

Abstract
It is well established that maladaptive innate immune responses to sterile tissue injury represent a fundamental mechanism of disease pathogenesis. In the context of cardiac ischemia reperfusion injury, neutrophils enter inflamed heart tissue, where they play an important role in potentiating tissue damage and contributing to contractile dysfunction. The precise mechanisms that govern how neutrophils are recruited to and enter the injured heart are incompletely understood. Using a model of cardiac transplant–mediated ischemia reperfusion injury and intravital 2-photon imaging of beating mouse hearts, we determined that tissue-resident CCR2+ monocyte–derived macrophages are essential mediators of neutrophil recruitment into ischemic myocardial tissue. Our studies revealed that neutrophil extravasation is mediated by a TLR9/MyD88/CXCL5 pathway. Intravital 2-photon imaging demonstrated that CXCL2 and CXCL5 play critical and nonredundant roles in guiding neutrophil adhesion and crawling, respectively. Together, these findings uncover a specific role for a tissue-resident monocyte-derived macrophage subset in sterile tissue inflammation and support the evolving concept that macrophage ontogeny is an important determinant of function. Furthermore, our results provide the framework for targeting of cell-specific signaling pathways in myocardial ischemia reperfusion injury.
Introduction
Myocardial ischemia reperfusion injury is a clinically relevant condition that contributes to morbidity in numerous patients. It can be encountered in the setting of reestablishing coronary arterial blood flow after transient interruptions, following the use of cardiopulmonary bypass for heart operations as well as after cardiac transplantation. This condition can trigger the death of cardiomyocytes, resulting in impaired contractility and loss of heart function (1). While several pathways contribute to ischemia reperfusion injury, neutrophilic infiltration into myocardial tissue is thought to play a critical role in promoting damage (2). Our previous work has shown that neutrophils infiltrate ischemic hearts immediately upon reperfusion (3). Once recruited, neutrophils can release various inflammatory and chemotactic mediators that cause cellular injury or help attract other leukocytes. Neutrophils can also plug small vessels at the sites of inflammation, thereby impairing blood flow (4). Moreover, graft-infiltrating neutrophils can augment alloimmune responses after heart transplantation (5). Their contribution to pathogenesis has been demonstrated by experimental studies where inhibiting neutrophilic adherence to endothelial cells protects against myocardial ischemia reperfusion injury and where their depletion promotes the survival of heart transplants (5, 6). A better mechanistic understanding of neutrophil trafficking into inflamed heart tissue could lead to the development of new therapeutics.
Recruitment of neutrophils from the vasculature into inflamed tissues is a multistep cascade that involves their interaction with endothelial cells. Sequential phases of this process include rolling, adhesion, crawling and transendothelial migration. These steps are regulated by secretion of inflammatory cytokines and chemokines, expression of selectins and adhesion molecules, and cytoskeletal remodeling of endothelial cells. In the case of sterile noninfectious inflammation such as ischemia reperfusion injury cell death, release of damage-associated molecular patterns and activation of innate immune pathways are early upstream events that are thought to trigger inflammatory responses (7). It is well established that the molecular cues that regulate neutrophil recruitment differ between various tissues, and it remains largely unknown which cells and pathways regulate this process in the heart (8, 9).
To define the upstream signals that orchestrate neutrophil trafficking during myocardial ischemia reperfusion injury, we took advantage of a mouse cardiac transplantation model. An essential advantage of this system is the ability to resolve the roles of resident (donor) and recruited (recipient) immune cell populations. Using our recently developed technique to intravitally image leukocyte trafficking in beating mouse hearts, we have uncovered a central mechanism that regulates neutrophil entry into injured myocardial tissue (3). We show that tissue-resident CCR2+ macrophages play a critical role in promoting the extravasation of neutrophils into hearts through TLR9/MyD88-mediated production of the chemokines CXCL2 and CXCL5.
Results
Heart-resident CCR2+ monocytes and monocyte-derived macrophages are critical to promote extravasation of neutrophils into cardiac tissue during ischemia reperfusion injury
Previous work from our group has demonstrated that adult mouse hearts harbor distinct macrophage populations (10, 11). We first set out to assess whether heart-resident monocytes and macrophages play a role in neutrophil recruitment after syngeneic heart transplantation, a model of sterile inflammation. We first treated B6 WT donor mice with clodronate liposomes 24 hours prior to organ harvest, a regimen that is known to deplete macrophages, and transplanted their hearts into syngeneic LysM-GFP neutrophil reporter hosts (12, 13). We initiated intravital 2-photon microscopy 2 hours after reperfusion. We have previously demonstrated that LysM-GFP nearly exclusively labels neutrophils in the donor heart at this time point (3). Following transplantation, recipient neutrophils are recruited to large coronary veins where they slow down, adhere to the vessel wall, crawl, and transmigrate into the myocardium when donor mice are treated with PBS liposomes (Figure 1A and Supplemental Video 1; supplemental material available online with this article; doi:10.1172/jci.insight.87315DS1) (3). Neutrophils form large clusters in the vessels, as well as in the graft tissue.
Figure 1. Intravital 2-photon imaging reveals impaired neutrophil tra_cking in heart grafts that lack monocyte-derived macrophages.

(A) Control PBS liposome–treated heart graft with neutrophil (green) arrest inside blood vessels (blood vessels appear red after injection of quantum dots), intravascular cluster formation, and extravasation (see Supplemental Video 1; n = 4 mice). Neutrophil tra_cking in hearts derived from (B) donors that received treatment with clodronate liposomes prior to organ harvest (see Supplemental Video 2; n = 4 mice). Relative time is displayed in hrs:min:sec. Scale bars: 50 μm. (C) Percentage of neutrophils that extravasated during imaging period was significantly higher in hearts derived from control PBS liposome–treated donors compared with heart grafts harvested from clodronate liposome–treated mice. (D) Neutrophil rolling velocities were comparable in coronary veins of cardiac grafts derived from control PBS liposome–treated and clodronate liposome–treated mice. (E) Intraluminal crawling velocities were significantly lower in hearts harvested from clodronate liposome–treated WT compared with PBS liposome–treated mice. *P < 0.05; **P < 0.01 (t test). Data in C, D, and E are derived from 4 mice for each experimental group. For D and E, symbols represent averages obtained from individual mice with over 30 neutrophils examined per mouse, horizontal bars denote means, and error bars denote ±SEM.
To investigate the requirement for resident cardiac macrophages in neutrophil recruitment, we treated donor animals with clondronate liposomes. We have previously demonstrated that a single dose of clondronate liposomes is sufficient to deplete resident cardiac macrophages (9). When donor animals are treated with clodronate liposomes prior to heart harvest, neutrophils slow down and adhere to the vessel wall but fail to extravasate efficiently (Figure 1, B and C, and Supplemental Video 2). While their rolling velocities are comparable with control conditions, neutrophils crawl significantly slower after treatment with clodronate liposomes (Figures 1, D and E).
