latex microspheres on which major histocompatibility complex (MHC) Class I (H-2Kb) dimers bearing 10 nM of cognate peptide (SIINFEKL) (Ag) are immobilized, along with 1 μg/ml of recombinant murine B7.1 (co-stimulation) and 2 ng/ml of rmIL-12 (cytokine). The cells are harvested at the indicated time points and evaluated for activation, clonal expansion, survival, metabolic status and differentiation by standard methodologies including flow cytometry, western blotting, PCR and metabolic flux analysis. UCP2 is inhibited (UCP2 siRNA or Genepin) after antigen stimulation.
UCP2 dampens mitochondrial ROS for CD8+ T cell clonal expansion
The mitochondrion is most commonly described as an energy (ATP)-producing organelle, but it is a metabolically active organelle that also generates reactive oxygen species (mROS). mROS play an essential role in antigen-induced T cell activation and proliferation [5], but the amount of mROS generated must be tightly regulated as overabundance of ROS can also result in cell death [17]. Because UCP2 is an integral mitochondrial protein that regulates ROS generation by affecting the electrochemical gradient across the inner mitochondrial membrane, we predicted a role for UCP2 in antigen-induced ROS generation by CD8+ T cells and thus conducted studies to establish its impact on CD8+ clonal expansion. Using genetic (si-UCP2) or pharmacological (Genepin) means to block UCP2 function, we studied the role of UCP2 in antigen-induced cytoplasmic (cROS) and/or mROS production by (5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester) CM-H2DCFDA and MitoSox staining respectively, followed by flow cytometry analysis. Our preliminary observations indicate that UCP2 inhibition in antigen-stimulated OT-1 cells produced a significant increase in mROS (as assessed by MitoSox fluorescence), but not cROS (CM-H2DCFDA fluorescence). The higher levels of mROS were associated with induction of expression of the apoptotic genes Bim, Bad and Bid, associated with reduced CD8+ T cell expansion. Importantly, quenching mROS with the mitochondria-targeted anti-oxidant MitoTempo (Enzo Life Sciences) reversed clonal contraction which was associated with the reversal of the apoptotic gene expression profile in CD8+ T cells. These observations are in line with previous studies that have demonstrated increased ROS production by macrophages obtained from UCP2 −/− mice [13,14]. However, they are the first set of observations that demonstrate a similar role for UCP2 in CD8+ T cells and suggest development of approaches to target UCP2-mediated ROS generation for clonal expansion of tumor antigen-specific CD8+ T cells ex vivo. As discussed above, UCP2 has also been shown to determine the fate of glycolysis-derived pyruvate for oxidation via the TCA cycle [18,19] and since TCA supplies NADH and FADH2 to support electron passage through the ETC for ROS generation, it is plausible that UCP2 controls the flux of substrate available for ETC-mediated mROS generation and thereby affects attrition of antigen-stimulated CD8+ T cells.
UCP2 balances metabolic reprogramming for CD8+ T cell differentiation
In cancer cells, UCP2 decreases glucose catabolism via glycolysis and fatty acid oxidation to support mitochondrial respiration [12]. Pyruvate generated by breakdown of glucose (glycolysis) has several metabolic fates (Fig. 2): a. It can undergo oxidation in the cytoplasm to form lactate; measured as ECAR by metabolic flux analysis, b. It is transported into the mitochondria via the pyruvate dehydrogenase (PDH) complex for conversion to Acetyl CoA, which is metabolized via the TCA cycle or c. The citrate generated by pyruvate is transported out from the mitochondria