

Abstract
De novo and acquired resistance to platinum therapy such as cisplatin (CDDP) is a clinical challenge in gastric cancer treatment. Aberrant expression and activation of Aurora kinase A (AURKA) and eukaryotic translation initiation factor 4E (eIF4E) are detected in several cancer types. Herein, we investigated the role of AURKA in CDDP resistance in gastric cancer. Western blot analysis demonstrated overexpression of AURKA and phosphorylation of eIF4E in acquired and de novo CDDP-resistant gastric cancer models. Inhibition of AURKA with MLN8237 (alisertib) alone or in combination with CDDP significantly suppressed viability of CDDP-resistant cancer cells (p<0.01). Additionally, inhibition or knockdown of AURKA decreased protein expression of p-eIF4E (S209), HDM2, and c-MYC in CDDP-resistant cell models. This was associated with a significant decrease in cap-dependent translation levels (p<0.01). In vivo tumor xenografts data corroborated these results and confirmed that inhibition of AURKA was sufficient to overcome CDDP resistance in gastric cancer. Our data demonstrate that AURKA promotes acquired and de novo resistance to CDDP through regulation of p-eIF4E (S209), c-MYC, HDM2, and cap-dependent translation. Targeting AURKA could be an effective therapeutic approach to overcome CDDP resistance in refractory gastric cancer and possibly other cancer types.
Keywords: Gastric cancer, Cisplatin, CDDP, Alisertib, MLN8237, MYC, HDM2, eIF4E
Graphical abstract
De novo and acquired resistance to cisplatin (CDDP) is a clinical challenge in gastric cancer treatment. Our data demonstrate that Aurora kinase A (AURKA) promotes acquired and de novo resistance to CDDP through activation of eukaryotic translation initiation factor 4E (p-eIF4E S209), c-MYC, HDM2, and cap-dependent translation. Targeting AURKA could be an effective therapeutic approach to overcome CDDP resistance in refractory gastric cancer and possibly other cancer types.
1. Introduction
Gastric cancer is currently the fourth most common malignancy worldwide and the third leading cause of cancer-related deaths in both males and females (Ferlay et al., 2015a). Resistance to chemotherapeutic agents is a major clinical problem in management of cancer patients. In fact, patients with gastric cancer exhibit poor response to neoadjuvant and adjuvant chemotherapy due to intrinsic and acquired drug resistance (Hohenberger and Gretschel, 2003; Jemal et al., 2011). Platinum-based regimens that include cisplatin (CDDP, cis-diamminedichloroplatinum) are employed to treat a variety of cancers such as ovarian, bladder, esophageal, and gastric cancers. Although platinum-based chemotherapeutic regimens have become standard approaches for treatment of patients with advanced gastric cancer, there is a relatively low rate of complete response (Van Cutsem et al., 2008). Therefore, there is an urgent need for the development of novel therapeutic approaches based on understanding the underlying molecular pathways mediating cisplatin resistance.
Aurora Kinase A (AURKA) is a serine/threonine cell cycle kinase, whose main physiological function in normal cells is related to centrosome and spindle assembly (Lens et al., 2010). We and others have reported amplification and over-expression of AURKA in several malignancies including gastric, esophageal, colon, breast, and ovarian cancers (Aradottir et al., 2015; Cirak et al., 2015; Dar et al., 2009; Dar et al., 2008b; Fang et al., 2011; Furukawa et al., 2006; Gritsko et al., 2003; Katayama et al., 1999; Katsha et al., 2015; Katsha et al., 2013; Zheng et al., 2016). Recent studies have shown that inhibition of AURKA can sensitize cancer cells to CDDP in gastric cancer (Sehdev et al., 2012) and its overexpression may predict platinum-resistance in epithelial ovarian cancer (Mignogna et al., 2016). It has been shown that AURKA can inhibit p53 through direct binding and activation of the E3-ubiquitine ligase, human double minute 2 (HDM2) (Sehdev et al., 2014). Notably, AURKA overexpression in cancer cells has also been implicated in activating a plethora of pro-survival oncogenic pathways including β-catenin, NF-κB and AKT (Dar et al., 2009; Dar et al., 2008b; Katsha et al., 2013). These studies suggest that AURKA overexpression in cancer cells has pro-survival and anti-apoptotic functions.
The eukaryotic translation initiation factor 4E (eIF4E), which is the rate-limiting component in formation of eIF4F complex (Jones et al., 1997), is involved in recognizing the 5′ 7-methyl guanosine cap of mRNAs (Mamane et al., 2004). Regulation of protein translation is important for the control of cell growth and proliferation. Aberrant activation of eIF4E has been linked to resistance to therapy and poor outcome in breast, melanoma, prostate, and gastric cancers (Chen et al., 2004; Graff et al., 2009; Pettersson et al., 2015; Zhan et al., 2015). Activity of eIF4E is dependent upon its phosphorylation on Ser209, leading to oncogenic transformation through translational control (Wendel et al., 2007). Enhanced-translation of growth promoting genes such as c-MYC, Cyclin D1, and VEGF has been associated with activation of eIF4E. Activation of eIF4E has been implicated in chemotherapeutic resistance in several cancer types (Kraljacic et al., 2011; Martinez-Marignac et al., 2013; Zhan et al., 2015). A recent study reported that inhibiting eIF4E might be a viable therapeutic approach to overcome resistance to vemurafenib, BRAF inhibitor, in melanoma (Zhan et al., 2015). Therefore, targeting eIF4E is considered a promising anticancer strategy to combat drug resistance (reviewed in (Siddiqui and Sonenberg, 2015).
In this study, we demonstrate that AURKA promotes acquired and de novo resistance to cisplatin in in vitro and in vivo gastric cancer cell models. We show that AURKA mediates phosphorylation of eIF4E to promote protein translation of pro-oncogenic downstream effectors such as c-MYC and HDM2. We propose targeting AURKA as an effective second line therapeutic approach in cisplatin-resistant cancers.
2. Materials and methods
2.1. Cell culture and reagents
Human gastric adenocarcinoma cell lines (AGS, SNU-1, MKN28, and MKN45) were maintained in Dulbecco’s modified Eagle’s medium (GIBCO, Carlsbad, CA). All cell lines were authenticated using short tandem repeat (STR) profiling (Genetica DNA Laboratories, Burlington, NC). The cell lines were supplemented with 10% fetal bovine serum (Invitrogen Life Technologies, Carlsbad, CA) and with 1% penicillin/streptomycin (GIBCO). The investigational Aurora Kinase A inhibitor alisertib, known as MLN8237, (Millennium Pharmaceuticals, Inc., Cambridge, MA) was used for in vitro and in vivo studies. The AURKA expression plasmid was generated as described previously (Dar et al., 2009). Specific antibodies against p-AURKA (T288), AURKA, p-eIF4E (S209), eIF4E, and β-actin were purchased from Cell Signaling Technology (Beverly, MA). Specific antibody against c-MYC was purchased from Santa Cruz Biotechnology (Dallas, TX). Transfection reagent LipoJet was purchased from SignaGen (Gaithersburg, MD). Cisplatin (CDDP) (APP Pharmaceuticals, LLC) was obtained from Vanderbilt University Medical Center pharmacy in a stock solution (2 mmol/L), prepared using 1X PBS.
2.2. Establishment of CDDP-resistant sublines from AGS cells
AGS CDDP-resistant cells were established by continuous exposure to CDDP starting at 0.1 μM and increasing in a stepwise manner to 10 μM for 6 months. Finally, we generated two AGS CDDP resistant Pools (Pools 1 & 2) from which we selected two highly resistant single clones (Clones 1 & 2) to be used in the study.
2.3. Western Blotting
Cells were scraped on ice, centrifuged, and pellets were re-suspended in RIPA lysis buffer. RIPA buffer was prepared following manufacturer’s instructions. Cell lysates were placed on ice. Protein concentration was determined using Bradford Protein Assay (Bio-Rad Laboratories, Hercules, CA). Proteins (25 μg) from each sample were subjected to SDS/PAGE and transferred onto nitrocellulose membranes using the semi-dry transfer protocol (Bio-Rad). After transfer, membranes were probed with each respective primary antibody overnight at 4°C. Following incubation, the membranes were probed with HRP-conjugated secondary antibodies (Cell Signaling). Protein bands were visualized using the commercial Immobilon Western Chemiluminescent HRP Substrate Kit (Millipore, Billerica, MA).