To define relevant donor-resident macrophage populations, we next transplanted B6 CD45.2+ WT hearts into congenic B6 CD45.1+ WT recipients and assessed donor macrophages 2 hours after reperfusion. We extended our previous observations and identified donor-derived MHCIIloCCR2− and MHCIIhiCCR2− macrophages and MHCIIhiCCR2+ macrophages, as well as MHCIIloCCR2+ monocytes, in hearts 2 hours after reperfusion (Figure 2, A and B) (11). Since our previous work has shown that CCR2+ monocyte–derived macrophages expand in the injured heart and express higher levels of inflammatory cytokines and chemokines than embryonic-derived CCR2− macrophages, we hypothesized that tissue-resident CCR2+ macrophages play a role in regulating neutrophil recruitment during ischemia reperfusion injury (11). To this end, we depleted cardiac CCR2+ monocytes and macrophages by treating B6 CCR2-DTR donor mice with diphtheria toxin (DT) prior to organ harvest (Supplemental Figure 1) and performed intravital 2-photon microscopy after transplantation of these hearts into syngeneic LysM-GFP recipients (Figure 1C and Supplemental Video 3). Compared with control DT–treated WT donor hearts that contain CCR2+ monocytes and macrophages, neutrophil recruitment into the cardiac tissue was markedly impaired when graft-resident donor CCR2+ monocytes and macrophages were absent (Figures 2, C and D, and Supplemental Videos 3 and 4). When donor hearts lack CCR2+ monocytes and macrophages, neutrophils roll with similar velocities as controls and adhere to the vessel wall; however, they crawl significantly slower, and their extravasation is impaired — similar to our findings in clodronate liposome–treated donor hearts (Figure 2, E–G). Thus, heart-resident CCR2+ monocytes and macrophages are critical to facilitate extravasation of neutrophils into injured myocardial tissue.
Figure 2. Intravital 2-photon imaging reveals impaired neutrophil tra_cking in heart grafts that lack CCR2+ monocytes and monocyte-derived macrophages.

(A) Dot plots depict flow cytometric analysis of macrophage/monocyte populations in B6 CD45.2+ heart grafts 2 hours after transplantation into congenic B6 CD45.1+ hosts. CCR2−MHCIIhi, CCR2−MHCIIlo, and CCR2+MHCIIhi macrophages and CCR2+MHCIIlo monocytes are present. Macrophage/monocyte populations are gated on donor hematopoietic (CD45.2+CD45.1−) and myeloid (CD11b+CD64+) cells. Plots are representative of 4 independent experiments with comparable results. (B) Quantification of donor monocyte/macrophage populations represented as percentage of CD45.2+CD11b+CD64+ cells based on gating depicted in A. Neutrophil (green) tra_cking in (C) control diphtheria toxin–treated (DT-treated) WT (see Supplemental Video 3; n = 4 mice)or (D) DT-treated CCR2-DTR (DT receptor) heart grafts (see Supplemental Video 4; n = 5 mice). Blood vessels appear red after injection of quantum dots (n = 4 mice). (E) Percentage of neutrophils that extravasated during imaging period was significantly higher in hearts derived from DT-treated WT donors compared with heart grafts harvested from DT-treated CCR2-DTR mice. (F) Neutrophil rolling velocities were comparable in coronary veins of cardiac grafts derived from DT-treated WT and DT-treated CCR2-DTR mice. (G) Intraluminal crawling velocities were significantly lower in hearts harvested from DT-treated CCR2-DTR compared with DT-treated WT mice. **P < 0.01 (t test). Data in E, F, and G are derived from 4 mice receiving hearts from DT-treated WT mice and 5 recipients of cardiac grafts derived from DT-treated CCR2-DTR donors. For F and G, symbols represent averages obtained from individual mice with over 30 neutrophils examined per mouse, horizontal bars denote means, and error bars denote ±SEM.
MyD88 signaling in heart-resident macrophages facilitates extravasation of neutrophils into heart tissue during ischemia reperfusion injury
Macrophages express pattern-recognition receptors and can respond to damage-associated molecular patterns. Previous studies have shown that several endogenous ligands that are released during sterile inflammation signal through MyD88 (7). Therefore, we next investigated whether MyD88 expression in donor hearts regulates the recruitment of neutrophils during ischemia reperfusion injury. We transplanted B6 MyD88–deficient hearts into syngeneic LysM-GFP recipients and imaged the grafts shortly after reperfusion. Similar to our observations in grafts that lack CCR2+ cells, neutrophils adhere to the vessel wall of coronary veins in MyD88-deficient hearts but do not enter the myocardial tissue efficiently during our imaging period (Figure 3, A–C, and Supplemental Videos 5 and 6). We further found that the rolling velocities of neutrophils were significantly higher and the crawling velocities significantly lower in MyD88-deficient compared with WT grafts (Figure 3, D and E). Notably, selectins and adhesion molecules that are known to regulate rolling and crawling of neutrophils were expressed at significantly lower levels in vascular endothelial cells in MyD88-deficient compared with WT hearts (Supplemental Figure 2). As various cells in hearts have the capacity to signal via MyD88, we next wanted to restrict MyD88 deficiency to heart-resident macrophages. For this purpose, we crossed B6 MyD88–floxed mice with B6 LysM-Cre animals. While both macrophages and neutrophils express lysozyme M, we have previously shown that resting hearts contain virtually no neutrophils (3). Intravital imaging of LysM-GFP hearts 2 hours after transplantation into syngeneic WT hosts demonstrated numerous spindle-shaped interstitial macrophages, many of which were adjacent to blood vessels (Supplemental Figure 3 and Supplemental Video 7). Neutrophil trafficking into hearts that lack MyD88 expression in lysozyme M–expressing cells was virtually identical to our observations in cardiac grafts that globally lack MyD88. Compared with control MyD88-floxed mice, rolling velocities were increased, crawling velocities were reduced, and extravasation of neutrophils was impaired when lysozyme M–expressing cells did not express MyD88 (Figures 3, F–J, and Supplemental Videos 8 and 9).
Figure 3. Intravital 2-photon imaging reveals impaired neutrophil extravasation into heart grafts that lack MyD88 expression.

Neutrophil tra_cking in (A) WT (see Supplemental Video 5; n = 4 mice) and (B) MyD88-deficient hearts (see Supplemental Video 6; n = 4 mice). Scale bars: 50 μm. (C) Percentage of neutrophils that entered myocardial tissue during imaging period was significantly lower when heart grafts were deficient in MyD88 compared with WT cardiac grafts. (D) Neutrophil rolling velocities were significantly higher in coronary veins of cardiac grafts that lacked expression of MyD88 when compared with WT hearts. (E) Intraluminal crawling velocities of neutrophils were significantly lower when hearts lacked expression of MyD88 when compared with WT cardiac grafts. Neutrophil tra_cking in (F) cardiac grafts derived from control MyD88-floxed mice (see Supplemental Video 8; n = 4 mice) and (G) mice that lack MyD88 selectively in Lysozyme M+ cells (see Supplemental Video 9; n = 4 mice). Relative time is displayed in hrs:min:sec. Scale bars: 50 μm. (H) Percentage of neutrophils that entered myocardial tissue during imaging period was significantly lower when heart grafts were deficient in MyD88 in Lysozyme M–expressing cells compared with control MyD88-floxed hearts. (I) Neutrophil rolling velocities were significantly higher in coronary veins of cardiac grafts that lacked expression of MyD88 in Lysozyme M–expressing cells compared with control MyD88-floxed hearts. (J) Intraluminal crawling velocities of neutrophils were significantly lower when hearts lacked expression of MyD88 in Lysozyme M–expressing cells compared with control MyD88-floxed hearts. *P < 0.05; **P < 0.01; ***P < 0.001 (t test). Data in C–E and H–J are derived from 4 mice for each experimental group. C–E and H–J represent averages obtained from individual mice with over 30 neutrophils examined per mouse, horizontal bars denote means, and error bars denote ±SEM.