to the cytoplasm where it is cleaved by ATP-citrate lyase to produce malonyl-CoA and Palmitic acid for de novo FAS [20,21]. The metabolic state (catabolic vs anabolic) of the cell may play a critical role in determining the fate of glycolysis-generated pyruvate, which is regulated by UCP2 [19]. In support of this, recent studies have shown that UCP2 restricts glycolysis and shunts pyruvate out of mitochondria, although mitochondrial respiration (OXPHOS) is only marginally affected [22,23]. This can be explained due to increased fatty acid oxidation and/or glutaminolysis to maintain mitochondrial respiration [24]. Given this role of UCP2 in regulating mitochondrial substrate utilization, we predicted that UCP2 plays an essential role in regulating the fate of pyruvate and affects mitochondrial metabolism for CD8+ T cell differentiation. Metabolic flux analysis revealed that inhibition of antigen-induced UCP2 enhances the ECAR/OCR ratio, due to increased glycolysis, but not a corresponding increase in mitochondrial respiration via the TCA cycle. Increases in glycolysis upon inhibition of UCP2 could be due to a number of factors including increased uptake of glucose via Glut1 or expression of key enzymes regulating the glycolytic flux. Although the exact mechanisms remain unclear, future studies directed at better understanding the role of UCP2 in mitochondrial metabolic activity in CD8+ T cells are clearly warranted, as the information obtained can be used to regulate mitochondrial metabolic activities for differentiation and clonal expansion. The anabolic processes that are supported by enhanced glycolysis are essential for differentiation of CD8+ T cells and our preliminary observations indicate that UCP2 plays a pivotal role in regulating choice of glucose oxidation pathway to favor catabolic vs anabolic pathways. In agreement with the changes in UCP2-mediated metabolic activities, inhibition of UCP2 decreases expression of CD62L and increases KLRG1 and CXCR3 as well as IFNγ, Gzmb, indicative of their enhanced differentiation into effector CD8+ T cells. To the best of our knowledge, these are the first observations indicating a role for antigen-induced UCP2 in CD8+ T cell differentiation and survival (CD62LloKLRG1HiIFNγHi) (Fig. 3). Collectively, these observations suggest that loss of UCP2 promotes terminally differentiated effector CD8+ T cells that are short-lived and supports the notion that antigen-induced UCP2 plays an essential role in metabolic reprogramming to regulate CD8+ T cell differentiation and clonal expansion. Ongoing studies directed at regulating UCP2 can benefit production of CD8+ T cells with varying effector functions and survival for testing their tumor efficacy by ACT.
Fig. 2.
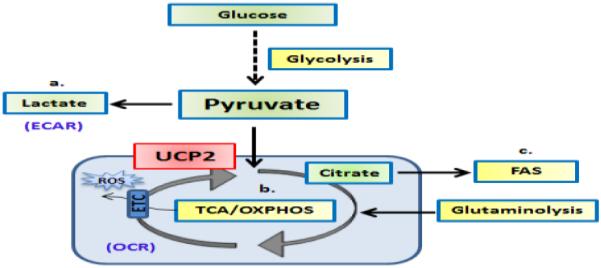
UCP2 controls the fate of pyruvate oxidation depending on the bioenergetic demands of the cell. Pyruvate generated from glycolysis can have several fates: a) It can be converted to lactate in the cytoplasm, b) It can be transported into the mitochondria for conversion to Acetyl CoA for further oxidation via the TCA cycle, or c) The citrate generated by pyruvate in the mitochondria is again transported out to the cytoplasm to produce malonyl-CoA and Palmitic acid for de novo fatty acid synthesis (FAS).
Fig. 3.
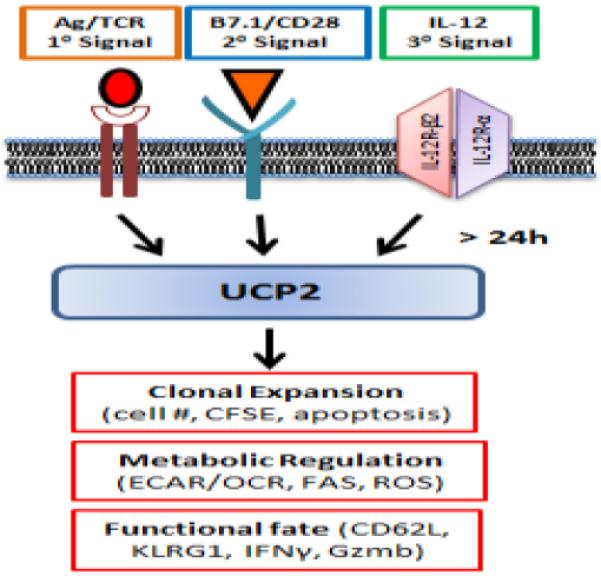
Antigen-induced UCP2 expression regulates CD8+ T cell clonal expansion, metabolic reprogramming and differentiation. Antigen stimulation of naïve CD8+ T cells with three requisite signals induces UCP2 expression by 24 h to achieve balanced metabolic reprogramming (lower ECAR/OCR), clonal expansion (lower apoptosis) of effector CD8+ T cells (CD62Llo KLRG1Hi IFNγInt).