2.4. AURKA and eIF4E silencing by small interfering RNA (siRNA)
Cells were seeded at 60% confluency in 10% FBS DMEM for 24h in p60 plates. AURKA or eIF4E were transiently silenced by using siAURKA and sieIF4E (Invitrogen) for a total of 48h. A negative siRNA control (Ambion, Austin, TX) was used in each experiment. Transfection of cells was achieved by using a LipoJet reagent (SignaGen) according to the manufacturer’s instructions. Following 24h transfection, medium was replaced with DMEM media, supplemented with 5% FBS and antibiotics for another 24h prior to harvesting. Validation of AURKA and eIF4E knockdown was assessed by qPCR and Western blot analyses.
2.5. AURKA overexpression
The expression plasmid for AURKA was generated as described previously (Dar et al., 2009). A synthetic Flag-tag sequence was added at the N-terminus of AURKA. The recombinant adenovirus expressing AURKA or control was generated as described previously (Katsha et al., 2013). The recombinant adenovirus was generated by co-transfecting Human embryonic kidney (HEK)-293 cells (American Tissue Culture Collection) with the shuttle and adenoviral backbone (pJM17) plasmids using the Calcium Phosphate Transfection Kit (Applied Biological Materials, Inc., Richmond, BC).
2.6. CellTiter-Glo Luminescence Assay
CellTiter-Glo Assay was used to determine IC50 and drug dose-response curves for each cell line following treatment with CDDP, alisertib (MLN8237), or combination. Cells were seeded at 2,000 cells/well in a 96-well plate. Cells were treated with CDDP with or without MLN8237 following a 12 × 2-fold serial dilution treatment in 5% FBS-DMEM medium. After 5 days, cell viability was measured using CellTiter-Glo Luminescence Assay (Promega, Madison, WI). The dose-response curves were fitted using GraphPad Prism 5, following a non-linear regression (four parameter, least squares fit) method. IC50 values were determined by a four-parameter non-linear regression method. The data were generated from at least three independent experiments.
2.7. RNA extraction and Real-Time RT-PCR
Cells were scraped, centrifuged, and total RNA was isolated using the RNeasy Mini kit (Qiagen, Germantown, MD), and cDNA synthesis was performed using an iScript cDNA Synthesis Kit (Bio-Rad). Specific Primers were designed using the online software Primer 3 (http://frodo.wi.mit.edu/primer3/). All primers were purchased from Integrated DNA Technologies (IDT) (Coralville, IA). c-MYC-F: CACCGAGTCGTAGTCGAGGT; c-MYC-R: TTTCGGGTAGTGGAAAACCA; AURKA-F: AGTTGGAGGTCCAAAACGTG; AURKA-R: TCCAAGTGGTGC ATATTCCA; HDM2-F: ACCTCACAGATTCCAGCTTCG; HDM2-R: TTTCATAGTATAAGTGTCTTTTT. The quantitative RT-PCR was performed using a Bio-Rad CFX Connect Real-time System, with the threshold cycle number determined by Bio-Rad CFX manager software version 3.0. Reactions were performed in triplicate and the threshold cycle numbers were averaged. The results of the genes were normalized to HPRT1 housekeeping gene.
2.8. Luciferase Reporter Assay
For the cap-dependent dual luciferase reporter assays, cells were transfected with 1 μg of a dual-Renilla-firefly-luciferase pcDNA3-rLuc-PolioIRES-fLuc reporter (a kind gift from Dr. John Blenis, Harvard Medical School), to measure cap-dependent/independent translation (Dos Santos et al., 2016). Cells were plated in 6-well plates, transfected using Lipofectamine 2000 reagent (Life Technologies), and treated with MLN8237 for 24h. The lysates were prepared in triplicate and the Dual-Luciferase Reporter Assay (Promega) was used following the manufacturer’s protocol. The rate of cap-dependent translation was defined as the ratio of Renilla/firefly luciferase activities.
For c-MYC transcription activity, cells were co-transfected with 250 ng of β-GAL and 1 μg of 4xEMS c-MYC reporter (a kind gift from Dr. Stephen R. Hann, Vanderbilt University School of Medicine) in 6-well plates using the PolyJet transfection reagent (SignaGen). As a positive control, parental cells were transfected with 500 ng of pcDNA-c-MYC plasmid in addition to 250 ng of β-GAL and 1 μg of 4×EMS c-MYC reporter Cells were treated with MLN8237 for 24h, and then lysed to measure luciferase activity using the Luciferase Assay kit (Promega). The c-MYC luciferase activities were normalized to β-GAL levels.
2.9. In vivo tumor xenograft
All animal work was approved by the Vanderbilt Institutional Animal Care and Use Committee. MKN45 cells (2 × 106) were suspended in 200 μl of DMEM and Matrigel mixture (50% DMEM supplemented with 10% FBS and 50% Matrigel) were injected into the flank regions of female 201 NIH III HO nude mice (Charles River Laboratories). We used 8 mice per group, the tumors were allowed to grow until 150 – 200 mm3 in size before starting treatment with CDDP (2.5mg/kg bodyweight, once a week, IP) alone, MLN8237 (40 mg/kg, 5 times/week, orally) alone, or the combination of CDDP and MLN8237 for 28 days. Tumor xenografts were measured every 3 days and tumor size was calculated according to the following formula: T vol = L × W2 × 0.5, in which T vol is tumor volume, L is tumor length and W is tumor width. For control group, mice were sacrificed when tumor reach 1000 mm3 in accordance with approved protocols. At the end of treatment, 3–6 the xenograft tumors from each group were collected and processed for Western blot (p-AURKA (T288), AURKA, p-eIF4E (S209), eIF4E, c-MYC). Immunohistochemically analysis was carried out on formalin fixed paraffin-embedded tissues to measure Ki-67 and cleaved caspase 3 protein expression levels. Ki-67 and cleaved caspase 3 protein expression levels were evaluated by ImageJ software. Relative integrated density indicates the quantification data of diaminobenzidine staining signal analyzed by ImageJ IHC Toolbox plugin (https://imagej.nih.gov/ij/plugins/ihc-toolbox/index.html) (Zhang et al., 2016).
2.10. Statistical Analysis
Data are presented as means +/− standard error of mean (SEM). Statistical significance of difference between control groups and treatment groups was determined using One-way ANOVA Test. Statistical analyses were carried out using GraphPad Prism 5 software, nonlinear regression (GraphPad Software Inc.). The correlation between two parameters was determined by two-tailed Student’s test. The differences were considered statistically significant when the P<0.05.
3. Results
3.1. Acquired resistance to CDDP correlates with high levels of AURKA and p-eIF4E proteins in gastric cancer cells
Cisplatin-based chemotherapeutic regimen is a standard approach for treatment of gastric cancer patients. Unfortunately, disease progression eventually occurs because of acquired drug resistance (reviewed by (Lu et al., 2016)). To determine the molecular mechanism that drives acquired resistance to CDDP in gastric cancer cells, we generated AGS cell models of acquired resistance to CDDP using a stepwise increase in CDDP concentrations starting at 0.1 μM to 10 μM for 6 months. At the end of CDDP treatments, in addition to CDDP-sensitive AGS Parental cells (control cells), we obtained the following CDDP-resistant AGS cell models: AGS CDDPR Pool 1, AGS CDDPR Pool 2, AGS CDDP Clone 1, and AGS CDDPR Clone 2. Short-term cell viability (5 days) in response to CDDP treatments was evaluated by CellTiter-Glo Assay. The CDDP IC50s of AGS CDDPR Pool 1 (15.4 μM), AGS CDDPR Pool 2 (14.2 μM), AGS CDDPR Clone 1 (17.5 μM), and AGS CDDPR Clone 2 (8.6 μM) were significantly higher than that of AGS Parental cells (4.9 μM, P<0.05) (Figure 1A), confirming that AGS CDDP-resistant cell models conferred the CDDP resistance phenotype. Using Western blot analysis, we found that CDDP-resistant AGS cells gained overexpression of AURKA, p-eIF4E (S209), c-MYC and HDM2 proteins, as compared to AGS Parental cells (Figure 1B).
Figure 1. CDDP-resistant cells express high levels of AURKA and p-eIF4E (S209) proteins.