We next wanted to assess how MyD88 signaling in macrophage subsets impacts the expression of neutrophil chemokines. For this purpose, we sorted donor CCR2− and monocyte-derived CCR2+ macrophages from B6 CD45.2+ WT or B6 CD45.2+ MyD88–deficient heart grafts 2 hours after transplantation into congenic B6 CD45.1+ recipients. In WT grafts, CXCL2 and CXCL5, but not CXCL1, were expressed at significantly higher levels in CCR2+ compared with CCR2− macrophages (Figure 4 and Supplemental Figure 4). The expression levels of CXCL2 and CXCL5, but not CXCL1, were significantly lower in CCR2+ macrophages that lacked MyD88. Thus, MyD88 signaling in CCR2+ macrophages regulates the expression of neutrophil chemokines, and MyD88 signaling in heart-resident macrophages is critical to promote the entry of neutrophils into injured myocardial tissue.
Figure 4. Expression levels of neutrophil chemoattractants are decreased in donor macrophage populations in MyD88-deficient heart grafts.
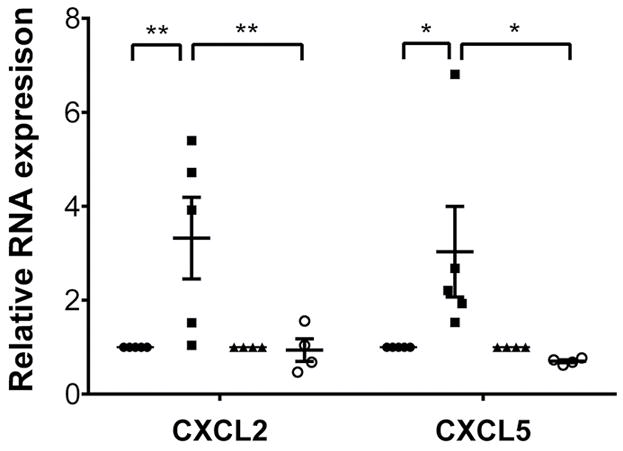
Expression levels of CXCL2 and CXCL5 in sorted donor-derived (CD45.2+CD45.1−CD11b+CD64+) CCR2+ and CCR2− macrophages examined 2 hours after transplantation of B6 CD45.2+ WT or B6 CD45.2+ MyD88–deficient hearts into congenic B6 CD45.1+ recipients. Results were normalized to 18s RNA and compared with the CCR2− macrophage population in each experimental group. *P < 0.05; **P < 0.01 (2-way ANOVA). Filled circles, WT CCR2− macrophages; filled squares, WT CCR2+ macrophages and monocyte-derived macrophages; filled triangles, MyD88-deficient CCR2− macrophages; open circles, MyD88-deficient CCR2+ macrophages and monocyte-derived macrophages. Graph represents at least 4 separate experiments per group where cells from 2 hearts were pooled for each experiment. Horizontal bars denote means, and error bars denote ±SEM.
Donor TLR9 expression regulates expression of neutrophil chemokines and neutrophil transendothelial migration
Having established that MyD88 signaling in donor macrophages regulates the entry of neutrophils into inflamed myocardial tissue, we next set out to examine which pathways upstream of MyD88 contribute to this process. Endogenous ligands released during sterile inflammation have been shown to signal via several pattern-recognition receptors that are upstream of MyD88. We evaluated the expression of the putative upstream receptors TLR2, TLR4, TLR7, and TLR9 in cardiac macrophages (Supplemental Figure 5). TLR2 and TLR9 were expressed at very high levels in both CCR2+ and CCR2− macrophages, while expression levels of TLR4 and TLR7 were comparatively low. To assess whether TLR2 expression in the donor regulates neutrophil trafficking, we transplanted B6 TLR2–deficient hearts into syngeneic LysM-GFP recipients. Under these conditions, neutrophil behavior was comparable to our observations in WT grafts (Figure 5, A and D–F, and Supplemental Video 10). We next transplanted B6 hearts that do not express TIRAP, an adaptor protein that is critical for TLR2 and TLR4 signaling via MyD88, into syngeneic LysM-GFP hosts. Neutrophil trafficking behavior in these hearts was similar to that in WT and TLR2-deficient grafts (Figures 5, B and D–F, and Supplemental Video 11). In stark contrast to TLR2 KO and TIRAP KO hearts, but similar to MyD88-deficient grafts, neutrophils roll faster, crawl slower, and fail to extravasate efficiently when donor hearts lack expression of TLR9 (Figures 5, C–F, and Supplemental Video 12). Furthermore, similar to our findings in MyD88-deficient cardiac macrophages, expression levels of CXCL2 and CXCL5, but not CXCL1, were significantly decreased in TLR9-deficient compared with WT CCR2+ macrophages (Figure 6 and Supplemental Figure 4). Together, these data indicate that TLR9 signaling in heart-resident cells promotes the expression of neutrophil chemokines and is a critical regulator of neutrophil extravasation.
Figure 5. Intravital 2-photon imaging demonstrates impaired neutrophil extravasation into TLR9-deficient heart grafts.

Neutrophil tra_cking in (A) TLR2-deficient (see Supplemental Video 10; n = 4 mice), (B) toll-interleukin 1 receptor domain containing adaptor protein–deficient (TIRAP-deficient; see Supplemental Video 11; n = 4 mice), and (C) TLR9-deficient (see Supplemental Video 12; n = 4 mice) cardiac grafts. White arrows in A and B point to sites of neutrophil extravasation. Relative time is displayed in hrs:min:sec. Scale bars: 50 μm. (D) Percentage of neutrophils that entered myocardial tissue during imaging period was comparable between WT, TLR2-deficient, and TIRAP-deficient hearts. However, the percentage of extravasated neutrophils was significantly lower in TLR9-deficient than WT cardiac grafts. (E) Neutrophil rolling velocities were significantly higher in coronary veins of TLR9-deficient cardiac grafts when compared with WT hearts. Rolling velocities in TLR2- or TIRAP-deficient hearts were comparable with WT cardiac grafts. (F) Intraluminal crawling velocities of neutrophils did not di_er significantly between WT, TLR2-deficient, and TIRAP-deficient hearts but were significantly lower than WT conditions when hearts lacked expression of TLR9. *P < 0.05; **P < 0.01 (one-way ANOVA). Data in D, E, and F are derived from 4 mice for each experimental group. For D, E, and F, symbols represent averages obtained from individual mice with over 30 neutrophils examined per mouse, horizontal bars denote means, and error bars denote ±SEM.
Figure 6. Expression levels of neutrophil chemoattractants are decreased in donor macrophage populations in TLR9-deficient heart grafts.