UCP2 as a molecular target for ACT of cancer with CD8+ T cells
Recent successes of ACT in the clinic have reignited interest for developing new approaches to ex vivo generate CD8+ T cells that can mediate durable anti-tumor efficacy. Several preclinical studies in murine models of cancer have shown that memory attributes can enhance anti-tumor efficacy of effector CD8+ T cells by ACT due to increased survival [25–27]. Because the ability of effector CD8+ T cells to traffic, target and kill tumor cells is essential for anti-tumor efficacy, we contend that a judicious balance between effector and memory attributes is essential for successful ACT. Moreover, the relative capacity of effector vs memory functions demonstrated by CD8+ T cells must be tailored for the type of malignancy, i.e. indolent vs aggressive cancers. For example for indolent ovarian cancer, it may be reasonable to produce CD8+ T cells with greater memory potential for persistence and durable protection. The identification of antigen-induced UCP2 as a regulator of CD8+ T cell functional fate provides a novel means to produce CD8+ T cells with varying effector and memory attributes and deploy them for ACT of cancer. Recent successes with the use of immune checkpoint blockade with antibodies targeting (cytotoxic T-lymphocyte-associated protein 4) CTLA-4 and/or the programmed cell death protein 1 pathway (PD-1/PD-L1) have translated to their rapid approval for treatment of advanced melanoma and other solid malignancies [28]. In preliminary studies, we have noted that effector CD8+ T cells generated with UCP2 inhibition also induce higher expression of check-point molecules like CTLA4 and PD-1. Although the role for CTLA4 and/or PD-1 in ACT of cancer has not been fully characterized, the prospect of CD8+ proliferative and/or functional exhaustion upon adoptive transfer would be counterproductive [29]. Because exhaustion of tumor antigen-specific CD8+ T cells occurs via a block in glycolysis/glutaminolysis and an increase in FAO [30], we contend that combinatorial strategies exploiting the interactive roles for UCP2-mediated metabolic regulation and check-point blockade would afford greater control of CD8+ T cell differentiation and tumor efficacy by ACT.
Conclusion
Molecular mechanisms regulating metabolic reprogramming by antigen stimulation of CD8+ T cells are not completely understood. Induction of UCP2 expression upon antigen activation dampens ROS generation in T cells thereby restricting apoptosis and resulting in enhanced clonal expansion. Moreover, antigen-induced UCP2 expression is essential for regulating glycolytic flux and mitochondrial respiration that restricts CD8+ T cell terminal effector differentiation. Collectively, these observations indicate that antigen-induced UCP2 expression balances differentiation and survival for clonal expansion of CD8+ T effector cells. The unraveling of the role of antigen stimulation-induced UCP2 expression offers a novel pharmacological means to regulate functional fate of CD8+ T cells which has far-reaching implications for effective ACT for cancer.
Acknowledgements
We thank all current and past members of the Shrikant laboratory for critical review, comments and discussions. This work was supported by the NIH National Cancer Institute (NCI) (P50CA159981-01A1-3, Protul A. Shrikant).
Abbreviations
- ACT
adoptive cell therapy
- CM-H2DCFDA
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester
- cROS
cytoplasmic reactive oxygen species
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- ECAR
extracellular acidification rate
- ETC
electron transport chain
- FAO
fatty acid oxidation
- FAS
fatty acid synthesis
- mROS
mitochondrial reactive oxygen species
- NAD+/NADH
nicotinamide adenine dinucleotide/dehydrogenase
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- PCR
polymerase chain reaction
- PD-1/PD-L1
programmed cell death protein 1/ligand
- PDH
pyruvate dehydrogenase
- ROS
reactive oxygen species
- SRC
spare respiratory capacity
- TCA
tricarboxylic acid cycle
- TCR
T cell receptor
- UCP2
uncoupling protein 2
Footnotes
This paper is a Focussed Research Review based on a presentation given at the Fourth International Conference on Cancer Immunotherapy and Immunomonitoring (CITIM 2015), held in Ljubljana, Slovenia, 27th –30th April 2015. It is part of a series of Focussed Research Reviews and meeting report in Cancer Immunology, Immunotherapy.
Conflict of Interest: The authors have no financial conflicts of interest.