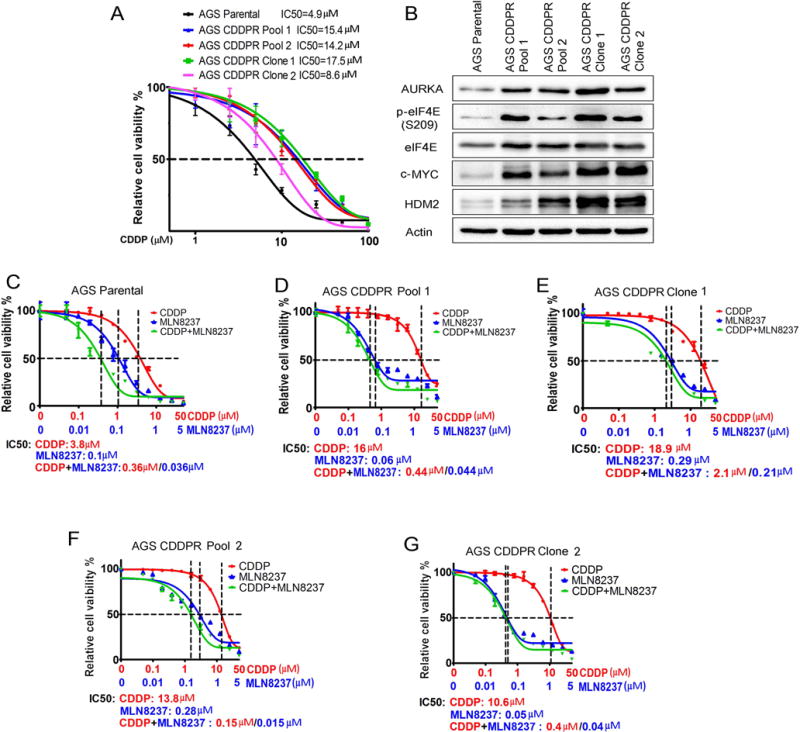
(A) AGS Parental, AGS CDDP-resistant (CDDPR) Pool 1, AGS CDDPR Pool 2, AGS CDDPR Clone 1, and AGS CDDPR Clone 2 cells in 96-well plates were treated with CDDP following a 8-point 2-fold serial dilution, starting at 100 μM concentration. Cell viability was measured using CellTiter-Glo assay. Dose-response was fitted using a three-parameter non-linear regression method. (B) Cell lysates from AGS Parental and CDDP resistant cell lines were subjected to Western blot analysis of the indicated proteins. Levels of AURKA, p-eIF4E (S209), c-MYC, and HDM2 proteins were substantially higher in AGS CDDP-resistant cells than AGS Parental cells. AGS Parental (C) AGS CDDPR Pool 1 (D) and AGS CDDPR Clone 1 (E) AGS CDDPR Pool 2 (F) and AGS CDDPR Clone 2 (G) cells in 96-well plates were treated with MLN8237 alone, CDDP alone, or CDDP + MLN8237 at a fixed ratio (10:1) following a 12-point 2-fold serial dilution. Cell viability was measured and dose response curves were generated as in (A). The mean value of IC50 of each drug treatment was generated from at least three independent experiments.
3.2. Cancer cells with acquired resistance to CDDP are sensitive to AURKA inhibition
To find out if targeting AURKA can reverse these signaling effects and restore therapeutic efficacy in CDDP-resistant cells, we inhibited AURKA with MLN8237, an investigational AURKA-specific inhibitor, alone or in combination with CDDP in AGS Parental and CDDPR cells. Using short-term cell viability (5 days) CellTiter-Glo assay, the results showed that MLN8237 alone significantly decreased cell survival (P<0.001) in AGS Parental cells, AGS CDDPR Pool 1, AGS CDDPR Pool 2, AGS CDDPR Clone 1, and AGS CDDPR Clone 2 cells in a dose-dependent manner. Interestingly, while the combination of MLN8237 and CDDP had an additive effect on parental cells (Figure 1C), the addition of CDDP to MLN8237 had minimal advantage over MLN8237 alone in CDDP-resistant cells (Figure 1D–1G). Collectively, the data indicated that inhibition of AURKA using MLN8237 is sufficient to decrease cell viability of AGS CDDP-resistant cells.
3.3. AURKA promotes CDDP acquired resistance through regulation of eIF4E, c-MYC, and HDM2
Based on our results indicating that AURKA is important for CDDP-acquired resistance (Figure 1), and the positive correlation between the protein e expression of AURKA, p-eIF4E (S209), c-MYC, and HDM2 in CDDP-resistant cells (Figure 1B), we examined if AURKA inhibition by MLN8237 can regulate these signaling molecules. Indeed, we observed a substantial reduction in the protein levels of p-eIF4E (S209), c-MYC, and HDM2 protein levels in AGS CDDPR Pool 1, AGS CDDPR Pool 2, AGS CDDPR Clone 1, and AGS CDDPR Clone 2 cells, as compared with non-treated cells (Figure 2). To validate the data indicating that inhibition of AURKA by MLN8237 leads to decreased protein expression of p-eIF4E (S209), c-MYC, and HDM2 in AGS CDDPR cells, we knocked down endogenous AURKA with siRNA and evaluated the expression of p-eIF4E and downstream effectors in these cells. Indeed, the Western blot data confirmed the downregulation of p-eIF4E (S209), c-MYC, and HDM2 proteins following the knockdown of AURKA (Figure 3A). The qRT-PCR data indicated that knocking down AURKA did not decrease the mRNA expression of c-MYC, and HDM2 in AGS CDDPR cells (Figure 3B). In the contrary, we observed some induction of mRNA expression level despite the reduction in their protein level, which may reflect a compensatory cellular feedback mechanism in an attempt to restore the protein expression back to its base level. Taken together, these results confirm that regulation of c-MYC and HDM2 in resistant cells is post-transcriptional. These results are also in line with phosphorylation of eIF4E (S209) which plays an important role in protein translation, suggesting that AURKA-mediated cap-dependent translation is likely a dominant mechanism. Therefore, we next investigated protein translation activity. We utilized a unique dual-Renilla-firefly-luciferase pcDNA3-rLuc-PolioIRES-fLuc reporter to measure cap-dependent/independent translation (Liu et al., 2013), followed by treatment with MLN8327. We found that the cap-dependent renilla activity is higher in CDDP-resistant cells (p<0.01), as compared to parental cells, and was significantly decreased after MLN8237 treatment in AGS CDDPR cells (P<0.01) (Figure 3C). We also measured the transcription activity of the induced high levels of c-MYC protein in resistant cells using the 4 × EMS c-MYC reporter, which contains 4 c-MYC binding sites (Boone et al., 2011), followed by treatment with MLN8327. The data indicated that inhibition of AURKA significantly decreased c-MYC transcription activity in AGS CDDPR cells (P<0.05) (Figure 3D).
Figure 2. Pharmacologic inhibition of AURKA with MLN8237 downregulates phosphorylation of eIF4E and protein expression of downstream effectors in CDDP-resistant cells.
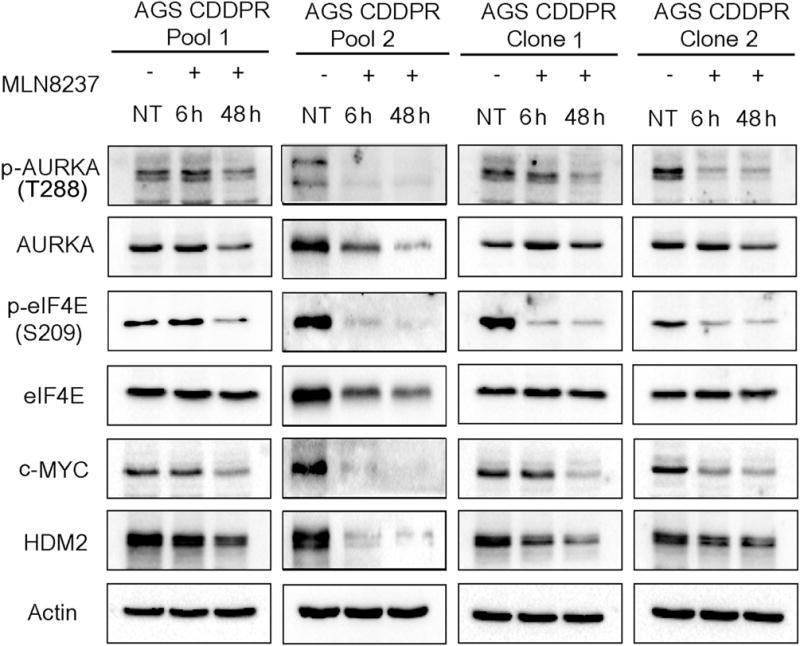
AGS CDDPR Pool 1, AGS CDDPR Pool2, AGS CDDPR Clone 1, and AGS CDDPR Clone 2 cells were treated with MLN8237 (0.5 μM), and cell lysates were subjected to Western blot analysis of the indicated proteins. The data showed that pharmacologic inhibition of AURKA with MLN8237, as indicated by decreased protein levels of p-AURKA (T288), reduced the protein levels of p-eIF4E (S209) and its protein translation targets, c-MYC and HDM2 in CDDP-resistant cells. Gel loading was normalized for equal β-actin.