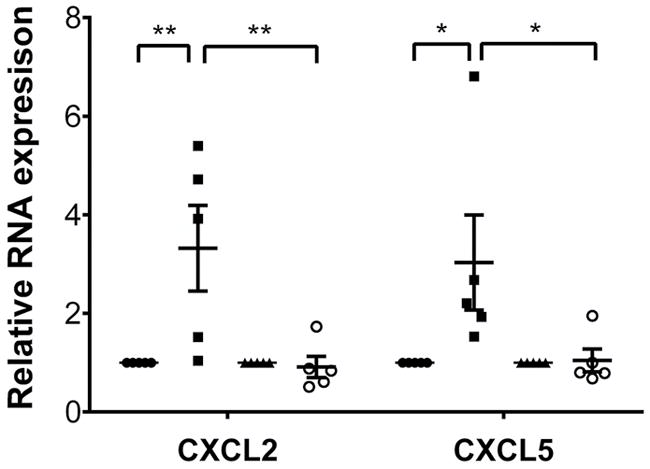
Expression levels of CXCL2 and CXCL5 in sorted donor-derived (CD45.2+CD45.1−CD11b+CD64+) CCR2+ and CCR2− macrophages examined 2 hours after transplantation of B6 CD45.2+ WT or B6 CD45.2+ MyD88–deficient hearts into congenic B6 CD45.1+ recipients. Results were normalized to 18s RNA and compared with the CCR2− macrophage population in each experimental group. *P < 0.05; **P < 0.01 (2-way ANOVA). Filled circles, WT CCR2− macrophages; filled squares, WT CCR2+ macrophages and monocyte-derived macrophages; filled triangles, TLR9-deficient CCR2− macrophages; open circles, TLR9-deficient CCR2+ macrophages and monocyte-derived macrophages. Graph represents at least 4 separate experiments per group where cells from 2 hearts were pooled for each experiment. Horizontal bars denote means, and error bars denote ±SEM.
Donor CXCL5 expression is critical to facilitate the extravasation of neutrophils into heart tissue during ischemia reperfusion injury
Having shown that TLR9 and MyD88 regulate the expression of CXCL2 and CXCL5 by CCR2+ cardiac macrophages, we next set out to evaluate these chemokines functionally. To this end, we isolated CCR2+ macrophages from B6 hearts and assessed their capacity to induce neutrophil chemotaxis in vitro. We found that inhibiting either CXCL2 or CXCL5 significantly attenuated neutrophil migration (Supplemental Figure 6). Blocking both CXCL2 and CXCL5 did not result in further inhibition of neutrophil chemotaxis. To evaluate the role of these chemokines in vivo, we first administered a CXCL2-blocking antibody to B6 LysM–GFP recipients of syngeneic WT heart grafts. Strikingly, when compared with mice that received isotype control antibodies, CXCL2 antibody–treated mice demonstrated increased rolling velocity and failure to adhere, crawl, and extravasate across the vascular endothelium into the graft tissue (Figures 7, A–E, and Supplemental Videos 13 and 14). Transplantation of CXCL5-deficient B6 hearts into syngeneic LysM-GFP hosts resulted in increased neutrophil rolling velocity, reduced crawling velocity, and diminished extravasation into myocardial tissue when compared with WT donor grafts — a trafficking behavior comparable with our observations in MyD88-deficient and TLR9-deficient grafts (Figures 7, F–J, and Supplemental Video 15). Thus, CXCL5 expression by donor cells plays a critical and nonredundant role in facilitating the migration of neutrophils into injured heart tissue.
Figure 7. CXCL2 and CXCL5 regulate neutrophil chemotaxis and tra_cking of neutrophils in heart grafts.

Neutrophil tra_cking in WT hearts after administration of (A) isotype control antibodies (see Supplemental Video 13; n = 4 mice) or (B) CXCL2-neutralizing antibody (see Supplemental Video 14; n = 4 mice). Relative time is displayed in hrs:min:sec. Scale bars: 50 μm. (C) Percentage of neutrophils that entered myocardial tissue during imaging period was significantly lower after neutralization of CXCL2 compared with mice that received isotype control antibodies. (D) Neutrophil rolling velocities were significantly higher in coronary veins of cardiac grafts after neutralization of CXCL2 compared with mice that received isotype control antibodies. (E) Due to a small number of adherent neutrophils, crawling velocities could not be evaluated when CXCL2 was neutralized. Neutrophil tra_cking in (F) WT and (G) CXCL5-deficient cardiac grafts (see Supplemental Videos 5 and 15, respectively; Isotype control, n = 4; CXCL2-neutralizing antibody, n = 4; CXCL5-deficient heart grafts, n = 5. Relative time is displayed in hrs:min:sec. Scale bars: 50 μm. (H) Percentage of neutrophils that entered myocardial tissue during imaging period was significantly lower when cells in the hearts were unable to produce CXCL5 compared with WT cardiac grafts. (I) Neutrophil rolling velocities were significantly higher in coronary veins of cardiac grafts when hearts lacked expression of CXCL5 compared with WT cardiac grafts. (J) Intraluminal crawling velocities of neutrophils were significantly lower when hearts lacked expression of CXCL5 compared with WT cardiac grafts. *P < 0.05; **P < 0.01; ***P < 0.001 (t test). Data in C–E and H–J are derived from 4–5 mice for each experimental group as indicated above. For C–E and H–J, symbols represent averages obtained from individual mice with over 30 neutrophils examined per mouse, horizontal bars denote means, and error bars denote ±SEM.
Discussion
Our work has identified several facets of neutrophil recruitment into inflamed heart tissue (Figure 8). We discovered that CCR2+ heart macrophages regulate the extravasation of neutrophils into inflamed myocardial tissue through MyD88. Interestingly, expression of TLR9, an innate immune receptor upstream of MyD88, and secretion of CXCL5 by heart-resident cells are critical to facilitate the transendothelial migration of neutrophils into injured hearts.
Figure 8. Model of CCR2+ macrophage–mediated neutrophil extravasation.