References
- 1.Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–1360. doi: 10.1084/jem.20151159. doi:10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annual review of cell and developmental biology. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. doi:10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 3.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. doi:10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. doi:10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, Bryce PJ, Chandel NS. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38(2):225–236. doi: 10.1016/j.immuni.2012.10.020. doi:10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med. 2002;195(1):59–70. doi: 10.1084/jem.20010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy MP, Siegel RM. Mitochondrial ROS fire up T cell activation. Immunity. 2013;38(2):201–202. doi: 10.1016/j.immuni.2013.02.005. doi:10.1016/j.immuni.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol. 2012;13(10):907–915. doi: 10.1038/ni.2386. doi:10.1038/ni.2386. [DOI] [PubMed] [Google Scholar]
- 9.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68–78. doi: 10.1016/j.immuni.2011.12.007. doi:10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Criscuolo F, Mozo J, Hurtaud C, Nubel T, Bouillaud F. UCP2, UCP3, avUCP, what do they do when proton transport is not stimulated? Possible relevance to pyruvate and glutamine metabolism. Biochim Biophys Acta. 2006;1757(9–10):1284–1291. doi: 10.1016/j.bbabio.2006.06.002. doi:10.1016/j.bbabio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Pecqueur C, Alves-Guerra C, Ricquier D, Bouillaud F. UCP2, a metabolic sensor coupling glucose oxidation to mitochondrial metabolism? IUBMB Life. 2009;61(7):762–767. doi: 10.1002/iub.188. doi:10.1002/iub.188. [DOI] [PubMed] [Google Scholar]
- 12.Pecqueur C, Bui T, Gelly C, Hauchard J, Barbot C, Bouillaud F, Ricquier D, Miroux B, Thompson CB. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 2008;22(1):9–18. doi: 10.1096/fj.07-8945com. doi:10.1096/fj.07-8945com. [DOI] [PubMed] [Google Scholar]
- 13.Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26(4):435–439. doi: 10.1038/82565. doi:10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 14.Bai Y, Onuma H, Bai X, Medvedev AV, Misukonis M, Weinberg JB, Cao W, Robidoux J, Floering LM, Daniel KW, Collins S. Persistent nuclear factor-kappa B activation in Ucp2−/− mice leads to enhanced nitric oxide and inflammatory cytokine production. J Biol Chem. 2005;280(19):19062–19069. doi: 10.1074/jbc.M500566200. doi:10.1074/jbc.M500566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kizaki T, Suzuki K, Hitomi Y, Taniguchi N, Saitoh D, Watanabe K, Onoe K, Day NK, Good RA, Ohno H. Uncoupling protein 2 plays an important role in nitric oxide production of lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci U S A. 2002;99(14):9392–9397. doi: 10.1073/pnas.142206299. doi:10.1073/pnas.142206299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rupprecht A, Brauer AU, Smorodchenko A, Goyn J, Hilse KE, Shabalina IG, Infante-Duarte C, Pohl EE. Quantification of uncoupling protein 2 reveals its main expression in immune cells and selective up-regulation during T-cell proliferation. PLoS One. 2012;7(8):e41406. doi: 10.1371/journal.pone.0041406. doi:10.1371/journal.pone.0041406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaminski M, Kiessling M, Suss D, Krammer PH, Gulow K. Novel role for mitochondria: protein kinase Ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol Cell Biol. 2007;27(10):3625–3639. doi: 10.1128/MCB.02295-06. doi:10.1128/mcb.02295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouillaud F. UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim Biophys Acta. 2009;1787(5):377–383. doi: 10.1016/j.bbabio.2009.01.003. doi:10.1016/j.bbabio.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Emre Y, Nubel T. Uncoupling protein UCP2: when mitochondrial activity meets immunity. FEBS Lett. 2010;584(8):1437–1442. doi: 10.1016/j.febslet.2010.03.014. doi:10.1016/j.febslet.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011;14(4):443–451. doi: 10.1016/j.cmet.2011.07.014. doi:10.1016/j.cmet.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joseph JW, Jensen MV, Ilkayeva O, Palmieri F, Alarcon C, Rhodes CJ, Newgard CB. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J Biol Chem. 2006;281(47):35624–35632. doi: 10.1074/jbc.M602606200. doi:10.1074/jbc.M602606200. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, Jung HJ, McCaffery JM, Kurland IJ, Reue K, Lee WN, Koehler CM, Teitell MA. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30(24):4860–4873. doi: 10.1038/emboj.2011.401. doi:10.1038/emboj.2011.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, Marmo R, Calcagnile VM, Palmieri L, Ricquier D, Paradies E, Scarcia P, Palmieri F, Bouillaud F, Fiermonte G. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci U S A. 2014;111(3):960–965. doi: 10.1073/pnas.1317400111. doi:10.1073/pnas.1317400111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42(3):406–417. doi: 10.1016/j.immuni.2015.02.002. doi:10.1016/j.immuni.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, Paulos CM, Muranski P, Restifo NP. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15(7):808–813. doi: 10.1038/nm.1982. doi:nm.1982 [pii] 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4(3):225–234. doi: 10.1038/ni889. doi:10.1038/ni889 ni889 [pii] [DOI] [PubMed] [Google Scholar]
- 27.Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, Mohney RP, Klebanoff CA, Lal A, Finkel T, Restifo NP, Gattinoni L. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479–4488. doi: 10.1172/JCI69589. doi:10.1172/jci69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi: 10.1016/j.ccell.2015.03.001. doi:10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. doi:10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, Li L, Boussiotis VA. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nature communications. 2015;6:6692. doi: 10.1038/ncomms7692. doi:10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]