Figure 3. Genetic knockdown of AURKA reduces phosphorylation of eIF4E and protein levels of its downstream effectors in CDDP-resistant cells.
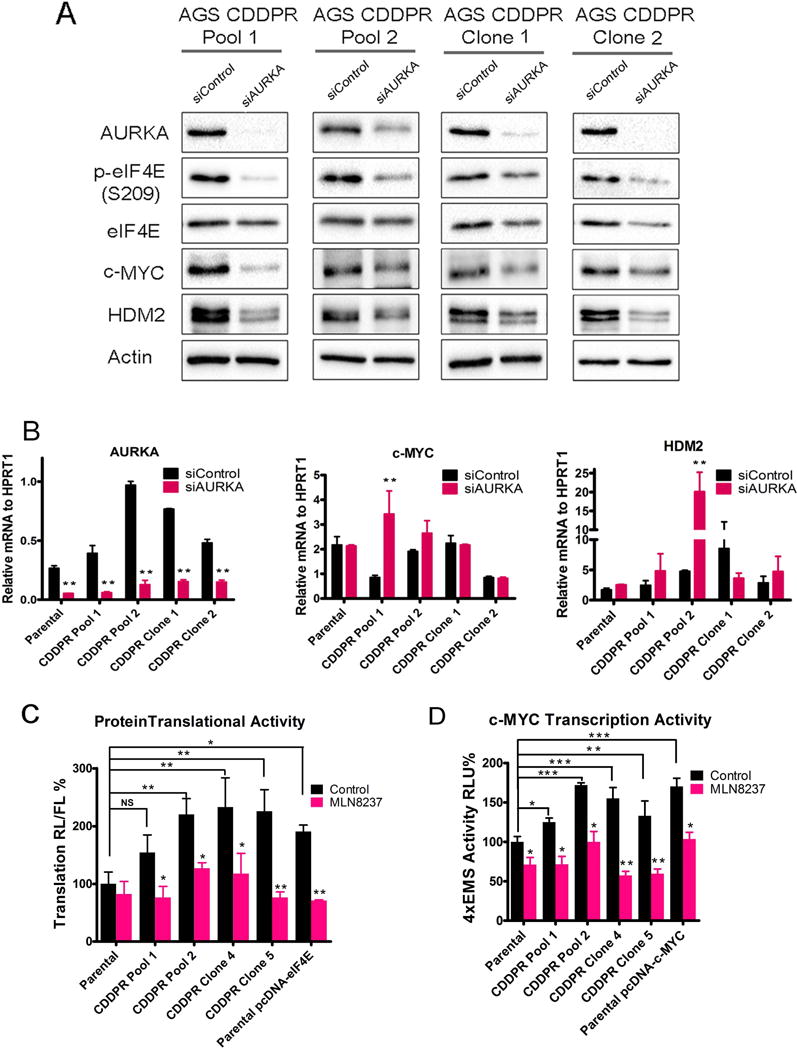
AGS CDDPR Pool 1, AGS CDDPR Pool 2, AGS CDDPR Clone 1, and AGS CDDPR Clone 2 cells were transfected with control siRNA (siControl) or siRNA specific for AURKA (siAURKA) for 48h. Cell lysates and total RNAs were subjected to Western blot (A) and Real-Time RT-PCR (B) analyses, respectively. The data showed that knockdown of AURKA led to decreased p-eIF4E (S209), c-MYC, and HDM2 protein levels, but did not decrease mRNA expression of the corresponding encoding genes in CDDP-resistant cells. Gel loading was normalized for equal β-actin. HPRT1 housekeeping gene was used as the internal control for PCR. (C) AGS Parental, AGS CDDPR Pool 1, AGS CDDPR Pool 2, AGS CDDPR Clone 1, and AGS CDDPR Clone 2 cells were transfected with luciferase reporter plasmid (pcDNA3-rLuc-Polio IRES-fLuc). Cells were treated with MLN8237 (0.5 μM) for 24h. The rate of cap-dependent translation was defined as the ratio of Renilla/firefly luciferase activities. (D) AGS Parental, AGS CDDPR Pool 1, AGS CDDPR Pool 2, AGS CDDPR Clone 1, and AGS CDDPR Clone 2 cells were transfected with 4×EMS c-MYC reporter. Cells were treated with MLN8237 (0.5 μM) for 24h. Luciferase activity was determined as described in Methods. The results showed that the basal level of cap-dependent translation activity and c-MYC transcription activity in AGS CDDPR cells is significantly higher than AGS Parental cells, *p<0.05. **p<0.01. **p<0.01. Additionally, treatment with MLN8237 significantly reduced cell cap-dependent translation or 4×EMS c-MYC Luciferase activity in comparison with the no treatment control group, *p<0.05.** p<0.01.
To test our hypothesis that AURKA-mediated acquired CDDP resistance is dependent on eIF4E and downstream effectors, we knocked down eIF4E or c-MYC in AGS CDDPR cells and assessed cell viability in response to CDDP. Our Western blot data showed, as expected, that knockdown of eIF4E downregulated the downstream effectors, c-MYC and HDM2 (Figure 4A). To confirm that the regulation of downstream effectors, c-MYC and HDM2, by AURKA is mediated by eIF4E, we transiently expressed AURKA with and without knocking down eIF4E in AGS cells. Western blot data showed that AURKA-induced c-MYC and HDM2 protein expression was abrogated following eIF4E knockdown (Figure 4B), confirming that AURKA regulates these proteins in an eIF4E-dependent manner. Notably, we observed that knockdown of eIF4E significantly sensitized AGS CDDPR Pool1 (p<0.05) and AGS CDDPR Clone 1 (p<0.01) cells to CDDP as indicated by a decrease in IC50 from 15.2 μM to 6 μM and from 18 μM to 5.3 μM, respectively (Figure 4C). In addition, our data indicated that knockdown of the downstream effector, c-MYC, significantly increased the sensitivity of AGS CDDPR Pool1 (p<0.01) and AGS CDDPR Clone 1 (p<0.05) cells to CDDP as indicated by a decrease in IC50 from 15.9 μM to 6.5 μM and from 20 μM to 10 μM, respectively (Figure 4D). Taken together, these results indicate that AURKA-mediated acquired CDDP resistance is dependent on eIF4E and its downstream targets in AGS gastric cancer cells and suggest that targeting AURKA can be an effective therapeutic approach in CDDP-resistant cells.
Figure 4. AURKA-mediated regulation of c-MYC and HDM2 requires eIF4E.
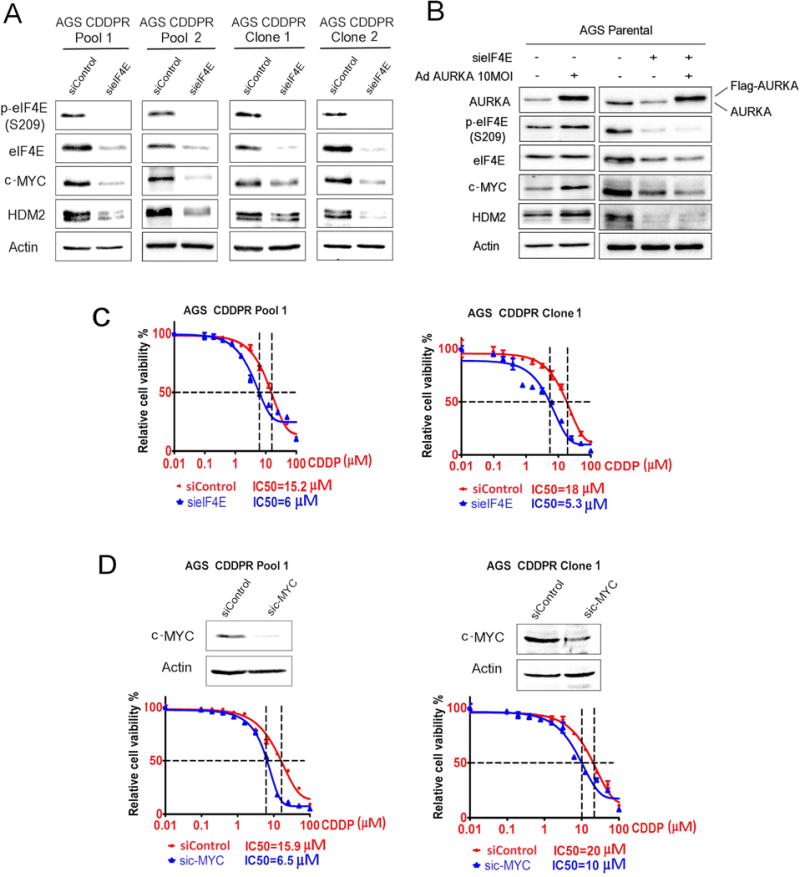
(A) AGS CDDPR Pool 1, AGS CDDPR Pool 2, AGS CDDPR Clone 1, and AGS CDDPR Clone 2 cells were transiently transfected with control siRNA (siControl) or siRNA specific for eIF4E (sieIF4E) for 48h. Cell lysates were subjected to Western blot analysis of the indicated proteins. Knocking down of eIF4E decreased c-MYC and HDM2 protein expression. (B) AGS Parental cells were transiently transfected by siControl + AdControl, siControl + AdAURKA, sieIF4E + AdControl, or sieIF4E + AdAURKA for 48h. Cell lysates were then subjected to Western blot analyses of the indicated proteins. The data showed that overexpression of AURKA increased protein levels of p-eIF4E (S209), c-MYC, and HDM2. Knockdown of eIF4E abrogates AURKA-induced upregulation of c-MYC and HDM2 protein expression. (C) AGS CDDPR Pool 1 and AGS CDDPR Clone 1 cells were transfected with siControl or sieIF4E for 48h and subjected to CellTiter-Glo viability assay. Knocking down of eIF4E significantly sensitized cells to CDDP (p<0.05), as indicated by 2- to 3-fold decrease in CDDP IC50. (D) AGS CDDPR Pool 1 and AGS CDDPR Clone 1 cells were transfected with siControl or sic-MYC for 48h and subjected to CellTiter-Glo viability assay. Knocking down of c-MYC significantly sensitized cells to CDDP (p<0.05), as indicated by 2-fold decrease in CDDP IC50.