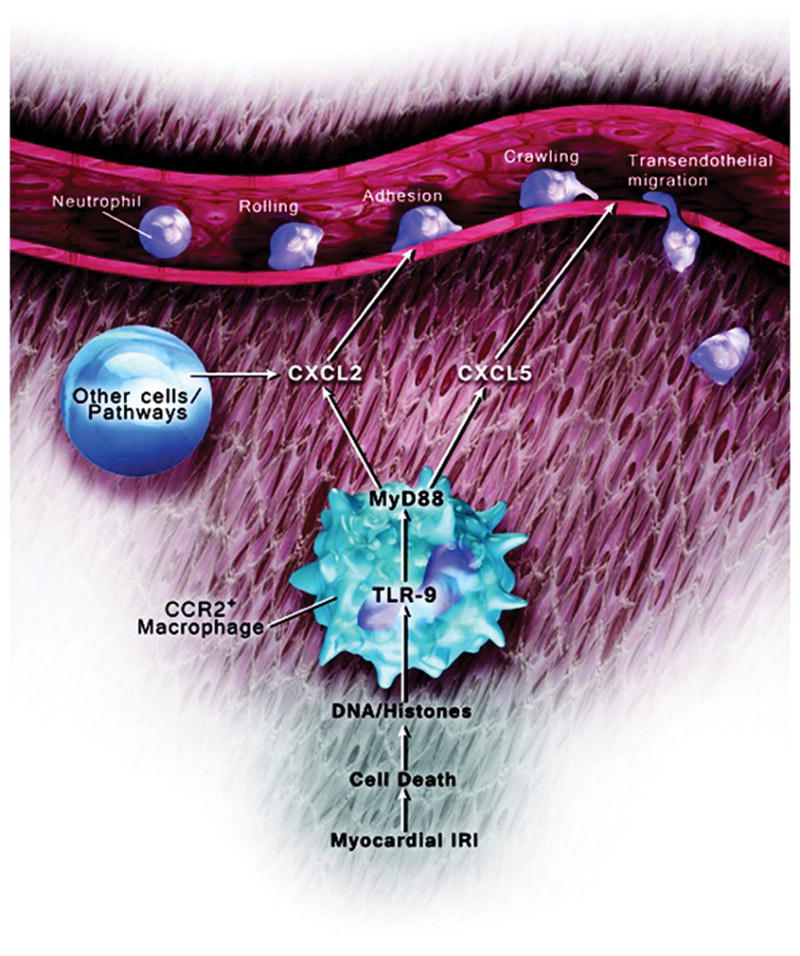
Tissue-resident CCR2+ macrophages regulate neutrophil tra_cking in injured hearts through TLR9/MyD88 pathway–dependent production of CXCL2 and CXCL5.
Previous studies have suggested that innate immune cells and pathways contribute to sterile myocardial inflammation, at least in part through regulating neutrophil infiltration. Such conclusions were drawn mainly based on associations with tissue levels of markers of neutrophil recruitment or activation such as myeloperoxidase. Intravital imaging has yielded important insights into the regulation of inflammatory responses to pathogens as well as sterile insults in a variety of tissues (14). Such imaging approaches were instrumental in defining individual steps of the leukocyte recruitment cascade (15). Importantly, such techniques have revealed that the cellular and molecular pathways that regulate leukocyte trafficking during inflammation differ between various tissues and organs (8). Our recently described approach to image leukocyte migratory behavior in beating hearts in real time has provided us with an experimental platform to dissect which cells and pathways regulate neutrophil recruitment into the inflamed myocardium (3).
In many organs, tissue-resident macrophages are sentinel cells that can promote or help resolve inflammation. For example, work from our group and others has demonstrated that alveolar macrophages contribute to pulmonary injury in the setting of sterile inflammation (16, 17). Alternatively, microglia plays a protective role after brain injury by modulating synapses (18). These seemingly divergent responses may be explained by the heterogeneity of macrophages with regard to their origin and polarization. Recent work by our group has shed some light on ontogeny and function of cardiac macrophages. At baseline, the adult murine heart contains CCR2− macrophages that are largely derived from embryonic progenitors, as well as CCR2+ monocytes and monocyte-derived macrophages (10). Transcriptional profiling and gene expression analyses indicated that CCR2+ macrophages play a role in generating inflammatory responses (10, 11). While previous studies have examined how the recruitment of monocyte subsets into the injured myocardium regulates inflammatory responses, it remains poorly understood how heart-resident macrophages impact sterile inflammation (19). Using an ischemia reperfusion injury model after syngeneic heart transplantation, we uncovered that CCR2+ cardiac macrophages play a critical role in promoting neutrophil extravasation into the injured myocardial tissue. This experimental model has several advantages, including its clinical relevance. Ischemia reperfusion injury after heart transplantation can be associated with immediate graft dysfunction and failure, which remain the leading causes of death within the first month (20). In addition, the use of a cardiac transplant model allowed us to deplete cell populations in the heart or selectively control the expression of innate immune molecules in heart-resident cells in a setting where the infiltrating leukocytes were WT. This is an important point, as it is often difficult to dissect the in vivo contributions of gene defects in cells that reside in the target organs versus circulatory leukocytes when using myocardial inflammatory models in hosts that globally lack the cells or pathways of interest (21).
Endogenous triggers of sterile inflammation, commonly referred to as damage-associated molecular patterns, can be of intra- or extracellular origin. A large number of the endogenous substances that are released during ischemia reperfusion injury signal through MyD88 pathways (22). Therefore, it is not surprising that inhibition of MyD88 attenuates ischemia reperfusion injury in a variety of experimental models. For example, systemic MyD88 deficiency protects against myocardial ischemia reperfusion injury that is induced through transient ligation of the left anterior descending coronary artery (23). Compared with WT animals, neutrophilic heart infiltration — assessed by IHC and tissue myeloperoxidase activity — is attenuated, and left ventricular function is preserved in MyD88-deficient mice. Of note, no differences were detected between WT and MyD88-deficient hearts when examining isolated cardiac preparations that were perfused with a solution free of leukocytes. Based on these observations and in apparent contrast to our findings, the authors speculated that MyD88 expression in circulating cells such as neutrophils mediates myocardial ischemia reperfusion injury. To our knowledge, how MyD88 deficiency impacts dynamic neutrophil trafficking during inflammation has not been previously described. Our observations do not exclude the possibility that MyD88 expression in neutrophils or other circulatory inflammatory cells plays a role in regulating neutrophilic migratory behavior, as our in vivo experiments examined WT recipients. However, we show that MyD88 deficiency in heart-resident macrophages alone is sufficient to impair the extravasation of neutrophils into the inflamed myocardial tissue. Consistent with previous work by many groups, we show that MyD88 regulates the expression of inflammatory mediators in hematopoietic cells. Interestingly, neutrophils roll faster and crawl slower when the donor hearts lack MyD88, which we speculate is related to alterations in expression levels of selectins and adhesion molecules. However, inflammatory pathways other than MyD88 are sufficient to mediate the adhesion of neutrophils to the surface of vascular endothelial cells in coronary veins.
Most studies examining MyD88-dependent pathways in myocardial injury have focused on the activation of TLR2 and TLR4 (24, 25). Examples of endogenous substances that have been shown to be released during ischemia-induced myocardial cell death and trigger inflammatory responses through these pathways include the DNA-binding protein high mobility group box 1 (HMGB1) and heat shock proteins (26). Our observation that neutrophil trafficking is comparable in TIRAP-deficient and WT heart grafts indicated that a pathway other than TLR2 or TLR4 accounted for the impairment in neutrophil extravasation when hearts do not express MyD88. Surprisingly, we found that TLR9 in ischemic hearts regulates the expression of neutrophil chemoattractants and is critical to promote the extravasation of neutrophils. TLR9 is an intracellular protein that signals via MyD88 and recognizes bacterial unmethylated CpG-DNA, as well as endogenous DNA and histones (27). In models of bacterial peritonitis sepsis and pressure overload induced by thoracic transverse aortic constriction, TLR9 expression mediates cardiac inflammation and heart dysfunction (28, 29). TLR9 expression in neutrophils can regulate their expression of inflammatory mediators, thereby contributing to hepatic ischemia reperfusion injury (30). Interestingly, in apparent contrast to the observations in our heart model, TLR9 expression did not have an impact on neutrophil recruitment to ischemic livers. We speculate that different tissues may release unique combinations of damage-associated molecular patterns, resulting in activation of varying receptors including TLRs. Our data suggest that dying cardiomyocytes release nuclear and mitochondrial DNA and histones molecules that activate tissue-resident CCR2+ monocyte-derived macrophages via TLR9/MyD88 signaling axis that results in the production of neutrophil chemoattractants.