3.4. AURKA enhances de novo CDDP resistance through regulation of eIF4E, c-MYC, and HDM2
We next investigated whether AURKA-eIF4E axis is also present in de novo CDDP resistance. We first screened a panel of gastric cancer cell lines for their sensitivity to CDDP and correlation with protein expression of AURKA, p-eIF4E, eIF4E, c-MYC, and HDM2. Our cell viability data in response to CDDP indicated various levels of sensitivity (IC50) of the following cell lines: AGS (4.9 μM), SNU-1 (0.9 μM), MKN28 (7.2 μM), and MKN45 (11.6 μM) (Figure 5A). Western blot data demonstrated high levels of AURKA in CDDP resistant cells (MKN28 and MKN45 cell lines) (Figure 5B). We next selected MKN45 cells, which exhibit the highest degree of CDDP resistance, relative to other cell lines, as a model of intrinsic de novo resistance to investigate whether targeting AURKA can achieve a therapeutic response. Cell viability data showed that MLN8237 alone or in combination with CDDP can significantly reduce cell viability as compared to CDDP alone (p<0.01, Figure 5C). Accordingly, Western blot data showed that inhibiting AURKA using MLN8237 (Figure 5D) or AURKA knockdown using siRNA (Figure 5E) downregulated protein expression of p-eIF4E (S209), c-MYC, and HDM2 in MKN45 cells, similar to acquired resistance cell models. To investigate if AURKA-mediated de novo CDDP resistance is dependent on eIF4E and c-MYC; we knocked down eIF4E or c-MYC in MKN45 cells and assessed cell viability in response to CDDP. Our data showed that knocking down either eIF4E or c-MYC significantly sensitized cells to CDDP (p<0.05), as indicated by approximately a 2-fold decrease in IC50 (Figure 5F). Western blot data indicated that knocking down of eIF4E markedly decreased c-MYC and HDM2 protein levels (Figure 5G). The knockdown of c-MYC in MKN45 cells was confirmed (Figure 5H). Collectively, our data demonstrate that AURKA-eIF4E axis promotes de novo CDDP resistance in MKN45 cells.
Figure 5. AURKA mediates de novo CDDP resistance through regulation of eIF4E and its downstream effectors.
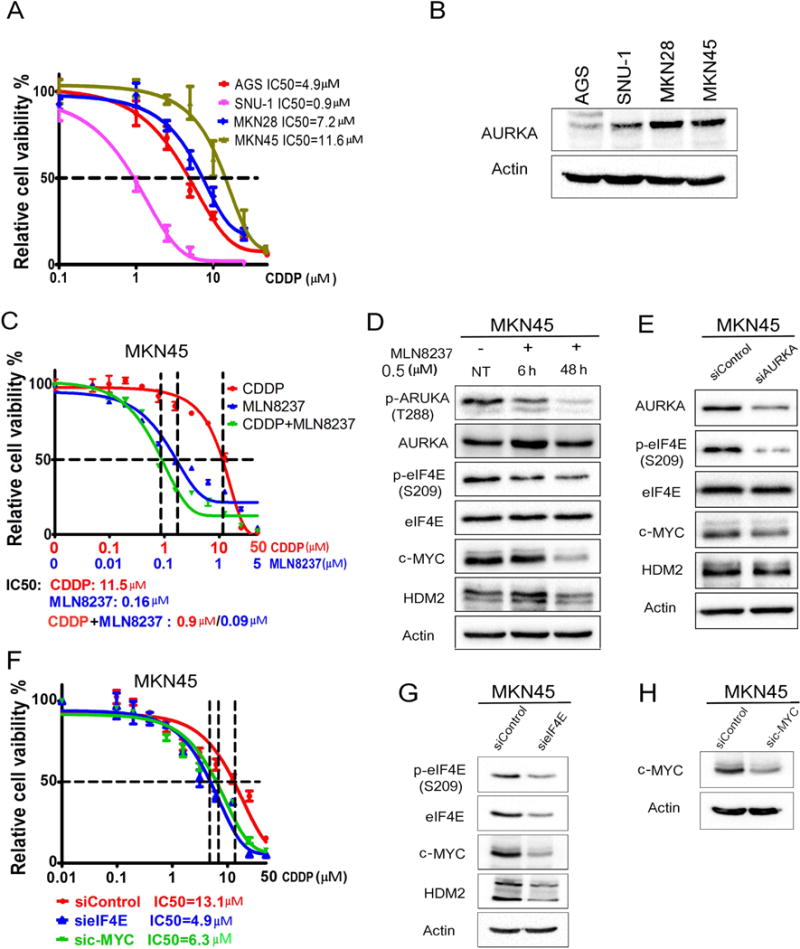
(A) Gastric cancer cells (AGS, SNU-1, MKN28, and MKN45) in 96-well plates were treated with CDDP following an 8-point 2-fold serial dilution, for 5 days. Cells were then subjected to CellTiter-Glo assay to determine cell viability. IC50 values were determined as described in Methods. (B) Protein extracts from the gastric cancer cell lines, shown in panel A, were subjected to Western blot analysis of the indicated proteins. The results indicated high protein levels of AURKA in cells lines with de novo CDDP resistance (MKN28 and MKN45). (C) CDDP-resistant MKN45 cells in 96-well plates were treated with CDDP, MLN8237, or CDDP + MLN8237 at a fixed ratio (10:1) following a 12-point 2-fold serial dilution of CDDP, for 5 days. The CellTiter-Glo assay results showed that treatment with MLN8237 alone or in combination with CDDP significantly reduced cell viability in comparison with the treatment with CDDP alone (P < 0.001). (D) MKN45 cells were treated with MLN8237 (0.5 μM) for 6h or 48h. (E) MKN45 cells were transiently transfected with siControl or siAURKA for 48h. Cell lysates were subjected to Western blot analysis of the indicated proteins. The results indicated that pharmacologic inhibition (D) or knockdown (E) of AURKA reduces protein levels of p-eIF4E (S209), c-MYC, and HDM2 in CDDP-resistant MKN45 cells. (F) MKN45 cells were transfected with siControl, sieIF4E, or sic-MYC for 48h and subjected to CellTiter-Glo viability assay. Knocking down of eIF4E or c-MYC significantly sensitized cells to CDDP (p<0.05), as indicated by 2-fold decrease in CDDP IC50. (G, H) Cell lysates were subjected to Western blot analysis of p-eIF4E (S209), eIF4E, c-MYC, and HDM2 proteins. Gel loading was normalized for equal β-actin.
3.5. MLN8237 alone or in combination with CDDP suppresses growth of resistant cells-derived xenografts in vivo
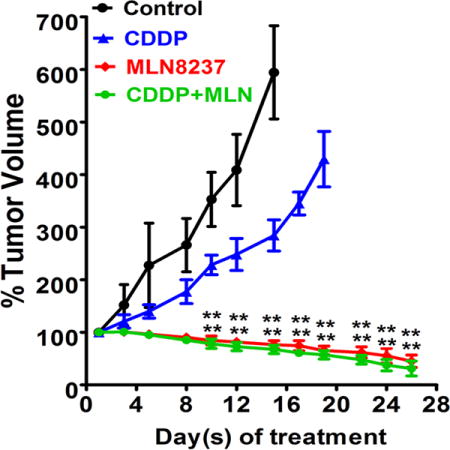
We next sought out to assess the in vivo efficacy of the MLN8237 alone or the combination of CDDP with MLN8237 using subcutaneous xenograft tumor models. The treatments were initiated after the tumor xenografts reached 150 – 200 mm3 in size; with at least 10 tumor xenografts per group. We treated the CDDP-resistant MKN45 cells-derived xenografts with CDDP alone, MLN8237 alone or in combination with CDDP, and examined the tumor growth rate and protein expression levels of eIF4E, p-eIF4E (S209), and c-MYC in xenografts. The data showed that CDDP treatment had a relatively limited negative effect on tumor growth, however, MLN8237 significantly reduced the rate of tumor growth following 4 weeks of treatments (P<0.01, Figure 6A, 6B). The addition of CDDP to MLN8237 did not enhance the anti-tumor efficacy of MLN8237 in CDDP-resistant cells. Western blot analyses data indicated that treatments with MLN8237 substantially decreased p-AURKA (T288), AURKA, p-eIF4E (S209), and c-MYC protein levels in xenografts (Figure 6C). Consistent with the decreased rate of tumor growth in response to treatment with MLN8237, the immuno-histochemical data analysis showed a significant decrease in Ki-67 (a marker of cell proliferation) and an increase in cleaved caspase-3 (a marker of apoptosis) in xenografts (P<0.001, Figure 6D). Taken together, our results strongly suggest that targeting AURKA using MLN8237 can induce tumor regression and achieve a therapeutic response in CDDP-resistant gastric cancer.
Figure 6. MLN8237 alone or in combination with CDDP effectively reduces resistant cells-derived xenograft growth in vivo.
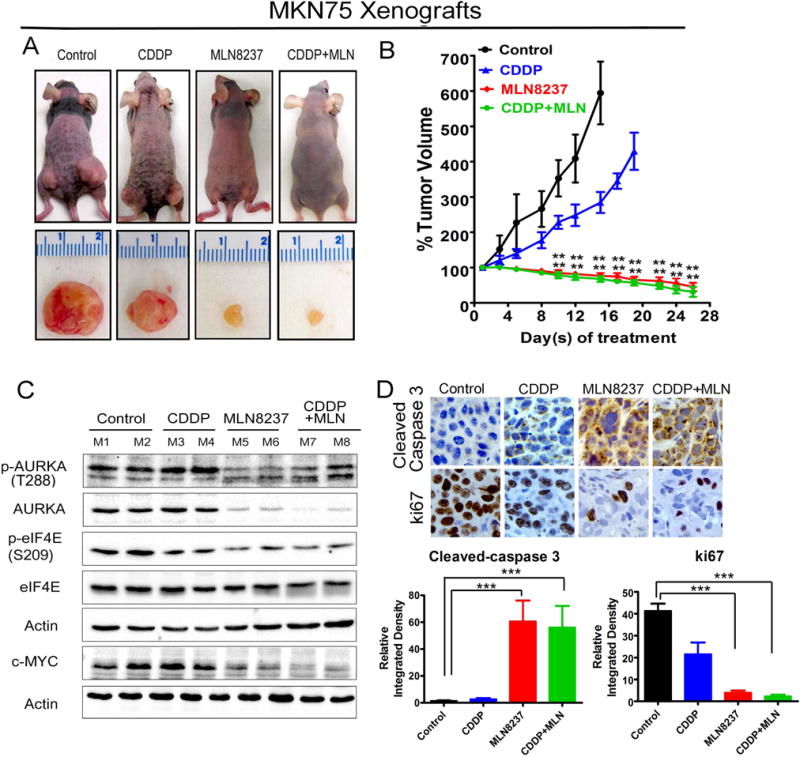
MKN45 cells-derived xenograft tumors (150–200 mm3 in size) were treated with CDDP (2.5 mg/kg, once a week) alone, MLN8237 (40 mg/kg, 5 times a week) alone or the combination of CDDP and MLN8237 for 28 days and tumor sizes were measured twice a week (A, B) The data indicated that MLN8237 alone or in combination with CDDP has a significant anti-tumor activity against resistant MKN45 cells-derived xenografts. (C) Tumor tissue lysates were subjected to Western blot analysis of the indicated proteins. (D) IHC analysis of MKN45 cells-derived xenografts showing ki67 and cleaved caspase-3 expressions in vivo. Relative integrated density indicates the quantification data of immunostaining signal which was calculated by Image J software; **p<0.01, ***p<0.001.
4. Discussion
Gastric cancer is the third cause of cancer-related deaths in the world. Resistance to chemotherapeutic agents is a major cause for gastric cancer recurrence and low patient survival rates (Ferlay et al., 2015b). Platinum therapy is a first-line chemotherapeutic agent for several cancer types, including gastric cancer. However, cancer cells eventually develop acquired resistance against cisplatin (CDDP) which adversely affects patients’ prognosis (Hainsworth et al., 1992; Kartalou and Essigmann, 2001). Therefore, identification of novel therapeutic strategies to target CDDP-resistant cancer cells has significant clinical implications for improving response and clinical outcome in gastric cancer patients. In this study, we established a link between AURKA, eIF4E, and CDDP resistance. Our data demonstrate that high levels of AURKA promote CDDP acquired and de novo resistance through eIF4E phosphorylation and up-regulation of its downstream effectors, c-MYC and HDM2 proteins. The results indicate that inhibiting AURKA can be an effective therapeutic approach to target CDDP-resistant gastric cancer cells.
Overexpression of AURKA has been associated with poor outcome and chemotherapeutic drug resistance (Sumi et al., 2011). At the same time aberrant activation of eIF4E, as a mechanism of drug resistance, has been described in several cancers (Huang et al., 2012; Liang et al., 2013). Our results indicated that AURKA inhibition or knockdown downregulated p-eIF4E (S209), c-MYC, and HDM2 proteins in acquired and de novo CDDP resistant cells. Overexpression and activation of c-MYC, by means of increased transcription, translation or protein stability, is associated with aggressive tumors and poor patient prognosis (Horiuchi et al., 2014; Schmidt, 2004). Of note, there are accumulating lines of evidence that show that AURKA can regulate c-MYC in cancer. We have previously shown that AURKA can regulate transcription of c-MYC through activation of β-catenin (Dar et al., 2009). Recent studies have shown that inhibition of AURKA can regulate phosphorylation and stability of MYC, suggesting targeting AURKA-MYC axis by AURKA inhibitors as a novel potentially effective therapeutic approach (Dauch et al., 2016; Richards et al., 2016; Silva et al., 2014). We investigated whether eIF4E and its downstream effector, c-MYC, mediate the function of AURKA and CDDP resistance. Our data demonstrate that knockdown of eIF4E or c-MYC significantly sensitized acquired or de novo resistant cells to CDDP. Therefore, our results add to the current knowledge and suggest a multifaceted regulation of c-MYC at different levels by AURKA in cancer. We and others have previously shown that AURKA can also regulate the key ubiquitin ligase involved in degradation of p53, HDM2, whereby overexpression of AURKA promoted HDM2 phosphorylation and enhanced its stability leading to degradation of p53 (Hsueh et al., 2013; Sehdev et al., 2014; Vilgelm et al., 2015). Collectively, our new findings in this report, add a significant novel perspective by showing that activation of AURKA-eIF4E axis is required for induction of c-MYC and HDM2 protein levels and CDDP resistance in gastric cancer cells. Of note, a recent study has shown that AURKA can mediate resistance to mTOR inhibition by everolimus (Katsha et al., 2017). This involved AURKA-dependent activation of eiF4E through inhibition of PP2A activity. Although, we did not investigate this signaling axis in details in CDDP resistance, it is possible that AURKA-mediated regulation of PP2A is involved in activation of eIF4E in CDDP resistance, too. Taken together, activation of AURKA-eIF4E axis in cancer cells could play a crucial role for resistance to several chemotherapeutics.