Whether neutrophil chemokines that interact with CXCR2 — such as CXCL1, CXCL2, and CXCL5 — are redundant or play distinct roles in various tissues during specific types of inflammation is not well understood. Our experiments have demonstrated conclusively that CXCL5 production by heart-resident cells is critical to mediate the extravasation of neutrophils into inflamed myocardial tissue. While our findings support the conclusion that CCR2+ cardiac macrophages produce CXCL2 and CXCL5 in a MyD88-dependent manner, these data do not exclude the possibility that CCR2+ macrophages may also interact with other cardiac resident cell types, such as endothelial cells, to regulate the production of neutrophil chemokines from additional sources. Interestingly, similar to our observations in MyD88- and TLR9-deficient grafts and our previous report describing blockade of Mac-1, the crawling velocity of neutrophils is decreased when hearts cannot produce CXCL5 (3). This may be related to the previously reported capacity of CXCL5 to promote expression of ICAM-1 on endothelial cells, possibly through secretion of proinflammatory cytokines (31, 32). In the absence of CXCL5, other ELR+ CXC chemokines such as CXCL1 or CXCL2 cannot rescue the neutrophil migratory defect.
This finding is different from our previous observations in lungs where CXCL2 was sufficient to mediate transendothelial migration of neutrophils during ischemia reperfusion injury (16). Our findings extend previous observations that have shown differences in tissue distribution of various neutrophil chemokines during inflammation and have suggested a role for CXCL5 in cardiac inflammation. Interestingly, after injection of LPS into mice, expression of CXCL5 is strongest in cardiac tissue, while expression of CXCL2 is highest in the lung (33). Furthermore, administration of anti-CXCL5 antibodies resulted in a substantial reduction in cardiac tissue myeloperoxidase levels in a rat myocardial ischemia reperfusion injury model induced by transient coronary ligation (34). Contrary to our observations, however, neutrophil influx into lungs infected with bacteria was enhanced when mice were deficient in CXCL5 (35). This was due to absent CXCL5 binding to proteins expressed on the surface of erythrocytes that have a high affinity for chemokines, thereby leading to higher gradients for CXCL1 and CXCL2 between tissue and blood attracting neutrophils into the infected lungs. It remains to be defined whether differences in target organs, the nature and severity of the inflammatory insult, the cells producing CXCL5, or local processing of CXCL5 contribute to dissimilarities in neutrophil recruitment in these models. To this end, previous studies have suggested that CXCL5 can be produced by hematopoietic as well as stromal cells, and we have shown that expression of this neutrophil chemokine is regulated by signaling through MyD88 in cardiac macrophages (36, 37).
While neutrophils adhere to the vascular endothelial cells when heart cells cannot produce CXCL5, their recruitment cascade is interrupted more upstream when CXCL2 is inhibited. Similar to our previous findings with inhibition of the ICAM-1 ligand LFA-1, adherence of neutrophils in coronary veins was virtually eliminated when we administered CXCL2-blocking antibodies. These findings are compatible with a model where presentation of CXCL2, but not CXCL5, on coronary vein endothelial cells enhances integrin affinity and promotes a firm arrest. Furthermore, CXCL2 blockade inhibiting neutrophil recruitment further upstream compared with macrophage-depleted or MyD88-deficient hearts raises the possibility that additional signaling pathways may regulate CXCL2 production in other donor cell populations.
In conclusion, using a recently described approach to image leukocyte trafficking in beating hearts in real time, we have uncovered cellular and molecular cues that regulate neutrophil extravasation into ischemic myocardium. Our observations extend the notion that requirements for leukocyte trafficking are impacted by the specific tissue and the nature of the inflammatory insult. Future studies will need to elucidate how other steps of neutrophil infiltration and the recruitment of other inflammatory cells are regulated in injured hearts.
Methods
Mice
C57BL/6 (B6) WT, B6 MyD88–deficient, B6 TLR9–deficient, B6 TIRAP–deficient, B6 CCR2-DTR, B6 MyD88–floxed (MyD88fl/fl), B6 LysM-Cre, and congenic B6 CD45.1+ mice were purchased from The Jackson Laboratories. B6 CXCL5–deficient mice and B6 LysM–GFP mice were provided by M. Miller, Washington University, who had originally obtained them from G.S. Worthen (University of Pennsylvania, Philadelphia, Pennsylvania, USA) and K. Ley (La Jolla Institute for Allergy and Immunology, La Jolla, California, USA), respectively (35). B6 MyD88fl/fl mice were intercrossed with LysM-Cre mice to generate animals that lacked MyD88 expression in lysozyme M–expressing cells. Select donor animals were treated with clodronate liposomes (200 μl i.v.) 24 hours prior to heart harvest (38). Control mice received PBS liposomes (200 μl) 24 hours before heart harvest. CCR2-expressing cells were depleted in donor hearts by injecting DT (250 ng i.p. 3, 2, and 1 days prior to heart harvest; Sigma-Aldrich) into CCR2-DTR mice. Some recipient mice were treated with CXCL2-neutraliizing or rat IgG2B isotype control antibodies (200 μg i.v. at the time of reperfusion, R&D Systems).
Heart transplantation
Heart grafts harvested from WT or various gene-deficient mice on a B6 background (CD45.2+) were transplanted into the right neck of B6 CD45.1+ or B6 LysM–GFP recipients following 1 hour of cold (4°C) ischemia as previously described (3). The recipient right common carotid artery and right external jugular vein were connected to the donor ascending aorta and the donor pulmonary artery, respectively. Grafts resumed a regular heartbeat immediately following reperfusion.
Flow cytometric cell sorting and analysis
Heart tissue was cut into small pieces and digested by placement into a RPMI 1640 solution containing Type 2 collagenase (1 mg/ml) (Worthington Biochemical Corporation) and 10 U/ml DNase (Sigma-Aldrich) at 37°C for 60 minutes. The digested tissue was then passed through a 70-μm cell strainer and treated with ACK lysing buffer. Cells were stained with fluorochrome-labeled anti-CD45.2 (clone 104, eBioscience), anti-CD45.1 (clone A20, BD Biosciences), anti-MHC class II (clone M5/114.15.2, BioLegend), anti CD11b (clone M1/70, BioLegend), anti-CCR2 (clone 575301, R&D Systems), anti-CD64 (clone X54-5/7.1, BioLegend), and anti-CD31 (clone MEC13.3, BD Biosciences).
Gene expression analysis
CD45.2−CD45.1−CD31+ vascular endothelial cells or donor-derived CD45.2+CD-11b+CD64+CCR2+ or CD45.2+CD11b+CD64+CCR2− macrophages/monocytes were sorted flow cytometrically from cardiac grafts 2 hours after transplantation of CD45.2+ hearts into CD45.1+ hosts. Cell sorting was performed using a Synergy sorting instrument (SY3200, Sony). For RNA isolation, macrophages were directly sorted into Quarter Load Threshold buffer, and RNA was purified using an RNeasy mini kit (Qiagen) based on the manufacturer’s protocol. RNA (1 ng) was reverse-transcribed using the iScript cDNA synthesis Kit (Bio-Rad), and the cDNA was amplified using a SsoAdvanced PreAmp Supermix (Bio-Rad). Amplified DNA was subjected to quantitative PCR (qPCR) using iQ SYBR Green Master Mix (Bio-Rad) in a Bio-Rad CFX-Connect. cDNA was diluted 1:5 in ddH2O and a 20 μl mixture containing 10 μl iQ SYBR Green Supermix, 1.0 μl forward and reverse primers, and 7.5 μl sterile water — as well as 2 μl of the diluted cDNA template — was generated. The real-time PCR was performed under the following conditions: 1 cycle at 95°C for 3 minutes, then 40 cycles at 95°C for 10 seconds, 56°C for 30 seconds, and 95°C for 1 minute, followed by 55°C for 1 minute. The intron-spanning primers were designed by using sequence information from Primer Bank (39). The Ct values were normalized to the endogenous control (18s RNA). Primer sequences of the genes we investigated in this study are CXCL1: 5′-ACTGCACCCAAAC-CGAAGTC-3′ (forward)/5′-TGGGGACACCTTTTAGCATCTT-3′ (reverse); CXCL2: 5′-CCAACCAC-CAGGCTACAGG-3′ (forward)/5′-GCGTCACACTCAAGCTCTG-3′ (reverse); CXCL5: 5′-GTTC-CATCTCGCCATTCATGC-3′ (forward)/5′-GCGGCTATGACTGAGGAAGG-3′ (reverse); CXCL5: 5′-GTTCCATCTCGCCATTCATGC-3′ (forward)/5′-GCGGCTATGACTGAGGAAGG-3′ (reverse); ICAM-1: 5′-GTGATGCTCAGGTATCCATCCA-3′ (forward)/5′-CACAGTTCTCAAAGCACAGCG-3′ (reverse); 5′-ATGAAGCCAGTGCATACTGTC-3′ (forward)/5′-CGGTGAATGTTTCAGATTGGAGT-3′ (reverse); ICAM-2: 5′-TGGTCCGAGAAGCAGATAGTAG-3′ (forward)/5′-GAGGCTGGTACACCCT-GATG-3′ (reverse)VCAM-1: 5′-TTGGGAGCCTCAACGGTACT-3′ (forward)/5′-GCAATCGTTTTG-TATTCAGGGGA-3′ (reverse); P-Selectin: 5′-CCCTGGCAACAGCCTTCAG-3′ (forward); 5′-GGGTCCT-CAAAATCGTCATCC-3′ (reverse); PECAM-1: 5′-ACGCTGGTGCTCTATGCAAG-3′ (forward)/5′-TCAGTTGCTGCCCATTCATCA-3′ (reverse); 18s RNA: 5′-GCTTGCTCGCGCTTCCT-TACCT-3′ (forward)/5′-TCACTGTACCGGCCGTGCGTA-3′ (reverse).
Neutrophil chemotaxis
Neutrophils were isolated from the BM of B6 mice by negative selection as previously described (40). Purified neutrophils (>95%) were resuspended in complete RPMI medium containing 10% FBS at a concentration of 106 cells per ml. One-hundred thousand neutrophils, suspended in 100 μl, were placed in the upper chamber of a transwell apparatus (6.5 mm, 3-μm pore size, polycarbonate membrane, Corning Costar Inc.). CCR2+CD11b+CD64+CD45+ monocytes and monocyte-derived macrophages (50,000/600μl), isolated from B6 hearts by flow cytometric sorting, were seeded in the lower chamber in the presence of 2.5 μg/ml of CXCL2-neutralizing antibodies (R&D Systems) and/or 2.5 μg/ ml of CXCL5-neutralizing antibodies (PeproTech). Neutrophil chemotaxis was carried out for 12 hours at 37°C in 5% CO2. Migrated neutrophils were fixed with 4% paraformaldehyde and then stained with 0.5% crystal violet for 30 minutes at room temperature. Nonmigrated cells from the upper chamber of the tran-swell insert were carefully removed using a cotton swab. Migrated neutrophils were quantified by counting 5 random fields under a 20× objective lens.
Two-photon microscopy
Time-lapse imaging was performed with a custom-built 2-photon microscope running ImageWarp version 2.1 acquisition software (A&B Software). LysM-GFP recipient mice were anesthetized with an i.p. injection of ketamine (50 mg/kg) and xylazine (10 mg/kg) and maintained with halved doses administered every hour. Mice were intubated orotracheally with a 20-gauge angiocatheter and ventilated with room air at a rate of 120 breaths per minute and with a tidal volume of 0.5 ml. For time-lapse imaging of leukocyte migration in heart grafts, we averaged 15 video-rate frames (0.5 seconds per slice) during the acquisition to match the ventilator rate and minimize movement artifacts. Each plane represents an image of 220 × 240 μm in the x and y dimensions. Twenty-one sequential planes were acquired in the z dimension (2.5 μm each) to form a Z stack. Each individual neutrophil was tracked from its first appearance in the imaging window and followed up to the time point at which it dislocated more than 20 μm from its starting position. To determine the percentage of extravasated neutrophils, we divided the number of extravascular neutrophils by the sum of intravascular and extravascular neutrophils. In 50 μl of PBS, 15 μl of 655-nm nontargeted Q-dots were injected i.v. to visualize blood vessels. Two-photon excitation produced a second harmonic signal from collagen within the myocardial tissue. Multidimensional rendering and manual cell tracking was done with Imaris (Bitplane). Data were transferred and plotted in GraphPad Prism 6.0 (Sun Microsystems Inc.) for creation of the graphs. Data have been pooled from 3–4 mice per experimental group.
Statistics
Values are presented as ±SEM. Significant differences between groups were assessed using an unpaired 2-tailed Student’s t test for 2 comparisons and ANOVA for multiple comparisons. A P value less than 0.05 was considered significant.
Study approval
All animal procedures were approved by the Animal Studies Committee at Washington University School of Medicine, St. Louis, Missouri, USA.
Supplementary Material
Acknowledgments
This work was supported by NIH grants 1P01AI116501, R01 HL113931, R01 HL094601, K08 HL123519, K08 HL 125940, and AG050096; Veteran’s Administration Merit Review grant 1I01BX002730; Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CHII2015-462); Foundation of Barnes-Jewish Hospital (8038-88); and a Burroughs Foundation Welcome Fund. We thank Eric T. Olson for assistance with graphic design for Figure 8. We thank Mark Miller and The In Vivo Imaging Core, Washington University, for assistance with 2 photon imaging, and we thank Scott Worthen, University of Pennsylvania, and Klaus Ley, La Jolla Institute for Allergy and Immunology, for originally providing CXCL5-deficient and LysM-GFP mice, respectively, to Mark Miller.
Footnotes
Author contributions
DK and KJL designed research studies and wrote the manuscript. WL and HMH designed research studies, conducted experiments, and analyzed data. RH conducted experiments. BTS analyzed data. DRG, AB, AEG, and ASK provided advice and discussion and helped design research studies. All authors reviewed the manuscript before submission.