A previous report indicated that AURKA was implicated in CDDP resistance in murine bone marrow Ba/F3 cells transformed by JAK2 V617F mutant (Sumi et al., 2011). Another study indicated that AURKA suppresses CDDP-induced apoptosis through phosphorylation of p73 and its sequestration in the cytoplasm in cancer cells (Katayama et al., 2012b). Of note, earlier reports have shown that AURKA can suppress p73 (Dar et al., 2008a; Katayama et al., 2012a) and activate STAT3, β-catenin, and NF-κB (Dar et al., 2009; Katsha et al., 2014; Katsha et al., 2013). Although we have not specifically investigated these pathways in the present study, it is plausible that targeting AURKA in CDDP-resistant cancer models may induce cancer cells death not only through suppression of eiF4E-c-MYC-HDM2, but also through reversing these signaling effects.
Conclusions
In summary, we present evidence for a novel mechanism by which AURKA promotes resistance to CDDP through activation of p-eIF4E, c-MYC, and HDM2. Targeting AURKA using MLN8237 may present a clinically relevant opportunity to treat CDDP-resistant tumors and enhance the therapeutic response in gastric cancer patients.
Acknowledgments
The authors thank Mr. Frank Revetta for assistance in immunohistochemistry and Ms. Michele Smith for proof reading the manuscript.
Funding
This study was supported by grants from the National Cancer Institute (R01CA131225), Department of Veterans Affairs, Vanderbilt SPORE in Gastrointestinal Cancer (P50 CA95103), Vanderbilt Ingram Cancer Center (P30 CA68485), and the Vanderbilt Digestive Disease Research Center (DK058404). The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute, Department of Veterans Affairs, or Vanderbilt University Medical Center.
Abbreviations
- AURKA
Aurora kinase A
- MLN8237
Alisertib
- CDDP
Cisplatin or cis-diamminedichloroplatinum
- eIF4E
The eukaryotic translation initiation factor 4E
- CDDPR
Cisplatin resistant
- HDM2
Human double minute 2
- siAURKA
small interfering RNA of AURKA
- siControl
small interfering RNA of scramble
- sieIF4E
small interfering RNA of eIF4E
Footnotes
Authors’ contributions
LW, and JA performed the main experiments, summarized the results, and wrote the manuscript. AK, and SH assisted in performing the experiments. AB assisted in interpretation and troubleshooting of the data. JE assisted in obtaining MLN8237 and in revising the manuscript. WER provided the concept, funding, supervision, and assisted in writing the manuscript. All authors read and approved the final manuscript.
Declarations
Competing interests
The authors declare no conflict of interest.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Availability of data and material
Not applicable
Competing interests
The authors declare that they have no competing interests
Contributor Information
Lihong Wang, Email: lihong.wang@Vanderbilt.Edu.
Janet Arras, Email: JA6VY@hscmail.mcc.virginia.edu.
Ahmed Katsha, Email: akatsha@gmail.com.
Saif Hamdan, Email: saif.a.hamdan@vanderbilt.edu.
Abbes Belkhiri, Email: abbes.belkhiri@vanderbilt.edu.
Jeffrey Ecsedy, Email: Jeffrey.Ecsedy@takeda.com.
Wael El-Rifai, Email: wael.el-rifai@vanderbilt.edu.
References
- Aradottir M, Reynisdottir ST, Stefansson OA, Jonasson JG, Sverrisdottir A, Tryggvadottir L, Eyfjord JE, Bodvarsdottir SK. Aurora A is a prognostic marker for breast cancer arising in BRCA2 mutation carriers. J Pathol Clin Res. 2015;1:33–40. doi: 10.1002/cjp2.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone DN, Qi Y, Li Z, Hann SR. Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:632–637. doi: 10.1073/pnas.1008848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CN, Hsieh FJ, Cheng YM, Lee PH, Chang KJ. Expression of eukaryotic initiation factor 4E in gastric adenocarcinoma and its association with clinical outcome. Journal of surgical oncology. 2004;86:22–27. doi: 10.1002/jso.20037. [DOI] [PubMed] [Google Scholar]
- Cirak Y, Furuncuoglu Y, Yapicier O, Aksu A, Cubukcu E. Aurora A overexpression in breast cancer patients induces taxane resistance and results in worse prognosis. J Buon. 2015;20:1414–1419. [PubMed] [Google Scholar]
- Dar AA, Belkhiri A, Ecsedy J, Zaika A, El-Rifai W. Aurora kinase A inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells. Cancer research. 2008a;68:8998–9004. doi: 10.1158/0008-5472.CAN-08-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AA, Belkhiri A, El-Rifai W. The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene. 2009;28:866–875. doi: 10.1038/onc.2008.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AA, Zaika A, Piazuelo MB, Correa P, Koyama T, Belkhiri A, Washington K, Castells A, Pera M, El-Rifai W. Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer. 2008b;112:1688–1698. doi: 10.1002/cncr.23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauch D, Rudalska R, Cossa G, Nault JC, Kang TW, Wuestefeld T, Hohmeyer A, Imbeaud S, Yevsa T, Hoenicke L, Pantsar T, Bozko P, Malek NP, Longerich T, Laufer S, Poso A, Zucman-Rossi J, Eilers M, Zender L. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nature medicine. 2016;22:744–753. doi: 10.1038/nm.4107. [DOI] [PubMed] [Google Scholar]
- Dos Santos EO, Carneiro-Lobo TC, Aoki MN, Levantini E, Basseres DS. Aurora kinase targeting in lung cancer reduces KRAS-induced transformation. Molecular cancer. 2016;15:12. doi: 10.1186/s12943-016-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z, Xiong Y, Li J, Liu L, Li M, Zhang C, Zhang W, Wan J. Copy-number increase of AURKA in gastric cancers in a Chinese population: a correlation with tumor progression. Medical oncology. 2011;28:1017–1022. doi: 10.1007/s12032-010-9602-4. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015a;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. Journal international du cancer. 2015b;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Sunamura M, Horii A. Molecular mechanisms of pancreatic carcinogenesis. Cancer science. 2006;97:1–7. doi: 10.1111/j.1349-7006.2005.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, McNulty AM, Parsons SH, Brail LH, Colligan BM, Koop JW, Hurst BM, Deddens JA, Neubauer BL, Stancato LF, Carter HW, Douglass LE, Carter JH. eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer research. 2009;69:3866–3873. doi: 10.1158/0008-5472.CAN-08-3472. [DOI] [PubMed] [Google Scholar]
- Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV, Cheng JQ. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9:1420–1426. [PubMed] [Google Scholar]
- Hainsworth JD, Johnson DH, Greco FA. Cisplatin-based combination chemotherapy in the treatment of poorly differentiated carcinoma and poorly differentiated adenocarcinoma of unknown primary site: results of a 12-year experience. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1992;10:912–922. doi: 10.1200/JCO.1992.10.6.912. [DOI] [PubMed] [Google Scholar]
- Hohenberger P, Gretschel S. Gastric cancer. Lancet. 2003;362:305–315. doi: 10.1016/s0140-6736(03)13975-x. [DOI] [PubMed] [Google Scholar]
- Horiuchi D, Anderton B, Goga A. Taking on challenging targets: making MYC druggable. American Society of Clinical Oncology educational book. American Society of Clinical Oncology Meeting. 2014:e497–502. doi: 10.14694/EdBook_AM.2014.34.e497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh KW, Fu SL, Chang CB, Chang YL, Lin CH. A novel Aurora-A-mediated phosphorylation of p53 inhibits its interaction with MDM2. Biochimica et biophysica acta. 2013;1834:508–515. doi: 10.1016/j.bbapap.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Huang S, Chen M, Shen Y, Shen W, Guo H, Gao Q, Zou X. Inhibition of activated Stat3 reverses drug resistance to chemotherapeutic agents in gastric cancer cells. Cancer letters. 2012;315:198–205. doi: 10.1016/j.canlet.2011.10.011. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Jones RM, MacDonald ME, Branda J, Altherr MR, Louis DN, Schmidt EV. Assignment of the human gene encoding eukaryotic initiation factor 4E (EIF4E) to the region q21–25 on chromosome 4. Somatic cell and molecular genetics. 1997;23:221–223. doi: 10.1007/BF02721373. [DOI] [PubMed] [Google Scholar]
- Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutation research. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- Katayama H, Ota T, Jisaki F, Ueda Y, Tanaka T, Odashima S, Suzuki F, Terada Y, Tatsuka M. Mitotic kinase expression and colorectal cancer progression. Journal of the National Cancer Institute. 1999;91:1160–1162. doi: 10.1093/jnci/91.13.1160. [DOI] [PubMed] [Google Scholar]
- Katayama H, Wang J, Treekitkarnmongkol W, Kawai H, Sasai K, Zhang H, Wang H, Adams HP, Jiang S, Chakraborty SN, Suzuki F, Arlinghaus RB, Liu J, Mobley JA, Grizzle WE, Sen S. Aurora kinase-A inactivates DNA damage-induced apoptosis and spindle assembly checkpoint response functions of p73. Cancer cell. 2012a;21:196–211. doi: 10.1016/j.ccr.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Wang J, Treekitkarnmongkol W, Kawai H, Sasai K, Zhang H, Wang H, Adams HP, Jiang S, Chakraborty SN, Suzuki F, Arlinghaus RB, Liu J, Mobley JA, Grizzle WE, Wang H, Sen S. Aurora kinase-A inactivates DNA damage-induced apoptosis and spindle assembly checkpoint response functions of p73. Cancer cell. 2012b;21:196–211. doi: 10.1016/j.ccr.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsha A, Arras J, Soutto M, Belkhiri A, El-Rifai W. AURKA regulates JAK2-STAT3 activity in human gastric and esophageal cancers. Mol Oncol. 2014;8:1419–1428. doi: 10.1016/j.molonc.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsha A, Belkhiri A, Goff L, El-Rifai W. Aurora kinase A in gastrointestinal cancers: time to target. Molecular cancer. 2015;14:106. doi: 10.1186/s12943-015-0375-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsha A, Soutto M, Sehdev V, Peng D, Washington MK, Piazuelo MB, Tantawy MN, Manning HC, Lu P, Shyr Y, Ecsedy J, Belkhiri A, El-Rifai W. Aurora kinase A promotes inflammation and tumorigenesis in mice and human gastric neoplasia. Gastroenterology. 2013;145:1312–1322. e1311–1318. doi: 10.1053/j.gastro.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsha A, Wang L, Arras J, Omar OM, Ecsedy JA, Belkhiri A, El-Rifai W. Activation of EIF4E by Aurora kinase A depicts a novel druggable axis in everolimus resistant cancer cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017 doi: 10.1158/1078-0432.CCR-16-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraljacic BC, Arguello M, Amri A, Cormack G, Borden K. Inhibition of eIF4E with ribavirin cooperates with common chemotherapies in primary acute myeloid leukemia specimens. Leukemia. 2011;25:1197–1200. doi: 10.1038/leu.2011.57. [DOI] [PubMed] [Google Scholar]
- Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nature reviews. Cancer. 2010;10:825–841. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- Liang S, Guo R, Zhang Z, Liu D, Xu H, Xu Z, Wang X, Yang L. Upregulation of the eIF4E signaling pathway contributes to the progression of gastric cancer, and targeting eIF4E by perifosine inhibits cell growth. Oncology reports. 2013;29:2422–2430. doi: 10.3892/or.2013.2397. [DOI] [PubMed] [Google Scholar]
- Liu J, Stevens PD, Eshleman NE, Gao T. Protein phosphatase PPM1G regulates protein translation and cell growth by dephosphorylating 4E binding protein 1 (4E-BP1) The Journal of biological chemistry. 2013;288:23225–23233. doi: 10.1074/jbc.M113.492371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Makala L, Wu D, Cai Y. Targeting translation: eIF4E as an emerging anticancer drug target. Expert reviews in molecular medicine. 2016;18:e2. doi: 10.1017/erm.2015.20. [DOI] [PubMed] [Google Scholar]
- Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N. eIF4E–from translation to transformation. Oncogene. 2004;23:3172–3179. doi: 10.1038/sj.onc.1207549. [DOI] [PubMed] [Google Scholar]
- Martinez-Marignac V, Shawi M, Pinedo-Carpio E, Wang X, Panasci L, Miller W, Pettersson F, Aloyz R. Pharmacological targeting of eIF4E in primary CLL lymphocytes. Blood cancer journal. 2013;3:e146. doi: 10.1038/bcj.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignogna C, Staropoli N, Botta C, De Marco C, Rizzuto A, Morelli M, Di Cello A, Franco R, Camastra C, Presta I, Malara N, Salvino A, Tassone P, Tagliaferri P, Barni T, Donato G, Di Vito A. Aurora Kinase A expression predicts platinum-resistance and adverse outcome in high-grade serous ovarian carcinoma patients. Journal of ovarian research. 2016;9:31. doi: 10.1186/s13048-016-0238-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson F, Del Rincon SV, Emond A, Huor B, Ngan E, Ng J, Dobocan MC, Siegel PM, Miller WH., Jr Genetic and pharmacologic inhibition of eIF4E reduces breast cancer cell migration, invasion, and metastasis. Cancer research. 2015;75:1102–1112. doi: 10.1158/0008-5472.CAN-14-1996. [DOI] [PubMed] [Google Scholar]
- Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, Bayliss R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2016 doi: 10.1073/pnas.1610626113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EV. The role of c-myc in regulation of translation initiation. Oncogene. 2004;23:3217–3221. doi: 10.1038/sj.onc.1207548. [DOI] [PubMed] [Google Scholar]
- Sehdev V, Katsha A, Arras J, Peng D, Soutto M, Ecsedy J, Zaika A, Belkhiri A, El-Rifai W. HDM2 regulation by AURKA promotes cell survival in gastric cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:76–86. doi: 10.1158/1078-0432.CCR-13-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehdev V, Peng D, Soutto M, Washington MK, Revetta F, Ecsedy J, Zaika A, Rau TT, Schneider-Stock R, Belkhiri A, El-Rifai W. The aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell death in esophageal adenocarcinoma cells. Molecular cancer therapeutics. 2012;11:763–774. doi: 10.1158/1535-7163.MCT-11-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui N, Sonenberg N. Signalling to eIF4E in cancer. Biochemical Society transactions. 2015;43:763–772. doi: 10.1042/BST20150126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A, Wang J, Lomahan S, Tran TA, Grenlin L, Suganami A, Tamura Y, Ikegaki N. Aurora kinase A is a possible target of OSU03012 to destabilize MYC family proteins. Oncology reports. 2014;32:901–905. doi: 10.3892/or.2014.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi K, Tago K, Kasahara T, Funakoshi-Tago M. Aurora kinase A critically contributes to the resistance to anti-cancer drug cisplatin in JAK2 V617F mutant-induced transformed cells. FEBS Lett. 2011;585:1884–1890. doi: 10.1016/j.febslet.2011.04.068. [DOI] [PubMed] [Google Scholar]
- Van Cutsem E, Van de Velde C, Roth A, Lordick F, Kohne CH, Cascinu S, Aapro M, European Organisation for, R., Treatment of Cancer -gastrointestinal cancer, g Expert opinion on management of gastric and gastro-oesophageal junction adenocarcinoma on behalf of the European Organisation for Research and Treatment of Cancer (EORTC)-gastrointestinal cancer group. European journal of cancer. 2008;44:182–194. doi: 10.1016/j.ejca.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, Weller KP, Horton LW, McClain CM, Ayers GD, Turner DC, Essaka DC, Stewart CF, Sosman JA, Kelley MC, Ecsedy JA, Johnston JN, Richmond A. Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer research. 2015;75:181–193. doi: 10.1158/0008-5472.CAN-14-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Teruya-Feldstein J, Pelletier J, Lowe SW. Dissecting eIF4E action in tumorigenesis. Genes & development. 2007;21:3232–3237. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Dahabieh MS, Rajakumar A, Dobocan MC, M’Boutchou MN, Goncalves C, Shiru LL, Pettersson F, Topisirovic I, van Kempen L, Del Rincon SV, Miller WH., Jr The role of eIF4E in response and acquired resistance to vemurafenib in melanoma. The Journal of investigative dermatology. 2015;135:1368–1376. doi: 10.1038/jid.2015.11. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Sun T, Kang P, Qian K, Deng B, Zhou J, Wang R, Jiang B, Li K, Liu F, Wu S, Tan Q. Combined analysis of rearrangement of ALK, ROS1, somatic mutation of EGFR, KRAS, BRAF, PIK3CA, and mRNA expression of ERCC1, TYMS, RRM1, TUBB3, EGFR in patients with non-small cell lung cancer and their clinical significance. Cancer chemotherapy and pharmacology. 2016;77:583–593. doi: 10.1007/s00280-016-2969-y. [DOI] [PubMed] [Google Scholar]
- Zheng F, Yue C, Li G, He B, Cheng W, Wang X, Yan M, Long Z, Qiu W, Yuan Z, Xu J, Liu B, Shi Q, Lam EW, Hung MC, Liu Q. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nature communications. 2016;7:10180. doi: 10.1038/ncomms10180. [DOI] [PMC free article] [PubMed] [Google Scholar]