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Dumont EA, et al. Cardiomyocyte death induced by myocardial ischemia and reperfusion: measurement with recombinant human annexin-V in a mouse model. Circulation. 2000;102(13):1564–1568. doi: 10.1161/01.cir.102.13.1564. [DOI] [PubMed] [Google Scholar]
- 2.Vinten-Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61(3):481–497. doi: 10.1016/j.cardiores.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Li W, et al. Intravital 2-photon imaging of leukocyte trafficking in beating heart. J Clin Invest. 2012;122(7):2499–2508. doi: 10.1172/JCI62970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatelain P, Latour JG, Tran D, de Lorgeril M, Dupras G, Bourassa M. Neutrophil accumulation in experimental myocardial infarcts: relation with extent of injury and effect of reperfusion. Circulation. 1987;75(5):1083–1090. doi: 10.1161/01.cir.75.5.1083. [DOI] [PubMed] [Google Scholar]
- 5.El-Sawy T, Belperio JA, Strieter RM, Remick DG, Fairchild RL. Inhibition of polymorphonuclear leukocyte-mediated graft damage synergizes with short-term costimulatory blockade to prevent cardiac allograft rejection. Circulation. 2005;112(3):320–331. doi: 10.1161/CIRCULATIONAHA.104.516708. [DOI] [PubMed] [Google Scholar]
- 6.Ma XL, Tsao PS, Lefer AM. Antibody to CD-18 exerts endothelial and cardiac protective effects in myocardial ischemia and reperfusion. J Clin Invest. 1991;88(4):1237–1243. doi: 10.1172/JCI115427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen H, Kreisel D, Goldstein DR. Processes of sterile inflammation. J Immunol. 2013;191(6):2857–2863. doi: 10.4049/jimmunol.1301539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petri B, Phillipson M, Kubes P. The physiology of leukocyte recruitment: an in vivo perspective. J Immunol. 2008;180(10):6439–6446. doi: 10.4049/jimmunol.180.10.6439. [DOI] [PubMed] [Google Scholar]
- 9.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10(6):427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 10.Epelman S, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40(1):91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavine KJ, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. 2014;111(45):16029–16034. doi: 10.1073/pnas.1406508111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170(3):818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu L, et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J Exp Med. 2014;211(7):1449–1464. doi: 10.1084/jem.20132126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330(6002):362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 15.Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203(12):2569–2575. doi: 10.1084/jem.20060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spahn JH, et al. DAP12 expression in lung macrophages mediates ischemia/reperfusion injury by promoting neutrophil extravasation. J Immunol. 2015;194(8):4039–4048. doi: 10.4049/jimmunol.1401415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maus UA, et al. Role of resident alveolar macrophages in leukocyte traffic into the alveolar air space of intact mice. Am J Physiol Lung Cell Mol Physiol. 2002;282(6):L1245–L1252. doi: 10.1152/ajplung.00453.2001. [DOI] [PubMed] [Google Scholar]
- 18.Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29(13):3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nahrendorf M, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russo MJ, et al. Factors associated with primary graft failure after heart transplantation. Transplantation. 2010;90(4):444–450. doi: 10.1097/TP.0b013e3181e6f1eb. [DOI] [PubMed] [Google Scholar]
- 21.Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol. 2012;52(5):1135–1144. doi: 10.1016/j.yjmcc.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117(10):2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng Y, et al. Innate immune adaptor MyD88 mediates neutrophil recruitment and myocardial injury after ischemia-reperfusion in mice. Am J Physiol Heart Circ Physiol. 2008;295(3):H1311–H1318. doi: 10.1152/ajpheart.00119.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathur S, Walley KR, Wang Y, Indrambarya T, Boyd JH. Extracellular heat shock protein 70 induces cardiomyocyte inflammation and contractile dysfunction via TLR2. Circ J. 2011;75(10):2445–2452. doi: 10.1253/circj.cj-11-0194. [DOI] [PubMed] [Google Scholar]
- 25.Tian J, et al. Extracellular HSP60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardio-vasc Res. 2013;98(3):391–401. doi: 10.1093/cvr/cvt047. [DOI] [PubMed] [Google Scholar]
- 26.Gupta S, Knowlton AA. HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Physiol Heart Circ Physiol. 2007;292(6):H3052–H3056. doi: 10.1152/ajpheart.01355.2006. [DOI] [PubMed] [Google Scholar]
- 27.Huang H, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology. 2011;54(3):999–1008. doi: 10.1002/hep.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lohner R, et al. Toll-like receptor 9 promotes cardiac inflammation and heart failure during polymicrobial sepsis. Mediators Inflamm. 2013;2013:261049. doi: 10.1155/2013/261049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology. 2010;51(2):621–632. doi: 10.1002/hep.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balamayooran G, et al. Role of CXCL5 in leukocyte recruitment to the lungs during secondhand smoke exposure. Am J Respir Cell Mol Biol. 2012;47(1):104–111. doi: 10.1165/rcmb.2011-0260OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vieira SM, et al. A crucial role for TNF-alpha in mediating neutrophil influx induced by endogenously generated or exogenous chemokines, KC/CXCL1 and LIX/CXCL5. Br J Pharmacol. 2009;158(3):779–789. doi: 10.1111/j.1476-5381.2009.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rovai LE, Herschman HR, Smith JB. The murine neutrophil-chemoattractant chemokines LIX, KC, and MIP-2 have distinct induction kinetics, tissue distributions, and tissue-specific sensitivities to glucocorticoid regulation in endotoxemia. J Leukoc Biol. 1998;64(4):494–502. doi: 10.1002/jlb.64.4.494. [DOI] [PubMed] [Google Scholar]
- 34.Chandrasekar B, Smith JB, Freeman GL. Ischemia-reperfusion of rat myocardium activates nuclear factor-KappaB and induces neutrophil infiltration via lipopolysaccharide-induced CXC chemokine. Circulation. 2001;103(18):2296–2302. doi: 10.1161/01.cir.103.18.2296. [DOI] [PubMed] [Google Scholar]
- 35.Mei J, et al. CXCL5 regulates chemokine scavenging and pulmonary host defense to bacterial infection. Immunity. 2010;33(1):106–117. doi: 10.1016/j.immuni.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madorin WS, Rui T, Sugimoto N, Handa O, Cepinskas G, Kvietys PR. Cardiac myocytes activated by septic plasma promote neutrophil transendothelial migration: role of platelet-activating factor and the chemokines LIX and KC. Circ Res. 2004;94(7):944–951. doi: 10.1161/01.RES.0000124395.20249.AE. [DOI] [PubMed] [Google Scholar]
- 37.Guha D, Klamar CR, Reinhart T, Ayyavoo V. Transcriptional Regulation of CXCL5 in HIV-1-Infected Macrophages and Its Functional Consequences on CNS Pathology. J Interferon Cytokine Res. 2015;35(5):373–384. doi: 10.1089/jir.2014.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kreisel D, et al. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A. 2010;107(42):18073–18078. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Spandidos A, Wang H, Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012;40(Database issue):D1144–D1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kreisel D, et al. Emergency granulopoiesis promotes neutrophil-dendritic cell encounters that prevent mouse lung allograft acceptance. Blood. 2011;118(23):6172–6182. doi: 10.1182/blood-2011-04-347823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.